
Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging


Abstract
:1. Introduction
2. Conventional Adrenocortical Carcinoma
2.1. Demographic Characteristics
2.2. Clinical Features
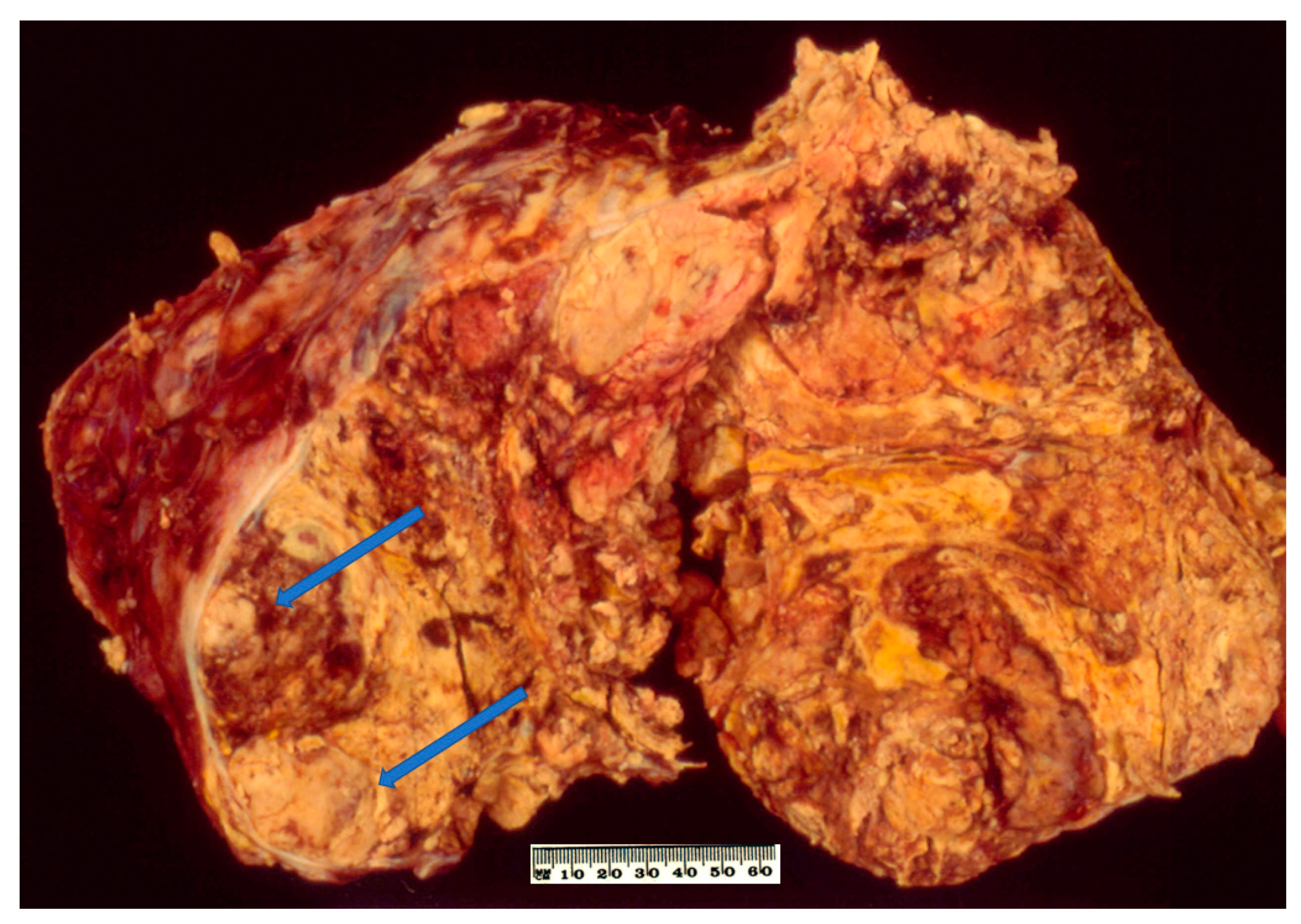
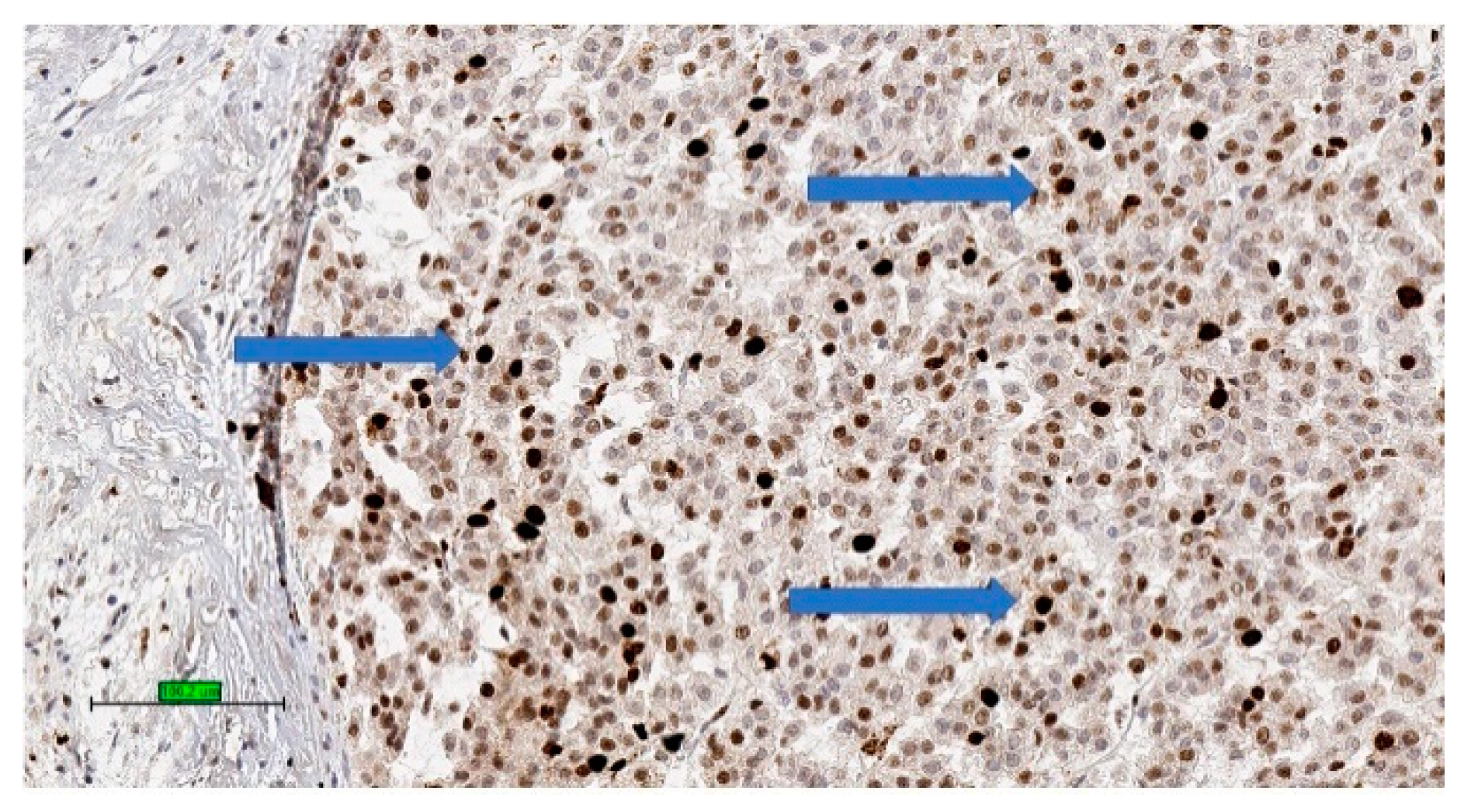
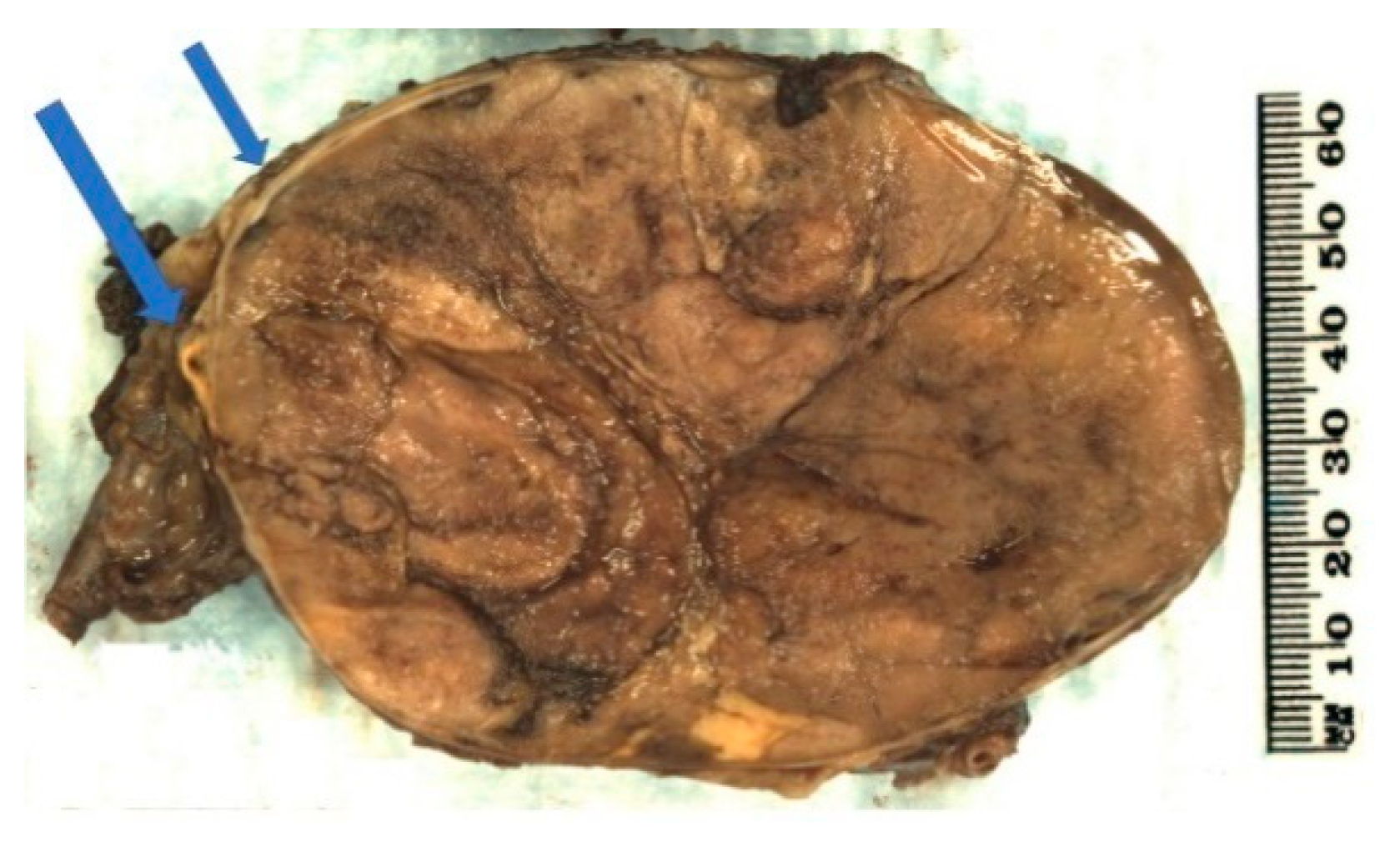
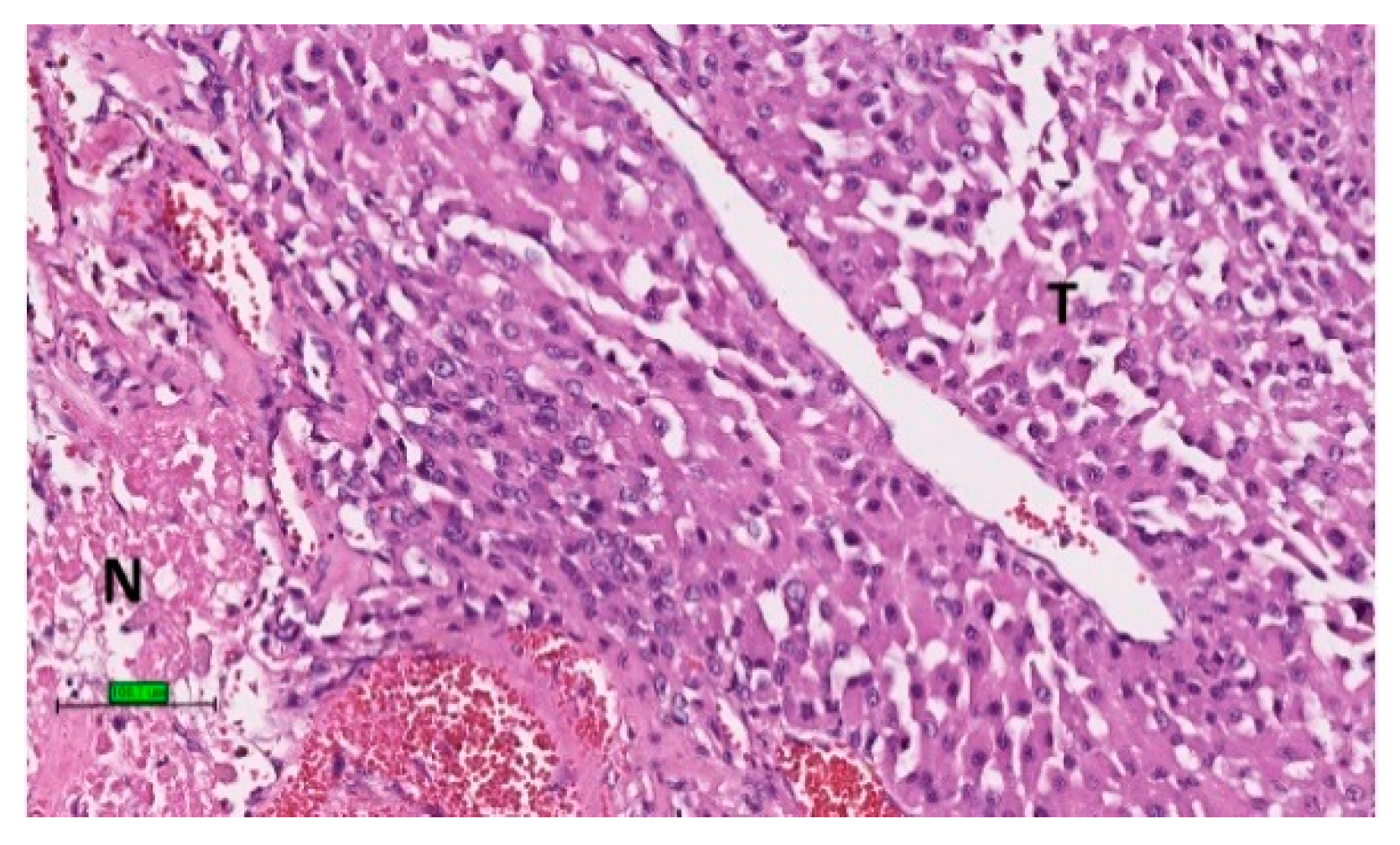
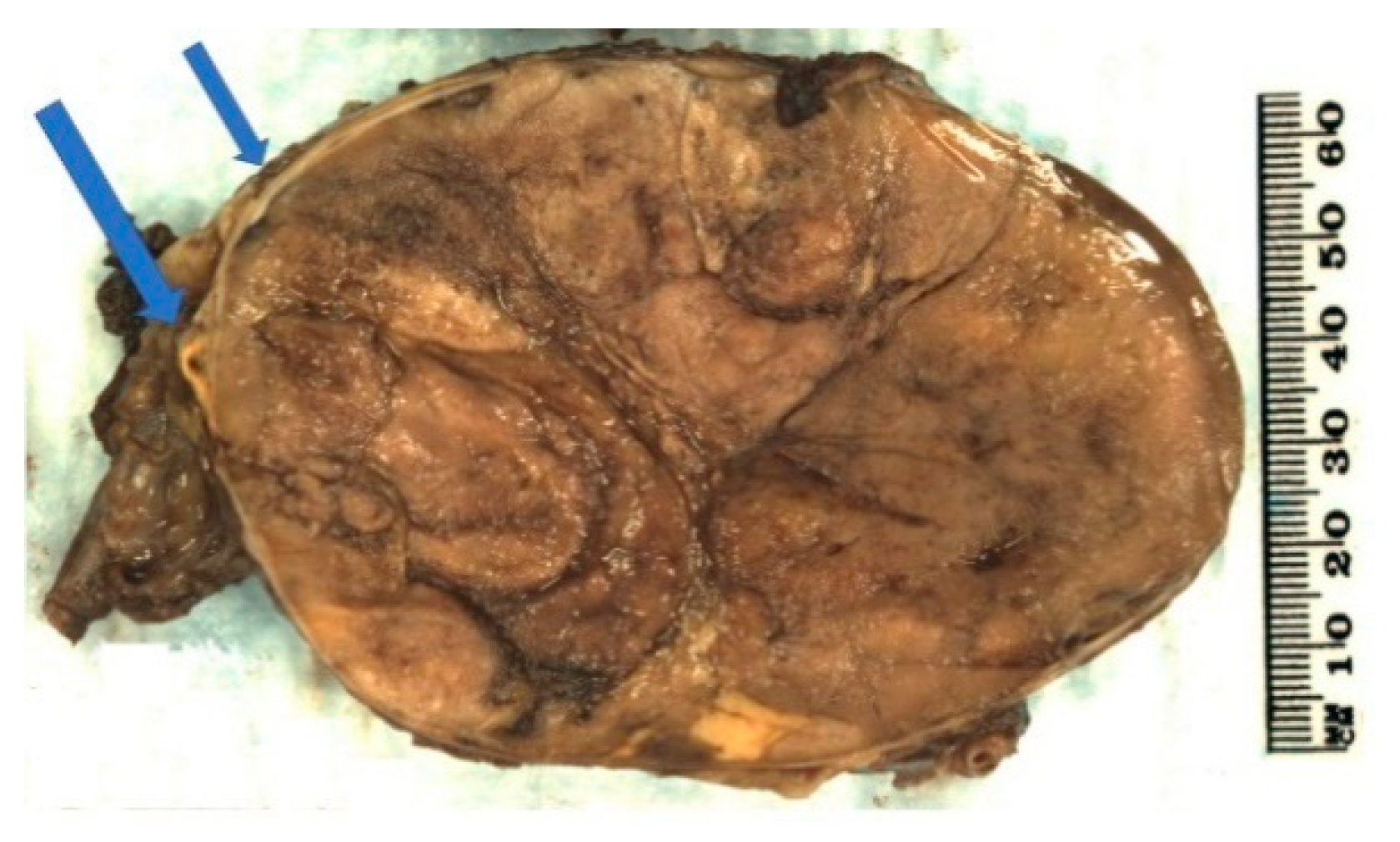
2.3. Pathology
2.4. Prognosis
3. Paediatric Adrenocortical Carcinoma
3.1. Demographic Characteristics
3.2. Clinical Features
3.3. Pathology
3.4. Prognosis
4. Oncocytic Adrenocortical Carcinoma
4.1. Demographic Characteristics
4.2. Clinical Features
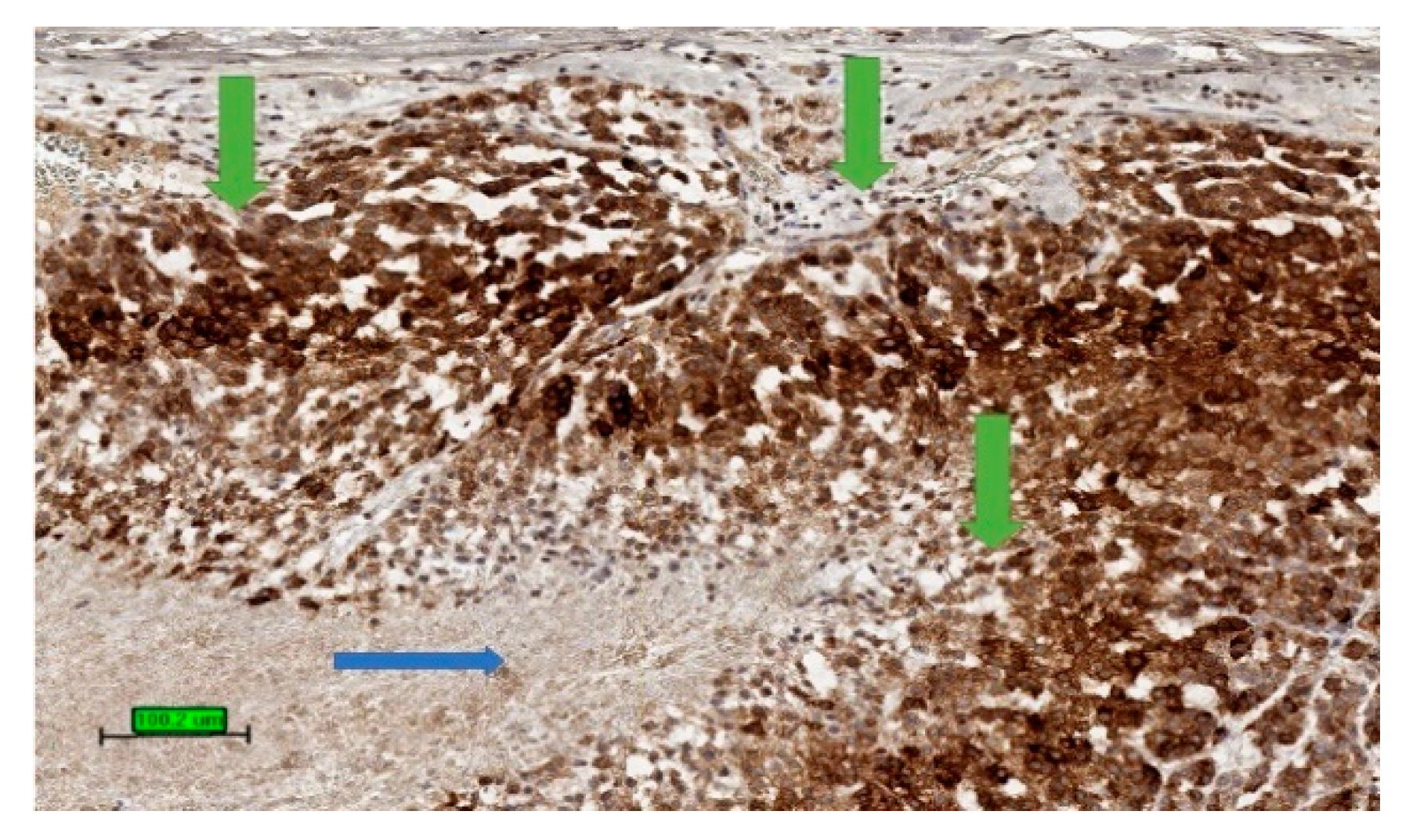
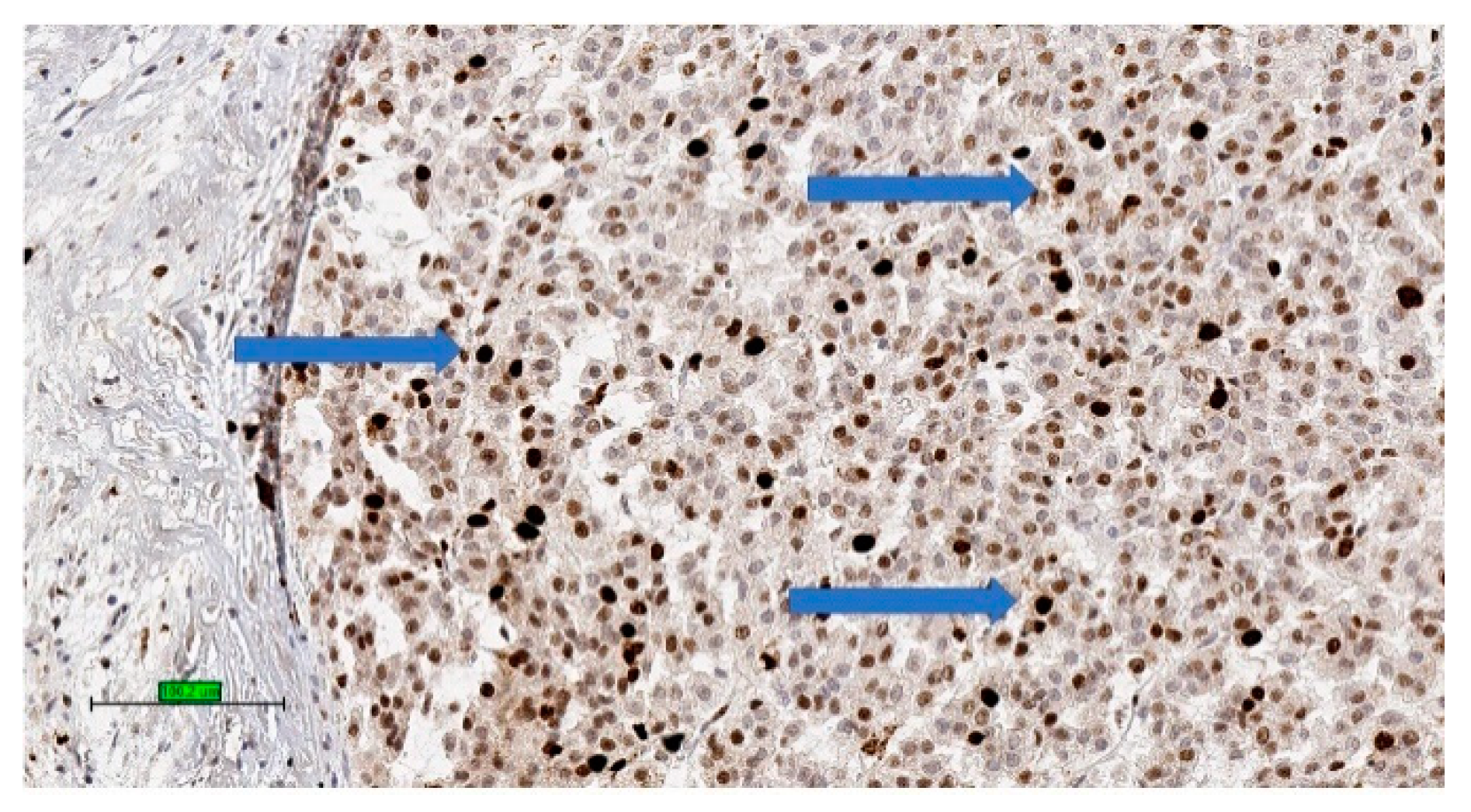
4.3. Pathology
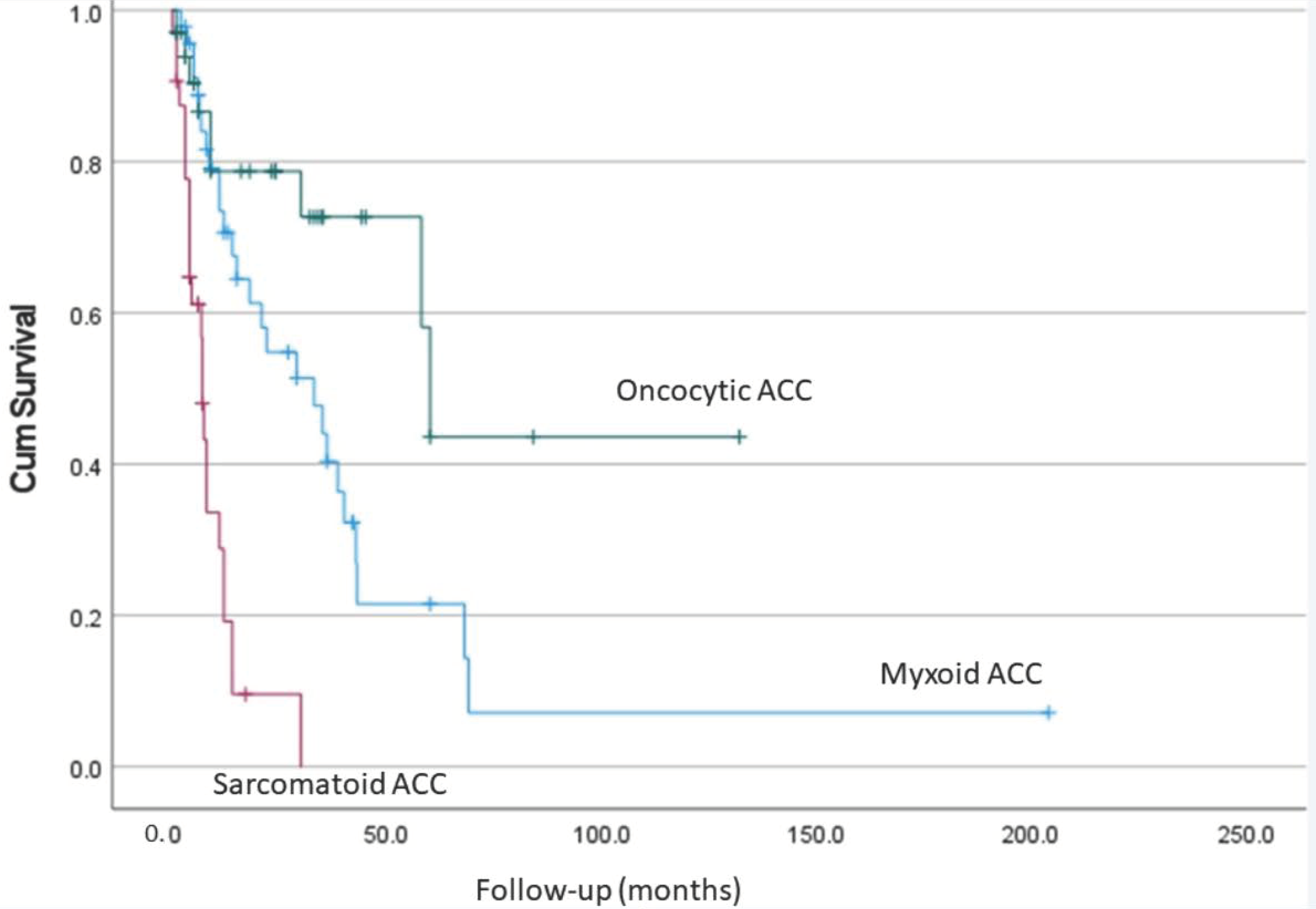
4.4. Prognosis
5. Myxoid Adrenocortical Carcinoma
5.1. Demographic Characteristics
5.2. Clinical Features
5.3. Pathology
5.4. Prognosis
6. Sarcomatoid Adrenocortical Carcinoma
6.1. Demographic Characteristics
6.2. Clinical Features
6.3. Pathology
6.4. Prognosis
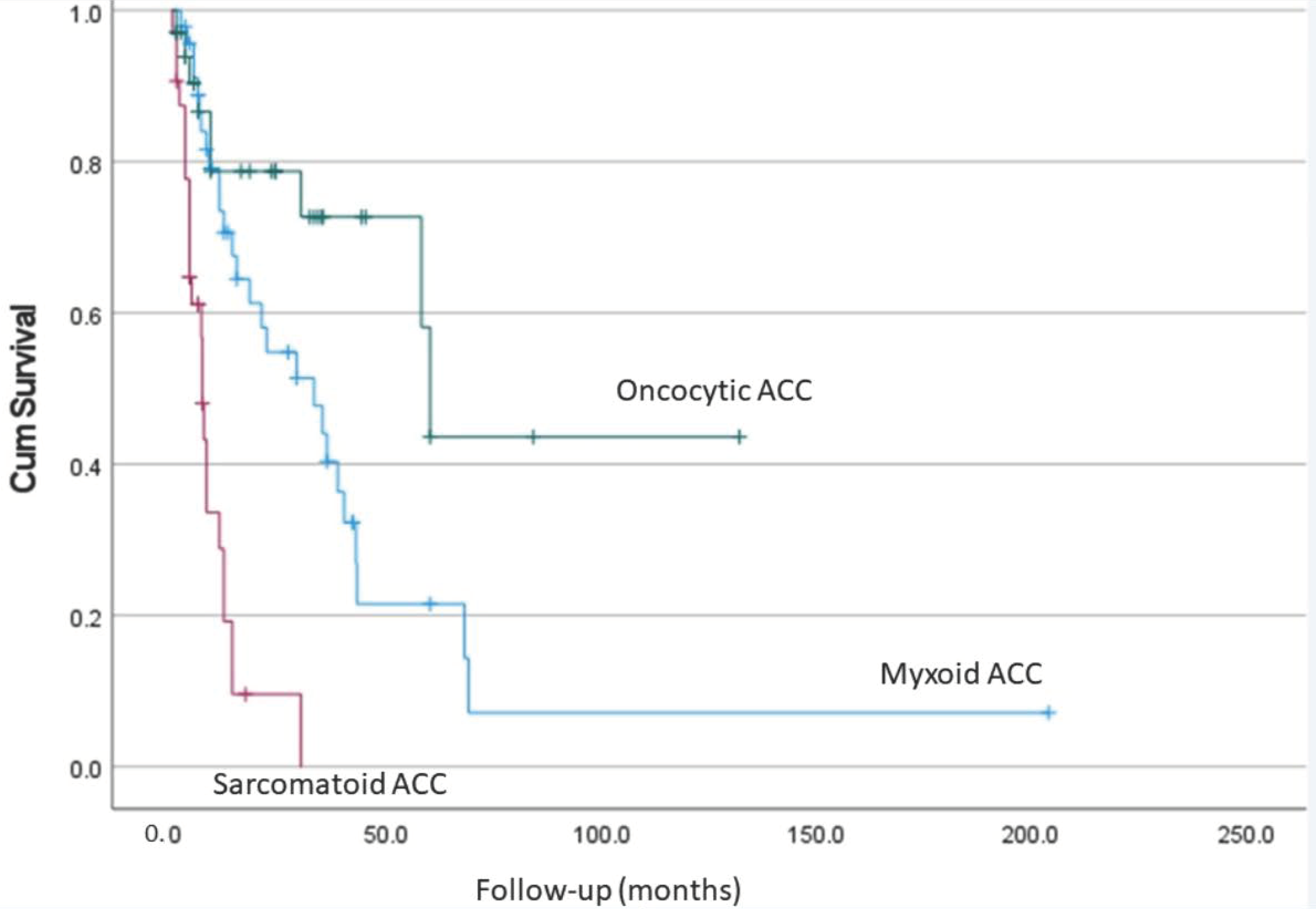
7. Comparison of Different Types of Adrenocortical Carcinoma
8. Tumour Staging
9. Pathological Reporting of Adrenocortical Carcinoma
10. Clinical Perspectives
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chandrasekar, T.; Goldberg, H.; Klaassen, Z.; Wallis, C.J.D.; Woon, D.T.S.; Herrera-Caceres, J.O.; Kulkarni, G.S.; Fleshner, N.E. The who, when, and why of primary adrenal malignancies: Insights into the epidemiology of a rare clinical entity. Cancer 2019, 125, 1050–1059. [Google Scholar] [CrossRef]
- Abe, I.; Lam, A.K. Anaplastic thyroid carcinoma: Updates on WHO classification, clinicopathological features and staging. Histol. Histopathol. 2020, 2021, 18277. [Google Scholar] [CrossRef]
- Lam, K.Y. Adrenal tumours in Chinese. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 421, 13–16. [Google Scholar] [CrossRef]
- Sharma, E.; Dahal, S.; Sharma, P.; Bhandari, A.; Gupta, V.; Amgai, B.; Dahal, S. The Characteristics and Trends in Adrenocortical Carcinoma: A United States Population Based Study. J. Clin. Med. Res. 2018, 10, 636–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichijo, T.; Ueshiba, H.; Nawata, H.; Yanase, T. A nationwide survey of adrenal incidentalomas in Japan: The first report of clinical and epidemiological features. Endocr. J. 2020, 67, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Rahane, C.S.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet. 2019, 230, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pinto, E.M.; Chen, X.; Easton, J.; Finkelstein, D.; Liu, Z.; Pounds, S.; Rodriguez-Galindo, C.; Lund, T.C.; Mardis, E.R.; Wilson, R.K.; et al. Genomic landscape of paediatric adrenocortical tumours. Nat. Commun. 2015, 6, 6302. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Huff, L.; Litman, T.; Im, K.; Edgerly, M.; Del Rivero, J.; Pittaluga, S.; Merino, M.; Bates, S.E.; Dean, M. Metastatic and recurrent adrenocortical cancer is not defined by its genomic landscape. BMC Med. Genom. 2020, 13, 165. [Google Scholar] [CrossRef]
- Ettaieb, M.H.T.; Kerkhofs, T.; Van Engeland, M.; Haak, H.R. Past, Present and Future of Epigenetics in Adrenocortical Carcinoma. Cancers 2020, 12, 1218. [Google Scholar] [CrossRef]
- Phan, A.T.; Grogan, R.H.; Rohren, E.; Perrier, N.D. Adrenal cortical carcinoma. In AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Edge, S., Greene, F., Byrd, D.R., Brookland, R.K., Washington, M.K., Gershenwald, J.E., Compton, C.C., Hess, K.R., Sullivan, D.C., Eds.; Springer: New York, NY, USA, 2016; Part XVII, Chapter 76; pp. 919–926. [Google Scholar]
- Lam, A.K.-Y. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef]
- Ettaieb, M.H.T.; Van Kuijk, S.M.J.; De Wit-Pastoors, A.; A Feelders, R.; Corssmit, E.P.M.; Eekhoff, E.M.W.; Van Der Valk, P.; Timmers, H.; Kerstens, M.N.; Klümpen, H.-J.; et al. Development and Internal Validation of a Multivariable Prediction Model for Adrenocortical-Carcinoma-Specific Mortality. Cancers 2020, 12, 2720. [Google Scholar] [CrossRef]
- Wanis, K.N.; Kanthan, R. Diagnostic and prognostic features in adrenocortical carcinoma: A single institution case series and review of the literature. World J. Surg. Oncol. 2015, 13, 117. [Google Scholar] [CrossRef] [Green Version]
- Sada, A.; Asaad, M.; Bews, K.A.; Thompson, G.B.; Young, W.F.; Bancos, I.; Farley, D.R.; Dy, B.M.; Lyden, M.L.; Habermann, E.B.; et al. Comparison between functional and non-functional adrenocortical carcinoma. Surgery 2020, 167, 216–223. [Google Scholar] [CrossRef]
- Jeong, Y.; Cho, S.C.; Cho, H.J.; Song, J.S.; Kong, J.S.; Park, J.W.; Ku, Y.H. Estrogen-secreting adrenocortical carcinoma. Yeungnam Univ. J. Med. 2019, 36, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umphonsathien, M.; Srichomkwun, P.; Buranasupkajorn, P.; Snabboon, T. The coexistence of Cushing syndrome and gynecomastia as the manifestations of adrenocortical carcinoma. Kaohsiung J. Med. Sci. 2018, 34, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xie, J.; Jiang, L.; Su, T.; Ye, L.; Zhou, W.; Jiang, Y.; Zhang, C.; Ning, G.; Wang, W. Feminizing Adrenocortical Carcinoma: The Source of Estrogen Production and the Role of Adrenal-Gonadal Dedifferentiation. J. Clin. Endocrinol. Metab. 2018, 103, 3706–3713. [Google Scholar] [CrossRef]
- Chentli, F.; Chabour, F.; Bouchibane, D.; Nouar, N. Feminizing Adrenocortical Carcinoma without Gynecomastia. Oman Med. J. 2017, 32, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Takenaka, Y.; Inoue, I.; Homma, K.; Hasegawa, T.; Sasano, H.; Awata, T.; Katayama, S. Feminizing Adrenocortical Carcinoma with Distinct Histopathological Findings. Intern. Med. 2016, 55, 3301–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, D.; Nyantoro, M.; Shedzad, H.; Robins, P.; Henley, D.; Ryan, S.; Nguyen, H.; Lisewski, D. Management of adrenocortical carcinoma in Western Australia: A perspective over 14 years. ANZ J. Surg. 2021, in press. [Google Scholar] [CrossRef]
- Karimi, F.; Dehghanian, A.; Fallahi, M.; Dalfardi, B. Pure Androgen-Secreting Adrenocortical Carcinoma Presenting with Hypoglycemia. Arch. Iran Med. 2019, 22, 527–530. [Google Scholar]
- Dilrukshi, M.D.S.A.; Wickramarachchi, A.W.; Abeyaratne, D.D.K.; Shine, B.; Jafar-Mohammadi, B.; Somasundaram, N.P. An Adrenocortical Carcinoma Associated with Non-Islet Cell Tumor Hypoglycemia and Aberrant ACTH Production. Case Rep. Endocrinol. 2020, 2020, 2025631. [Google Scholar] [CrossRef]
- De Nattes, T.; Moreau-Grangé, L.; Vezzosi, D.; Haddoux, J.; Hie, M.; Guerrot, D.; Grangé, S. Adrenocortical carcinoma complicated by renal thrombotic microangiopathy, a case-series. BMC Nephrol. 2020, 21, 35. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, S.; Amano, R.; Onoda, N.; Noda, S.; Hirata, K.; Asano, Y.; Kurata, K.; Miura, K.; Yamazoe, S.; Kimura, K.; et al. Nonfunctional adrenocortical carcinoma initially presenting as retroperitoneal hemorrhage. BMC Surg. 2015, 15, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symeonidis, D.; Chatzinikolaou, I.; Koukoulis, G.; Mamaloudis, I.; Tepetes, K. Adrenocortical Carcinoma Presenting with Signs of Acute Abdomen. Case Rep. Surg. 2013, 2013, 132726. [Google Scholar] [CrossRef]
- Jagtap, S.V.; Desai, S.; Halder, S.; Jagtap, S.S.; Badwe, A.S. Adrenocortical Carcinoma Presenting as A Rupture and Extensive Retroperitoneal Haemorrhage. J. Clin. Diagn. Res. 2014, 8, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Kamilaris, C.D.; Hannah-Shmouni, F.; Stratakis, C.A. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef] [PubMed]
- Simonds, W.F.; Varghese, S.; Marx, S.J.; Nieman, L.K. Cushing’s syndrome in multiple endocrine neoplasia type 1. Clin. Endocrinol. 2012, 76, 379–386. [Google Scholar] [CrossRef]
- Domènech, M.; Grau, E.; Solanes, A.; Izquierdo, A.; Del Valle, J.; Carrato, C.; Pineda, M.; Dueñas, N.; Pujol, M.; Lázaro, C.; et al. Characteristics of Adrenocortical Carcinoma Associated With Lynch Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 318–325. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Brondani, V.B.; Helena, V.P.; Charchar, H.L.S.; Zerbini, M.C.N.; Leite, L.A.S.; Hoff, A.O.; Latronico, A.C.; Mendonca, B.B.; Diz, M.D.P.E.; et al. Clinical spectrum of Li-Fraumeni syndrome/Li-Fraumeni-like syndrome in Brazilian individuals with the TP53 p.R337H mutation. J. Steroid Biochem. Mol. Biol. 2019, 190, 250–255. [Google Scholar] [CrossRef]
- Menon, R.K.; Ferrau, F.; Kurzawinski, T.R.; Rumsby, G.; Freeman, A.; Amin, Z.; Korbonits, M.; Chung, T.-T.L.L. Adrenal cancer in neurofibromatosis type 1: Case report and DNA analysis. Endocrinol. Diabetes Metab. Case Rep. 2014, 2014, 140074. [Google Scholar] [CrossRef] [Green Version]
- Morin, E.; Mete, O.; Wasserman, J.D.; Joshua, A.M.; Asa, S.L.; Ezzat, S. Carney Complex with Adrenal Cortical Carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E202–E206. [Google Scholar] [CrossRef]
- Anselmo, J.; Medeiros, S.; Carneiro, V.; Greene, E.; Levy, I.; Nesterova, M.; Lyssikatos, C.; Horvath, A.; Carney, J.A.; Stratakis, C.A. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J. Clin. Endocrinol. Metab. 2012, 97, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traill, Z.; Tuson, J.; Woodham, C. Adrenal carcinoma in a patient with Gardner’s syndrome: Imaging findings. Am. J. Roentgenol. 1995, 165, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Sharma, A.; Sharma, D.; Sankhwar, S. Incidentally detected adrenocortical carcinoma in familial adenomatous polyposis: An unusual presentation of a hereditary cancer syndrome. BMJ Case Rep. 2018, 2018, bcr2018226799. [Google Scholar] [CrossRef] [Green Version]
- Shiroky, J.S.; Lerner-Ellis, J.P.; Govindarajan, A.; Urbach, D.R.; Devon, K.M. Characteristics of Adrenal Masses in Familial Adenomatous Polyposis. Dis. Colon Rectum 2018, 61, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Pakalniskis, M.G.; Ishigami, K.; Pakalniskis, B.L.; Fujita, N. Adrenal collision tumour comprised of adrenocortical carcinoma and myelolipoma in a patient with congenital adrenal hyperplasia. J. Med. Imaging Radiat. Oncol. 2020, 64, 67–68. [Google Scholar] [CrossRef]
- Łebek-Szatańska, A.; Nowak, K.M.; Samsel, R.; Roszkowska-Purska, K.; Zgliczyński, W.; Papierska, L. Adrenocortical carcinoma associated with giant bilateral myelolipomas in classic congenital adrenal hyperplasia. Pol. Arch. Intern. Med. 2019, 129, 549–550. [Google Scholar] [CrossRef]
- Varma, T.; Panchani, R.; Goyal, A.; Maskey, R. A case of androgen-secreting adrenal carcinoma with non-classical congenital adrenal hyperplasia. Indian J. Endocrinol. Metab. 2013, 17, S243–S245. [Google Scholar] [CrossRef]
- Yokoyama, H.; Adachi, T.; Tsubouchi, K.; Tanaka, M.; Sasano, H. Non-functioning adrenocortical carcinoma arising in an adrenal rest: Immunohistochemical study of an adult patient. Tohoku J. Exp. Med. 2013, 229, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salle, L.; Mas, R.; Teissier-Clément, M.-P. Ectopic adrenocortical carcinoma of the ovary: An unexpected outcome. Ann. Endocrinol. 2020, 81, 516–518. [Google Scholar] [CrossRef]
- Chentli, F.; Terki, N.; Azzoug, S. Ectopic adrenocortical carcinoma located in the ovary. Eur. J. Endocrinol. 2016, 175, K17–K23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, F.J.; Scheithauer, B.W.; Erickson, L.A.; Jenkins, R.B.; Giannini, C. Ectopic Low-grade Adrenocortical Carcinoma in the Spinal Region: Immunohistochemical and molecular cytogenetic study of a pediatric case. Am. J. Surg. Pathol. 2009, 33, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Permana, H.; Darmawan, G.; Ritonga, E.; Kusumawati, M.; Miftahurachman, M.; Soetedjo, N.N. An Interesting Case of Hepatic Adrenocortical Carcinoma. Acta Med. Indones 2018, 50, 257–259. [Google Scholar] [PubMed]
- Zhou, D.-K.; Liu, Z.-H.; Gao, B.-Q.; Wang, W. Giant nonfunctional ectopic adrenocortical carcinoma on the anterior abdominal wall: A case report. World J. Clin. Cases 2019, 7, 2075–2080. [Google Scholar] [CrossRef]
- Wright, J.P.; Montgomery, K.W.; Tierney, J.; Gilbert, J.; Solórzano, C.C.; Idrees, K. Ectopic, retroperitoneal adrenocortical carcinoma in the setting of Lynch syndrome. Fam. Cancer 2018, 17, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Lee, S.-E.; Kim, J.H.; Kim, J.H. Characteristics of adrenocortical carcinoma in South Korea: A registry-based nationwide survey. Endocr. Connect. 2020, 9, 519–529. [Google Scholar] [CrossRef]
- Chatzoulis, G.; Passos, I.; Bakaloudi, D.-R.; Giannakidis, D.; Koumpoulas, A.; Ioannidis, K.; Tsifountoudis, I.; Pappas, D.; Spyridopoulos, P. Correction to: Giant nonfunctioning adrenal tumors: Two case reports and review of the literature. J. Med. Case Rep. 2018, 12, 361. [Google Scholar] [CrossRef] [Green Version]
- Uruc, F.; Urkmez, A.; Yuksel, O.H.; Sahin, A.; Verit, A. Androgen secreting giant adrenocortical carcinoma with no metastases: A case report and review of the literature. Can. Urol. Assoc. J. 2015, 9, E644–E647. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.M.; Medeiros, L.J.; Vickery, A.L., Jr. Pathologic Features of Prognostic Significance in Adrenocortical Carcinoma. Am. J. Surg. Pathol. 1989, 13, 202–206. [Google Scholar] [CrossRef]
- Shekhar, S.; Gubbi, S.; Papadakis, G.Z.; Nilubol, N.; Hannah-Shmouni, F. Adrenocortical carcinoma and pulmonary embolism from tumoral extension. Endocrinol. Diabetes Metab. Case Rep. 2019, 2019, 19-0095. [Google Scholar] [CrossRef]
- Weiss, L.M. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am. J. Surg. Pathol. 1984, 8, 163–169. [Google Scholar] [CrossRef]
- Aubert, S.; Wacrenier, A.; Leroy, X.; Devos, P.; Carnaille, B.; Proye, C.; Wemeau, J.L.; Lecomte-Houcke, M.; Leteurtre, E. Weiss System Revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am. J. Surg. Pathol. 2002, 26, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Duregon, E.; Fassina, A.; Volante, M.; Nesi, G.; Santi, R.; Gatti, G.; Cappellesso, R.; Ciaramella, P.D.; Ventura, L.; Gambacorta, M.; et al. The Reticulin Algorithm for Adrenocortical Tumor Diagnosis: A multicentric validation study on 245 unpublished cases. Am. J. Surg. Pathol. 2013, 37, 1433–1440. [Google Scholar] [CrossRef]
- Volante, M.; Bollito, E.; Sperone, P.; Tavaglione, V.; Daffara, F.; Porpiglia, F.; Terzolo, M.; Berruti, A.; Papotti, M. Clinicopathological study of a series of 92 adrenocortical carcinomas: From a proposal of simplified diagnostic algorithm to prognostic stratification. Histopathology 2009, 55, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissier, F.; Aubert, S.; Leteurtre, E.; Al Ghuzlan, A.; Patey, M.; Decaussin, M.; Doucet, L.; Gobet, F.; Hoang, C.; Mazerolles, C.; et al. Adrenocortical Tumors: Improving the practice of the Weiss system through virtual microscopy: A National Program of the French Network INCa-COMETE. Am. J. Surg. Pathol. 2012, 36, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Arola, J.; Salmenkivi, K.; Liu, J.; Kahri, A.I.; Heikkilá, P. p53 and Ki67 in adrenocortical tumors. Endocr. Res. 2000, 26, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Soon, P.S.; Gill, A.J.; Benn, D.E.; Clarkson, A.; Robinson, B.G.; McDonald, K.L.; Sidhu, S.B. Microarray gene expression and immunohistochemistry analyses of adrenocortical tumors identify IGF2 and Ki-67 as useful in differentiating carcinomas from adenomas. Endocr. Relat. Cancer 2009, 16, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Duregon, E.; Cappellesso, R.; Maffeis, V.; Zaggia, B.; Ventura, L.; Berruti, A.; Terzolo, M.; Fassina, A.; Volante, M.; Papotti, M. Validation of the prognostic role of the "Helsinki Score” in 225 cases of adrenocortical carcinoma. Hum. Pathol. 2017, 62, 1–7. [Google Scholar] [CrossRef]
- Pennanen, M.; Heiskanen, I.; Sane, T.; Remes, S.; Mustonen, H.; Haglund, C.; Arola, J. Helsinki score—A novel model for prediction of metastases in adrenocortical carcinomas. Hum. Pathol. 2015, 46, 404–410. [Google Scholar] [CrossRef]
- Lam, K.-Y.; Lo, C.-Y. Metastatic tumours of the adrenal glands: A 30-year experience in a teaching hospital. Clin. Endocrinol. 2002, 56, 95–101. [Google Scholar] [CrossRef]
- Lam, A.K. Update on paragangliomas and pheochromocytomas. Turk Patoloji Derg. 2015, 31 (Suppl. 1), 105–112. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.H.; Patel, S.; Wei, L.; Phay, J.E.; Shirley, L.A.; Kirschner, L.S.; Schmidt, C.; Abdel-Misih, S.; Brock, P.; Shah, M.H.; et al. Metastatic Adrenocortical Carcinoma: A Single Institutional Experience. Horm. Cancer 2019, 10, 161–167. [Google Scholar] [CrossRef]
- Kovecsi, A.; Jung, I.; Bara, T.J.; Azamfirei, L.; Kovacs, Z.; Gurzu, S. First Case Report of a Sporadic Adrenocortical Carcinoma With Gastric Metastasis and a Synchronous Gastrointestinal Stromal Tumor of the Stomach. Medicine 2015, 94, e1549. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.; Schedelbeck, U.; Pulzer, A.; Bluemel, C.; Wild, V.; Fassnacht, M.; Steger, U. A case report of a solitary pancreatic metastasis of an adrenocortical carcinoma. BMC Surg. 2015, 15, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satter, E.K.; Barnette, D.J. Adrenocortical carcinoma with delayed cutaneous metastasis. J. Cutan. Pathol. 2008, 35, 677–680. [Google Scholar] [CrossRef]
- Souteiro, P.; Donato, S.; Costa, C.; Pereira, C.A.; Simões-Pereira, J.; Oliveira, J.; Belo, S.; Santos, A.P.; Cardoso, H.; Leite, V.; et al. Diagnosis, treatment, and survival analysis of adrenocortical carcinomas: A multicentric study. Hormons 2020, 19, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Dominici, A.; Rizzo, M.; Travaglini, F.; Nesi, G.; Franchi, A. Non-functioning adrenal cortical carcinoma presenting with metastasis to the tongue. J. Oral Pathol. Med. 2003, 32, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.R.; Kar, A.; Goodspeed, A.E.; Pozdeyev, N.; Somerset, H.; Raeburn, C.D.; Tan, A.C.; Leong, S.; Wierman, M.E.; Kiseljak-Vassiliades, K. Leptomeningeal Metastasis from Adrenocortical Carcinoma: A Case Report. J. Endocr. Soc. 2020, 4, bvaa017. [Google Scholar] [CrossRef]
- Burotto, M.; Tageja, N.; Rosenberg, A.Z.; Mahalingam, S.; Quezado, M.; Velarde, M.; Edgerly, M.; Fojo, T. Brain metastasis in patients with adrenocortical carcinoma: A clinical series. J. Clin. Endocrinol. Metab. 2015, 100, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkachenko, R.; Golovko, A.; Kuryk, O. A case report of late local relapse of adrenocortical carcinoma 18 years after adrenalectomy. Exp. Oncol. 2018, 40, 251–253. [Google Scholar] [CrossRef]
- Bergeat, D.; Rayar, M.; Beuzit, L.; Sandri, G.B.L.; Dagher, J.; Merdrignac, A.; Tanguy, L.; Boudjema, K.; Sulpice, L.; Meunier, B. An unusual case of adrenocortical carcinoma with liver metastasis that occurred at 23 years after surgery. Hepatobiliary Surg. Nutr. 2016, 5, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Beuschlein, F.; Weigel, J.; Saeger, W.; Kroiss, M.; Wild, V.; Daffara, F.; Libé, R.; Ardito, A.; Al Ghuzlan, A.; Quinkler, M.; et al. Major Prognostic Role of Ki67 in Localized Adrenocortical Carcinoma After Complete Resection. J. Clin. Endocrinol. Metab. 2015, 100, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Chehade, M.; Bullock, M.; Glover, A.R.; Hutvagner, G.; Sidhu, S.B. Key MicroRNA’s and Their Targetome in Adrenocortical Cancer. Cancers 2020, 12, 2198. [Google Scholar] [CrossRef]
- Billon, E.; Finetti, P.; Bertucci, A.; Niccoli, P.; Birnbaum, D.; Mamessier, E.; Bertucci, F. PDL1 expression is associated with longer postoperative, survival in adrenocortical carcinoma. OncoImmunology 2019, 8, e1655362. [Google Scholar] [CrossRef] [Green Version]
- Duregon, E.; Volante, M.; Giorcelli, J.; Terzolo, M.; Lalli, E.; Papotti, M.G. Diagnostic and prognostic role of steroidogenic factor 1 in adrenocortical carcinoma: A validation study focusing on clinical and pathologic correlates. Hum. Pathol. 2013, 44, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, A.M.F.; Soares, I.C.; Mariani, B.M.D.P.; Nishi, M.Y.; Bezerra-Neto, J.E.; Charchar, H.D.S.; Brondani, V.B.; Tanno, F.; Srougi, V.; Chambo, J.L.; et al. Sterol O-Acyl Transferase 1 as a Prognostic Marker of Adrenocortical Carcinoma. Cancers 2020, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Gao, W.-C.; Chen, S.-S.; Bai, L.; Luo, L.; Zheng, X.-G.; Luo, Y. Primary site surgery for metastatic adrenocortical carcinoma improves survival outcomes: An analysis of a population-based database. OncoTargets Ther. 2017, 10, 5311–5315. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Zheng, J.; Cai, J.; Wu, S.; Diao, X.; Xie, W.; Chen, X.; Liao, C.; Yu, H.; Fan, X.; et al. A nomogram for individualized estimation of survival among adult patients with adrenocortical carcinoma after surgery: A retrospective analysis and multicenter validation study. Cancer Commun. 2019, 39, 80. [Google Scholar] [CrossRef] [Green Version]
- McAteer, J.P.; Huaco, J.A.; Gow, K.W. Predictors of survival in pediatric adrenocortical carcinoma: A Surveillance, Epidemiology, and End Results (SEER) program study. J. Pediatr. Surg. 2013, 48, 1025–1031. [Google Scholar] [CrossRef]
- Picard, C.; Orbach, D.; Carton, M.; Brugieres, L.; Renaudin, K.; Aubert, S.; Berrebi, D.; Galmiche, L.; Dujardin, F.; Leblond, P.; et al. Revisiting the role of the pathological grading in pediatric adrenal cortical tumors: Results from a national cohort study with pathological review. Mod. Pathol. 2019, 32, 546–559. [Google Scholar] [CrossRef]
- Balmant, N.V.; de Souza Reis, R.; de Oliveira Santos, M.; de Camargo, B.; Gatta, G. Rare cancers in childhood and adolescence in Brazil: First report of data from 19 population-based cancer registries. Cancer 2019, 125, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.E.J.; Gerber, V.K.Q.; Ibañez, H.C.; Melanda, V.S.; Parise, I.Z.S.; Watanabe, F.M.; Pianovski, M.A.D.; Fiori, C.M.C.M.; Fabro, A.L.M.R.; Silva, D.B.D.; et al. Penetrance of the TP53 R337H Mutation and Pediatric Adrenocortical Carcinoma Incidence Associated with Environmental Influences in a 12-Year Observational Cohort in Southern Brazil. Cancers 2019, 11, 1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zekri, W.; Hammad, M.; Rashed, W.M.; Ahmed, G.; Elshafie, M.; Adly, M.H.; Elborai, Y.; Abdalla, B.; Taha, H.; Elkinaae, N.; et al. The outcome of childhood adrenocortical carcinoma in Egypt: A model from developing countries. Pediatr. Hematol. Oncol. 2020, 37, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-J.; Yu, T.-T.; Ma, J.; Zhou, Y.; Xu, M.; Gao, Y.-J. Composite Adrenocortical Carcinoma and Neuroblastoma in an Infant with a TP53 Germline Mutation: A case report and literature review. J. Pediatr. Hematol. Oncol. 2019, 41, 399–401. [Google Scholar] [CrossRef]
- Chauhan, D.S.; Rahar, S.; Singh, S.; Ahuja, A.; Bhardwaj, M.; Sen, A. Adrenocortical carcinoma in a 6-month-old infant. J. Indian Assoc. Pediatr. Surg. 2020, 25, 310–313. [Google Scholar] [CrossRef]
- Kwon, A.; Choi, Y.; Jung, J.W.; Suh, J.; Kim, H.-S. Using Etomidate in a 2-month-old Infant with Cushing Syndrome due to Adrenocortical Carcinoma. J. Clin. Res. Pediatr. Endocrinol. 2021, in press. [Google Scholar] [CrossRef]
- Gupta, N.; Rivera, M.; Novotny, P.; Rodriguez, V.; Bancos, I.; Lteif, A. Adrenocortical Carcinoma in Children: A Clinicopathological Analysis of 41 Patients at the Mayo Clinic from 1950 to 2017. Horm. Res. Paediatr. 2018, 90, 8–18. [Google Scholar] [CrossRef]
- Malhotra, S.; Waikar, A.R.; Singh, P.; Guarini, L.; Jacobson-Dickman, E.; Motaghedi, R.; Kazachkova, I. Rare Presentation of Adrenocortical Carcinoma in a 4-Month-Old Boy. World J. Oncol. 2017, 8, 81–85. [Google Scholar] [CrossRef]
- Takeuchi, T.; Yoto, Y.; Ishii, A.; Tsugawa, T.; Yamamoto, M.; Hori, T.; Kamasaki, H.; Nogami, K.; Oda, T.; Nui, A.; et al. Adrenocortical carcinoma characterized by gynecomastia: A case report. Clin. Pediatr. Endocrinol. 2018, 27, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-T.; Shieh, J.-J.; Chang, J.H.-M.; Chang, S.-W.; Chen, T.-C.; Hsu, W.-H. Early detection of adrenocortical carcinoma in a child with Li-Fraumeni syndrome. Pediatr. Blood Cancer 2009, 52, 541–544. [Google Scholar] [CrossRef]
- Choong, S.S.; Latiff, Z.A.; Mohamed, M.; Lim, L.L.; Chen, K.S.; Vengidasan, L.; Razali, H.; Abdul Rahman, E.J.; Ariffin, H. Malaysian Society of Paediatric Haematology-Oncology. Childhood adrenocortical carcinoma as a sentinel cancer for detecting families with germline TP53 mutations. Clin Genet. 2012, 82, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Han, R.; Ye, L.; Xie, J.; Tao, B.; Sun, F.; Zhuo, R.; Chen, X.; Deng, X.; Ye, C.; et al. Adrenocortical carcinoma in patients with MEN1: A kindred report and review of the literature. Endocr. Connect. 2019, 8, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.S.; Fleitz, J.M.; Kleinschmidt-DeMasters, B.K. Pediatric Adrenal Cortical Carcinoma: Brain Metastases and Relationship to NF-1, Case Reports and Review of the Literature. J. Neuro-Oncol. 2005, 75, 127–133. [Google Scholar] [CrossRef]
- Rauch, L.B.; Erdman, S.H.; Aldrink, J.H.; Ranalli, M.A.; Prasad, V.; Hoffman, R.P. Fatal Extraintestinal Adrenal Malignancy in a 12-year-old Girl with Familial Adenomatous Polyposis. J. Pediatr. Gastroenterol. Nutr. 2014, 58, e19–e20. [Google Scholar] [CrossRef]
- Eltan, M.; Arslan Ates, E.; Cerit, K.; Menevse, T.S.; Kaygusuz, S.B.; Eker, N.; Bagci, P.; Ergelen, R.; Turan, S.; Bereket, A.; et al. Adrenocortical carcinoma in atypical Beckwith-Wiedemann syndrome due to loss of methylation at imprinting control region 2. Pediatr. Blood Cancer 2020, 67, e28042. [Google Scholar] [CrossRef] [PubMed]
- Cöktü, S.; Spix, C.; Kaiser, M.; Beygo, J.; Kleinle, S.; Bachmann, N.; Kohlschmidt, N.; Prawitt, D.; Beckmann, A.; Klaes, R.; et al. Cancer incidence and spectrum among children with genetically confirmed Beckwith-Wiedemann spectrum in Germany: A retrospective cohort study. Br. J. Cancer 2020, 123, 619–623. [Google Scholar] [CrossRef]
- Gulack, B.C.; Rialon, K.L.; Englum, B.R.; Kim, J.; Talbot, L.J.; Adibe, O.O.; E Rice, H.; Tracy, E.T. Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB). J. Pediatr. Surg. 2016, 51, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Wieneke, J.A.; Thompson, L.D.; Heffess, C.S. Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients. Am. J. Surg. Pathol. 2003, 27, 867–881. [Google Scholar] [CrossRef]
- Jehangir, S.; Nanjundaiah, P.; Sigamani, E.; Burad, D.; Manipadam, M.T.; Lea, V.; Ly, T.; Holland, A.J.A. Pathological prognostication of paediatric adrenocortical tumours: Is a gold standard emerging? Pediatr. Blood Cancer 2019, 66, e27567. [Google Scholar] [CrossRef]
- Chatterjee, G.; DasGupta, S.; Mukherjee, G.; Sengupta, M.; Roy, P.; Arun, I.; Datta, C.; Mishra, P.K.; Banerjee, S.; Chatterjee, U. Usefulness of Wieneke criteria in assessing morphologic characteristics of adrenocortical tumors in children. Pediatr. Surg. Int. 2015, 31, 563–571. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Islam, N.; Roy, P.; Datta, C.; Mishra, P.K.; Banerjee, S.; Chaudhuri, M.K.; Chatterjee, U. Weineke criteria, Ki-67 index and p53 status to study pediatric adrenocortical tumors: Is there a correlation? J. Pediatr. Surg. 2016, 51, 1795–1800. [Google Scholar] [CrossRef]
- Jarzembowski, J.A. New prognostic indicators in pediatric adrenal tumors: Neuroblastoma and adrenal cortical tumors, can we predict when these will behave badly? Surg. Pathol. Clin. 2020, 13, 625–641. [Google Scholar] [CrossRef]
- Kerkhofs, T.M.; Ettaieb, M.H.; Verhoeven, R.H.; Kaspers, G.J.; Tissing, W.J.; Loeffen, J.; Van den Heuvel-Eibrink, M.M.; De Krijger, R.R.; Haak, H.R. Adrenocortical carcinoma in children: First population-based clinicopathological study with long-term follow-up. Oncol. Rep. 2014, 32, 2836–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambaiti, E.; Duci, M.; De Corti, F.; Gamba, P.; Dall’Igna, P.; Ghidini, F.; Virgone, C. Clinical prognostic factors in pediatric adrenocortical tumors: A meta-analysis. Pediatr. Blood Cancer 2021, e28836, in press. [Google Scholar] [CrossRef]
- Weiss, L.M.; Bertagna, X.; Chrousos, G.P.; Kawashima, A.; Kleihues, P.; Koch, C.A.; Giordano, T.J.; Medeiros, L.J.; Merino, M.J.; Ordonez, N.G.; et al. Adrenal cortical carcinoma. In WHO Third Edition Word Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Endocrine Organs, 3rd ed.; DeLellis, R.A., Lloyd, R.V., Heitz, P.U., Eng, C., Eds.; IARC: Lyon, France, 2004; Chapter 3; pp. 139–142. [Google Scholar]
- Kanitra, J.J.; Hardaway, J.C.; Soleimani, T.; Koehler, T.J.; McLeod, M.K.; Kavuturu, S. Adrenocortical oncocytic neoplasm: A systematic review. Surgery 2018, 164, 1351–1359. [Google Scholar] [CrossRef]
- Renaudin, K.; Smati, S.; Wargny, M.; Al Ghuzlan, A.; Aubert, S.; Leteurtre, E.; Patey, M.; Sibony, M.; Sturm, N.; Tissier, F.; et al. Clinicopathological description of 43 oncocytic adrenocortical tumors: Importance of Ki-67 in histoprognostic evaluation. Mod. Pathol. 2018, 31, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Vatrano, S.; Volante, M.; Duregon, E.; Giorcelli, J.; Izzo, S.; Rapa, I.; Votta, A.; Germano, A.; Scagliotti, G.; Berruti, A.; et al. Detailed genomic characterization identifies high heterogeneity and histotype-specific genomic profiles in adrenocortical carcinomas. Mod. Pathol. 2018, 31, 1257–1269. [Google Scholar] [CrossRef] [Green Version]
- el-Naggar, A.K.; Evans, D.B.; Mackay, B. Oncocytic adrenal cortical carcinoma. Ultrastruct Pathol. 1991, 15, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Paulose, K.P. Oncocytic variant of adrenal carcinoma presenting as Cushing’s syndrome. J. Assoc. Phys. India 1998, 46, 235–237. [Google Scholar]
- Krishnamurthy, S.; Ordóñez, N.G.; Shelton, T.O.; Ayala, A.G.; Sneige, N. Fine-needle aspiration cytology of a case of oncocytic adrenocortical carcinoma. Diagn Cytopathol. 2000, 22, 299–303. [Google Scholar] [CrossRef]
- Kurek, R.; Von Knobloch, R.; Feek, U.; Heidenreich, A.; Hofmann, R. Local recurrence of an oncocytic adrenocortical carcinoma with ovary metastasis. J. Urol. 2001, 166, 985. [Google Scholar] [CrossRef]
- Hoang, M.P.; Ayala, A.G.; Albores-Saavedra, J. Oncocytic adrenocortical carcinoma: A morphologic, immunohistochemical and ultrastructural study of four cases. Mod. Pathol. 2002, 15, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Bisceglia, M.; Ludovico, O.; Di Mattia, A.; Ben-Dor, D.; Sandbank, J.; Pasquinelli, G.; Lau, S.K.; Weiss, L.M. Adrenocortical oncocytic tumors: Report of 10 cases and review of the literature. Int. J. Surg. Pathol. 2004, 12, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Park, S.; Kim, S.R.; Suh, Y.L. Oncocytic adrenocortical carcinomas: A pathological and immunohistochemical study of four cases in comparison with conventional adrenocortical carcinomas. Pathol. Int. 2004, 54, 603–610. [Google Scholar] [CrossRef]
- Tanaka, K.; Kumano, Y.; Kanomata, N.; Takeda, M.; Hara, I.; Fujisawa, M.; Kawabata, G.; Kamidono, S. Oncocytic adrenocortical carcinoma. Urology 2004, 64, 376–377. [Google Scholar] [CrossRef]
- Gołkowski, F.; Buziak-Bereza, M.; Huszno, B.; Bałdys-Waligórska, A.; Stefańska, A.; Budzyński, A.; Okoń, K.; Chrzan, R.; Urbanik, A. The unique case of adrenocortical malignant and functioning oncocytic tumour. Exp. Clin. Endocrinol. Diabetes 2007, 115, 401–404. [Google Scholar] [CrossRef]
- Ali, A.E.; Raphael, S.J. Functional oncocytic adrenocortical carcinoma. Endocr. Pathol. 2007, 18, 187–189. [Google Scholar] [CrossRef]
- Argyriou, P.; Zisis, C.; Alevizopoulos, N.; Kefaloyannis, E.M.; Gennatas, C.; Petraki, C.D. Adrenocortical oncocytic carcinoma with recurrent metastases: A case report and review of the literature. World J. Surg. Oncol. 2008, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, H.; Kawamura, H.; Matsuzaki, M.; Yokoyama, E.; Kitajima, M.; Onizuka, S.; Yamakawa, M. Oncocytic adrenocortical carcinoma. Ann. Diagn. Pathol. 2010, 14, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Mwandila, M.; Waller, H.; Stott, V.; Mercer, P. A case of a testosterone-secreting oncocytic adrenocortical carcinoma. N. Z. Med. J. 2010, 123, 80–82. [Google Scholar] [PubMed]
- Wong, D.D.; Spagnolo, D.V.; Bisceglia, M.; Havlat, M.; McCallum, D.; Platten, M.A. Oncocytic adrenocortical neoplasms—A clinicopathologic study of 13 new cases emphasizing the importance of their recognition. Hum. Pathol. 2011, 42, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Tirkes, T.; Gokaslan, T.; McCrea, J.; Sandrasegaran, K.; Hollar, M.A.; Akisik, F.; Lall, C. Oncocytic neoplasms of the adrenal gland. AJR Am. J. Roentgenol. 2011, 196, 592–596. [Google Scholar] [CrossRef]
- Kalra, S.; Manikandan, R.; Srinivas, B.H. Oncocytic adrenocortical carcinoma—A rare pathological variant. BMJ Case Rep. 2015, 2015, bcr2014208818. [Google Scholar] [CrossRef] [Green Version]
- Carré, J.; Grunenwald, S.; Vezzosi, D.; Mazerolles, C.; Bennet, A.; Meduri, G.; Caron, P. Virilizing oncocytic adrenocortical carcinoma: Clinical and immunohistochemical studies. Gynecol. Endocrinol. 2016, 32, 662–666. [Google Scholar] [CrossRef]
- Ertan, Y.; Argon, A.; Özdemir, M.; Yürekli, B.P.S.; Dökümcü, Z.; Makay, Ö. Oncocytic adreno cortical tumors: Pathological features of 16 cases and review of the literature. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 237–244. [Google Scholar] [CrossRef]
- Sumner, E.; Acar, B.C.; Acker, M.R. Oncocytic adrenocortical carcinoma: A rare adrenal tumor subtype. Can. J. Urol. 2017, 24, 8865–8867. [Google Scholar]
- Fonseca, D.; Murthy, S.S.; Tagore, K.R.; Rao, B.V.; Thamminedi, S.R.; Raju, K.V.V.N.; Sharma, R.; Challa, S. Diagnosis of adrenocortical tumors by reticulin algorithm. Indian J. Endocrinol. Metab. 2017, 21, 734–737. [Google Scholar] [CrossRef]
- Al Balooshi, B.; Miyanath, S.; Elhennawy, A.; Saeedi, Y.; Tirmazy, S.H.; Muhasin, M.; Ray, B.; Al Sharhan, M.; Hotait, H.; Houcinat, Y.; et al. Adrenocortical oncocytic carcinoma and papillary thyroid carcinoma incidentally detected in an asymptomatic patient by F-18 FDG PET/CT. Asia Ocean J. Nucl. Med. Biol. 2018, 6, 179–185. [Google Scholar] [CrossRef]
- Kaur, R.J.; Pichurin, P.N.; Hines, J.M.; Singh, R.J.; Grebe, S.K.; Bancos, I. Adrenal cortical carcinoma associated with Lynch syndrome: A case report and review of literature. J. Endocr. Soc. 2019, 3, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.K.; Khalil, M.; Pasieka, J.; Kong, S.; Xu, Y.; Harvey, A. Oncocytic subtypes of adrenal cortical carcinoma: Aggressive in appearance yet more indolent in behavior? Surgery 2019, 166, 524–533. [Google Scholar] [CrossRef]
- Lehr, I.; Gillis, C.; French, C.; Simmonds, A.; Organ, M. Images—Oncocytic adrenocortical carcinoma: A rare tumor variant. Can. Urol. Assoc. J. 2020, 14, E45. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Yasuda, M.; Nakano, Y.; Yoshida, K.; Umeda, Y.; Yagi, T.; Yamazaki, Y.; Sasano, H.; Otsuka, F. A rare case of oncocytic adrenocortical carcinoma clinically presented as an incidentaloma. Endocr. J. 2020, 67, 883–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akin, O.; Atas, E.; Atasoy, İ.A.; Durmaz, N.; Kartal, Ö. Two subsequent metachroneus solid tumors: Oncocytic variant adrenocortical carcinoma and rhabdomyosarcoma of childhood: Case report and literature review. J. Clin. Res. Pediatr. Endocrinol 2021, in press. [Google Scholar] [CrossRef]
- Wadhwani, N.; Mais, D.; Kaushik, D.; Kitano, M. A case of adrenocortical oncocytic carcinoma arising in ectopic adrenal tissue: A multidisciplinary diagnostic challenge. Ecancermedicalscience 2020, 14, 1135. [Google Scholar] [CrossRef]
- Kasem, K.; Lam, A.K. Adrenal oncocytic phaeochromocytoma with putative adverse histologic features: A unique case report and review of the literature. Endocr. Pathol. 2014, 25, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.K.; Harriman, B.B.; Toker, C. Myxoid adrenal cortical carcinoma: A light and electron microscopic study. Arch. Pathol. Lab. Med. 1979, 103, 635–638. [Google Scholar]
- Forsthoefel, K.F. Myxoid adrenal cortical carcinoma. A case report with differential diagnostic considerations. Arch. Pathol. Lab. Med. 1994, 118, 1151–1153. [Google Scholar]
- Brown, F.M.; Gaffey, T.A.; Wold, L.E.; Lloyd, R.V. Myxoid neoplasms of the adrenal cortex: A rare histologic variant. Am. J. Surg. Pathol. 2000, 24, 396–401. [Google Scholar] [CrossRef]
- Izumi, M.; Serizawa, H.; Iwaya, K.; Takeda, K.; Sasano, H.; Mukai, K. A case of myxoid adrenocortical carcinoma with extensive lipomatous metaplasia. Arch. Pathol. Lab. Med. 2003, 127, 227–230. [Google Scholar] [CrossRef]
- Suresh, B.; Kishore, T.A.; Albert, A.S.; Joy, A. Myxoid adrenal cortical carcinoma—A rare variant of adrenocortical carcinoma. Indian J. Med. Sci. 2005, 59, 505–507. [Google Scholar] [CrossRef] [Green Version]
- Karim, R.Z.; Wills, E.J.; McCarthy, S.W.; Scolyer, R.A. Myxoid variant of adrenocortical carcinoma: Report of a unique case. Pathol. Int. 2006, 56, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.S.; Chen, J.H.; Lin, L.W. Myxoid adrenal cortical carcinoma presenting as primary hyperaldosteronism: Case report and review of the literature. Int. J. Surg. Pathol. 2011, 19, 803–807. [Google Scholar] [CrossRef]
- Papotti, M.; Volante, M.; Duregon, E.; Delsedime, L.; Terzolo, M.; Berruti, A.; Rosai, J. Adrenocortical tumors with myxoid features: A distinct morphologic and phenotypical variant exhibiting malignant behavior. Am. J. Surg. Pathol. 2010, 34, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, J.; Liang, Z.; Gao, J.; Zeng, X.; Liu, T. Myxoid adrenocortical neoplasms: A study of the clinicopathologic features and EGFR gene status of ten Chinese cases. Am. J. Clin. Pathol. 2011, 136, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Weissferdt, A.; Phan, A.; Suster, S.; Moran, C.A. Myxoid adrenocortical carcinoma: A clinicopathologic and immunohistochemical study of 7 cases, including 1 case with lipomatous metaplasia. Am. J. Clin. Pathol. 2013, 139, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzu, S.; Szentirmay, Z.; Bara, T.; Bara, T.; Jung, I., Jr. Myxoid variant of adrenocortical carcinoma: A report of two illustrative cases and a brief review of the literature. Pathology 2014, 46, 83–85. [Google Scholar] [CrossRef]
- Mandal, P.K.; Sinha, M.G. Cytodiagnosis of myxoid adrenocortical carcinoma and role of immunocytochemistry to differentiate it from renal cell carcinoma. J. Cytol. 2014, 31, 111–113. [Google Scholar] [CrossRef]
- Sung, T.Y.; Choi, Y.M.; Kim, W.G.; Lee, Y.M.; Kim, T.Y.; Shong, Y.K.; Kim, W.B.; Song, D.E. Myxoid and sarcomatoid variants of adrenocortical carcinoma: Analysis of rare variants in single tertiary care center. J. Korean Med. Sci. 2017, 32, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Yasuda, M.; Hasegawa, K.; Yamazaki, Y.; Sasano, H.; Otsuka, F. A novel case of myxoid variant of adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1. Endocr. J. 2019, 66, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.; Thalody, K.; Kapur, U.; Nguyen, T.T. Robot-assisted laparoscopic adrenalectomy for rare myxoid adrenocortical carcinoma. Case Rep. Urol. 2019, 2019, 9794345. [Google Scholar] [CrossRef] [Green Version]
- Raparia, K.; Ayala, A.G.; Sienko, A.; Zhai, Q.J.; Ro, J.Y. Myxoid adrenal cortical neoplasms. Ann. Diagn. Pathol. 2008, 12, 344–348. [Google Scholar] [CrossRef]
- Solcia, E.; Klöppel, G.; Sobin, L.H. In collaboration with 9 Pathologists from 4 countries. Tumours of the adrenal cortex. In World Health Organization International Histological Classification of Tumours: Histological Typing of Endocrine Tumours, 2nd ed.; Solcia, E., Klöppel, G., Sobin, L.H., Eds.; In Collaboration with 9 Pathologists from 4 Countries; Springer: Berlin, Germany, 2000; Chapter 2.2.1; p. 33. [Google Scholar]
- Okazumi, S.; Asano, T.; Ryu, M.; Nagashima, T.; Odaka, M.; Isono, K.; Nishizawa, T. Surgical resection of adrenal carcinoma extending into the vena cava, rightatrium and ventricle: Case report and review of the literature. Nihon Geka Gakkai Zasshi 1987, 88, 231–238. (In Japanese) [Google Scholar]
- Collina, G.; Maldarizzi, F.; Betts, C.M.; Eusebi, V. Primary sarcomatoid carcinoma of the adrenal gland. First case report. Virchows Arch. A Pathol. Anat. Histopathol. 1989, 415, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Decorato, J.W.; Gruber, H.; Petti, M.; Levowitz, B.S. Adrenal carcinosarcoma. J. Surg. Oncol. 1990, 45, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Fischler, D.F.; Nunez, C.; Levin, H.S.; McMahon, J.T.; Sheeler, L.R.; Adelstein, D.J. Adrenal carcinosarcoma presenting in a woman with clinical signs of virilization. a case report with immunohistochemical and ultrastructural findings. Am. J. Surg. Pathol. 1992, 16, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Barksdale, S.K.; Marincola, F.M.; Jaffe, G. Carcinosarcoma of the adrenal cortex presenting with mineralocorticoid excess. Am. J. Surg. Pathol. 1993, 17, 941–945. [Google Scholar] [CrossRef]
- Lee, M.S.; Park, I.A.; Chi, J.G.; Ham, E.K.; Lee, K.C.; Lee, C.W. Adrenal carcinosarcoma—A case report. J. Korean Med. Sci. 1997, 12, 374–377. [Google Scholar] [CrossRef]
- Sturm, N.; Moulai, N.; Laverrière, M.H.; Chabre, O.; Descotes, J.L.; Brambilla, E. Primary adrenocortical sarcomatoid carcinoma: Case report and review of literature. Virchows Arch. 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Coli, A.; Di Giorgio, A.; Castri, F.; Destito, C.; Marin, A.W.; Bigotti, G. Sarcomatoid carcinoma of the adrenal gland: A case report and review of literature. Pathol. Res. Pract. 2010, 206, 59–65. [Google Scholar] [CrossRef]
- Feng, Y.C.; Yang, Z.G.; Chen, T.W.; Su, X.Y.; Deng, W.; Wang, Q.L. Adrenal sarcomatoid carcinoma: A rare case depicted on multi-detector row computed tomography. Indian J. Med. Sci. 2010, 64, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Desimone, M.; Rao, H.R.; Huang, G.J.; Seethala, R.R. Adrenocortical carcinosarcoma: A case report and review of the literature. Diagn. Pathol. 2010, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Rossi, G.; Fiocchi, F.; Giacometti, M.; Fontana, A.; Gibertini, M.C.; Roncucci, L.; Luppi, G.; Torricelli, P.; Rossi, A.; et al. Primary adrenal gland carcinosarcoma associated with metastatic rectal cancer: A hitherto unreported collision tumor. Tumori 2011, 97, 27e–30e. [Google Scholar] [CrossRef]
- Yan, J.J.; Sun, A.J.; Ren, Y.; Hou, C. Primary adrenocortical sarcomatoid carcinoma: Report of a case. Can Urol. Assoc. J. 2012, 6, E189–E191. [Google Scholar] [CrossRef]
- Thway, K.; Olmos, D.; Shah, C.; Flora, R.; Shipley, J.; Fisher, C. Oncocytic adrenal cortical carcinosarcoma with pleomorphic rhabdomyosarcomatous metastases. Am. J. Surg. Pathol. 2012, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.S.; Grignon, D.J.; Ulbright, T.M.; Idrees, M.T. A case report of adrenocortical carcinosarcoma with oncocytic and primitive neuroectodermal-like features. Hum. Pathol. 2013, 44, 1947–1955. [Google Scholar] [CrossRef]
- Shaikh, A.S.; Bakhshi, G.D.; Khan, A.S.; Jamadar, N.M.; Nirmala, A.K.; Raza, A.A. Primary adrenal sarcomatoid carcinoma. Clin. Pract. 2014, 4, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, D.; Boyd, C.; Eatock, F. Adrenal sarcomatoid carcinoma: A case report and review of the literature. Ulst. Med. J. 2014, 83, 89–92. [Google Scholar]
- Wei, Y.B.; Gao, Y.L.; Wu, H.T.; Ou-Yang, S.F.; Xu, T.; Mao, D.F.; Yang, J.R. Rare incidence of primary adrenocortical carcinosarcoma: A case report and literature review. Oncol. Lett. 2015, 9, 153–158. [Google Scholar] [CrossRef]
- Turhan Iyidir, O.; Cerit, E.T.; Özkan, Ç.; Altınova, E.; Çimen, A.R.; Sözen, S.; Kerem, M.; Aktürk, M.; Memiş, L.; Törüner, B.; et al. A case report of bilateral adrenal sarcomatoid carcinoma. Case Rep. Surg. 2016, 2016, 3768258. [Google Scholar] [CrossRef]
- Ishikawa, N.; Nagase, M.; Takami, S.; Araki, A.; Ishikawa, N.; Koike, C.; Shiina, H.; Maruyama, R. A case report of bilateral sarcomatoid carcinoma of adrenal glands with adrenal insufficiency. Int. J. Surg. Pathol. 2016, 24, 743–748. [Google Scholar] [CrossRef]
- Papathomas, T.G.; Duregon, E.; Korpershoek, E.; Restuccia, D.F.; van Marion, R.; Cappellesso, R.; Sturm, N.; Rossi, G.; Coli, A.; Zucchini, N.; et al. Sarcomatoid adrenocortical carcinoma: A comprehensive pathological, immunohistochemical, and targeted next-generation sequencing analysis. Hum. Pathol. 2016, 58, 113–122. [Google Scholar] [CrossRef]
- Zhu, C.; Zheng, A.; Mao, X.; Shi, B.; Li, X. Primary adrenal sarcomatoid carcinoma metastatic to the lung: Case report and review of the literature. Oncol. Lett. 2016, 11, 3117–3122. [Google Scholar] [CrossRef] [PubMed]
- Saeger, W.; Mohren, W.; Behrend, M.; Iglauer, P.; Wilczak, W. Sarcomatoid adrenal carcinoma: Case report with contribution to pathogenesis. Endocr. Pathol. 2017, 28, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Sabrine, D.; Zakia, B.; Kaoutar, Z. Adrenocortical sarcomatoid carcinoma: A case report and review of the literature. J. Surg. Case Rep. 2020, 2020, rjaa211. [Google Scholar] [CrossRef]
- Audenet, F.; Méjean, A.; Chartier-Kastler, E.; Rouprêt, M. Adrenal tumours are more predominant in females regardless of their histological subtype: A review. World J. Urol. 2013, 31, 1037–1043. [Google Scholar] [CrossRef]
- Lam, K.Y.; Chan, A.C.L.; Lo, C.Y. Morphological analysis of adrenal glands: A prospective analysis. Endocr. Pathol. 2001, 12, 33–38. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Goh, G.; Healy, J.M.; Fonseca, A.L.; Scholl, U.I.; Stenman, A.; Kunstman, J.W.; Brown, T.C.; Overton, J.D.; Mane, S.M.; et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, E493–E502. [Google Scholar] [CrossRef]
- Libé, R.; Borget, I.; Ronchi, C.L.; Zaggia, B.; Kroiss, M.; Kerkhofs, T.; Bertherat, J.; Volante, M.; Quinkler, M.; Chabre, O.; et al. ENSAT network. Prognostic factors in stage III-IV adrenocortical carcinomas (ACC): An European Network for the Study of Adrenal Tumor (ENSAT) study. Ann. Oncol. 2015, 26, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.; Byrd, D.R.; Compton, C.C.; Fritz, A.G.; Greene, F.; Trotti, A. Adrenal gland. In AJCC Cancer Staging Manual, 7th ed.; Edge, S., Byrd, D.R., Compton, C.C., Fritz, A.G., Greene, F., Trotti, A., Eds.; Springer: New York, NY, USA, 2010; Part IX, Chapter 47; pp. 515–520. [Google Scholar]
- Poorman, C.E.; Ethun, C.G.; Postlewait, L.M.; Tran, T.B.; Prescott, J.D.; Pawlik, T.M.; Wang, T.S.; Glenn, J.; Hatzaras, I.; Shenoy, R.; et al. A Novel T-Stage classification system for adrenocortical carcinoma: Proposal from the us adrenocortical carcinoma study group. Ann. Surg. Oncol. 2018, 25, 520–527. [Google Scholar] [CrossRef]
- Sandrini, R.; Ribeiro, R.C.; DeLacerda, L. Childhood adrenocortical tumors. J. Clin. Endocrinol. Metab. 1997, 82, 2027–2031. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Berney, D.; de Krijger, R.R.; Erickson, L.; Fassnacht, M.; Mete, O.; Papathomas, T.; Papotti, M.; Sasano, H.; Thompson, L.D.R.; et al. Data set for reporting of carcinoma of the adrenal cortex: Explanations and recommendations of the guidelines from the International Collaboration on Cancer Reporting. Hum. Pathol 2021, in press. [Google Scholar] [CrossRef]
- Lam, A.; Chong, G.; Dahlstrom, J.; McNicol, A.M.; Gill, A.; Sullivan, L. Adrenal Gland Tumours Structured Protocol, 1st ed.; Royal College of Pathologists: Surry Hills, NSW, Australia, 2013. [Google Scholar]
- Lam, A.; Dahlstrom, J.; Gupta, R.; Turchini, J.; Gill, A.J.; Schulte, K.M.; Elliott, M.; Clifton-Bligh, R.; Nolan, C.J. Carcinomas of the Adrenal Cortex Structured Reporting Protocol, 1st ed.; Royal College of Pathologists: Surry Hills, NSW, Australia, 2020; ISBN 978-1-76081-418-2; Available online: https://www.rcpa.edu.au/getattachment/7c2ed1b4-a38c-49a8-8d87-5c669049738f/Protocol-Adrenal-gland-tumours.aspx (accessed on 1 February 2021).
- Vural, V.; Kılınç, E.M.; Sarıdemir, D.; Gök, İ.B.; Hüseynov, A.; Akbarov, A.; Yaprak, M. Association between tumor size and malignancy risk in hormonally inactive adrenal incidentalomas. Cureus 2020, 12, e6574. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, N.; Avsar, E.; Tazegul, G.; Sari, R.; Altunbas, H.; Balci, M.K. Clinical characteristics and follow-up results of adrenal incidentaloma. Exp. Clin. Endocrinol. Diabetes 2020. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.; Lo, C.Y.; Lam, K.Y. Surgical implications of underestimation of adrenal tumour size by computed tomography. Br. J. Surg. 1999, 86, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.; Scarsbrook, A.; Abbas, A.; Fraser, S.; Limumpornpetch, P.; Dineen, R.; Stewart, P.M. Adrenal Incidentaloma. Endocr. Rev. 2020, 41, 775–820. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, A.; Samsel, R.; Papierska, L.; Roszkowska-Purska, K.; Nowak, K.; Jodkiewicz, Z.; Kasperlik-Załuska, A. Adrenal tumour bigger than 5 cm—What could it be? An analysis of 139 cases. Endokrynol. Pol. 2017, 68, 411–415. [Google Scholar] [CrossRef] [Green Version]
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci. Statements 2002, 19, 1–25.
- Kahramangil, B.; Kose, E.; Remer, E.M.; Reynolds, J.P.; Stein, R.; Rini, B.; Siperstein, A.; Berber, E. A modern assessment of cancer risk in adrenal incidentalomas: Analysis of 2219 patients. Ann. Surg. 2021, in press. [Google Scholar] [CrossRef]
- Vanderveen, K.A.; Thompson, S.M.; Callstrom, M.R.; Young WFJr Grant, C.S.; Farley, D.R.; Richards, M.L.; Thompson, G.B. Biopsy of pheochromocytomas and paragangliomas: Potential for disaster. Surgery 2009, 146, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Delivanis, D.A.; Erickson, D.; Atwell, T.D.; Natt, N.; Maraka, S.; Schmit, G.D.; Eiken, P.W.; Nathan, M.A.; Young, W.F., Jr.; Bancos, I. Procedural and clinical outcomes of percutaneous adrenal biopsy in a high-risk population for adrenal malignancy. Clin. Endocrinol. 2016, 85, 710–716. [Google Scholar] [CrossRef]
- Gupta, R.K.; Majumdar, K.; Saran, R.K.; Srivastava, S.; Sakhuja, P.; Batra, V.V. Role of endoscopic ultrasound-guided fine-needle aspiration in adrenal lesions: Analysis of 32 patients. J. Cytol. 2018, 35, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Altieri, B.; Ronchi, C.L.; Kroiss, M.; Fassnacht, M. Next-generation therapies for adrenocortical carcinoma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101434. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Conventional | Oncocytic | Myxoid | Sarcomatoid | |
|---|---|---|---|---|---|
| Adult | Paediatric | ||||
| Number of cases | ~8000 | ~200 | 56 | 47 | 28 |
| Mean Age | 47 to 55 | 5 (median = 4) | 48 | 48 | 56 |
| Most common age group (decade) | sixth/seventh | first (<5) | fourth | fifth | sixth/seventh |
| Male to Female | 1 to 1.4 | 1 to 2 | 1 to 1.1 | 1 to 1 | 1 to 1.3 |
| Functioning | half | 85% | half | 57% | 11% |
| Most common hormone produced | Cortisol | Sex hormones | Sex hormones | Cortisol | - |
| Laterality | left adrenal | left adrenal | left adrenal | left adrenal | right adrenal |
| Left to right ratio | 1.2 to 1 | 1.4 to 1 | 1.6 to 1 | 1.5 to 1 | 1 to 1.4 |
| Bilateral | 1% | - | none | none | 7% (n = 2) |
| Size (median/maximum) | 100–120 mm/280 mm | 95 mm/200 mm | 10 mm/285 mm | 100 mm/300 mm | 127 mm/240 mm |
| Weight (median [range]) | 528 g (38–4000) | 276 g (20–1046) | 552 g (50–5720) | 450 g (38.5–3200) | 620 g (20–6500) |
| Metastases | ~ one third | 31% | 13% | 68% | 75% |
| (26 to 35%) | |||||
| Median survival | 17–35 months | - | 60 months | 29 months | 7 months |
| Prognostic Grouping | 7th Edition | 8th Edition |
|---|---|---|
| Stage I | T1 N0 M0 | T1 N0 M0 |
| Stage II | T2 N0 M0 | T2 N0 M0 |
| Stage III | T3 N0 M0 | T3 N0 M0 |
| T1/2 N1 M0 | T1/2 N1 M0 | |
| T4 N0 M0 | ||
| T3/4 N1 M0 | ||
| Stage IV | T4 N0 M0 | |
| T3/4 N1 M0 | ||
| Any T N M1 | Any T N M1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, A.K.-y. Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging. Biomedicines 2021, 9, 175. https://doi.org/10.3390/biomedicines9020175
Lam AK-y. Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging. Biomedicines. 2021; 9(2):175. https://doi.org/10.3390/biomedicines9020175
Chicago/Turabian StyleLam, Alfred King-yin. 2021. "Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging" Biomedicines 9, no. 2: 175. https://doi.org/10.3390/biomedicines9020175
APA StyleLam, A. K.-y. (2021). Adrenocortical Carcinoma: Updates of Clinical and Pathological Features after Renewed World Health Organisation Classification and Pathology Staging. Biomedicines, 9(2), 175. https://doi.org/10.3390/biomedicines9020175