
ALK2 Receptor Kinase Association with FKBP12.6 Is Structurally Conserved with the ALK2-FKBP12 Complex


Abstract

1. Introduction
2. Experimental Section
2.1. Plasmids
2.2. Immunoprecipitation
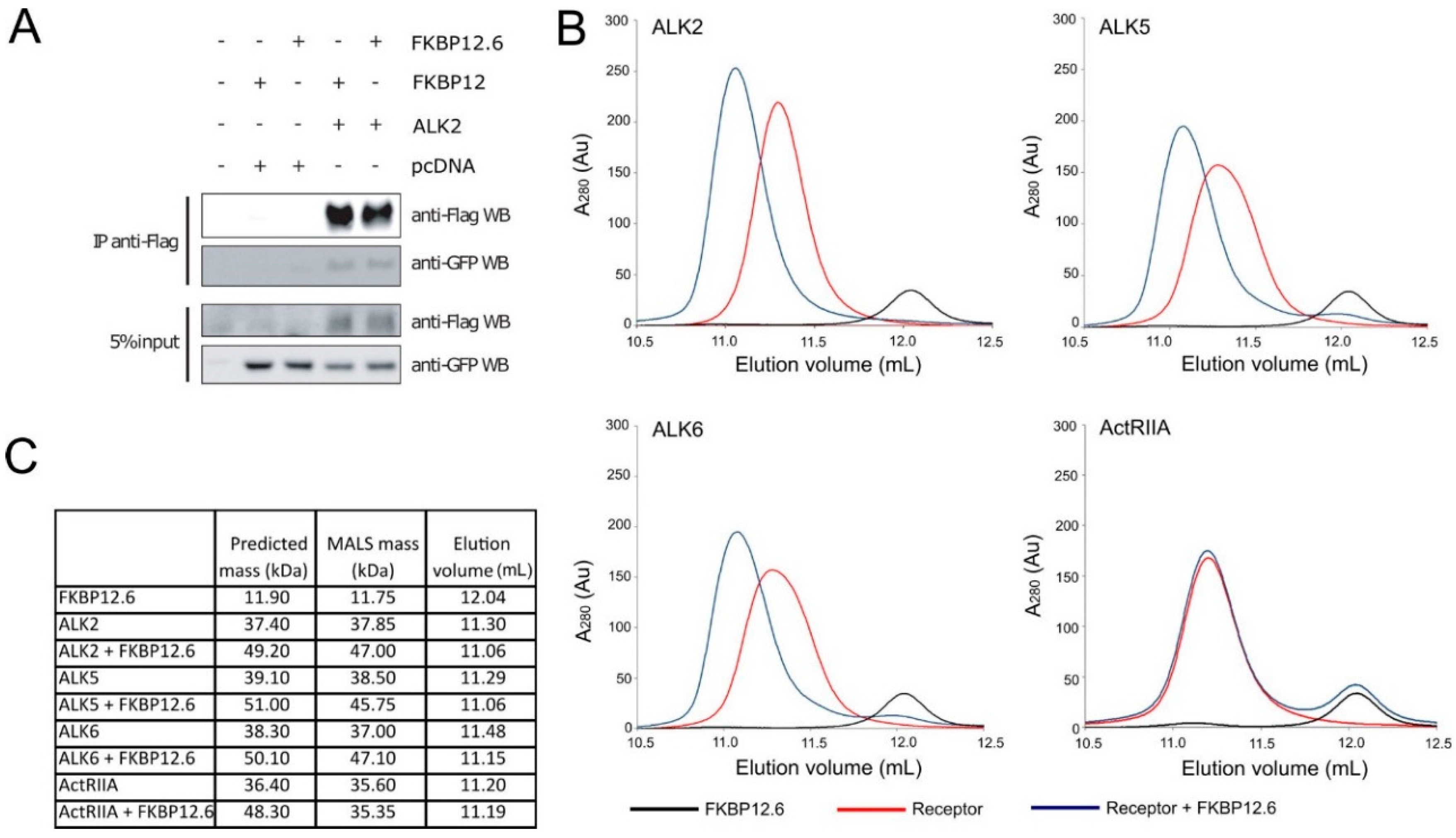
2.3. SEC-MALS
2.4. Protein Expression and Purification
2.5. Crystallization
2.6. Structure Determination
2.7. Accession Numbers
3. Results
3.1. FKBP12.6 Binds to Type I Receptors
3.2. Structure Determination of ALK2 Complexed with FKBP12.6
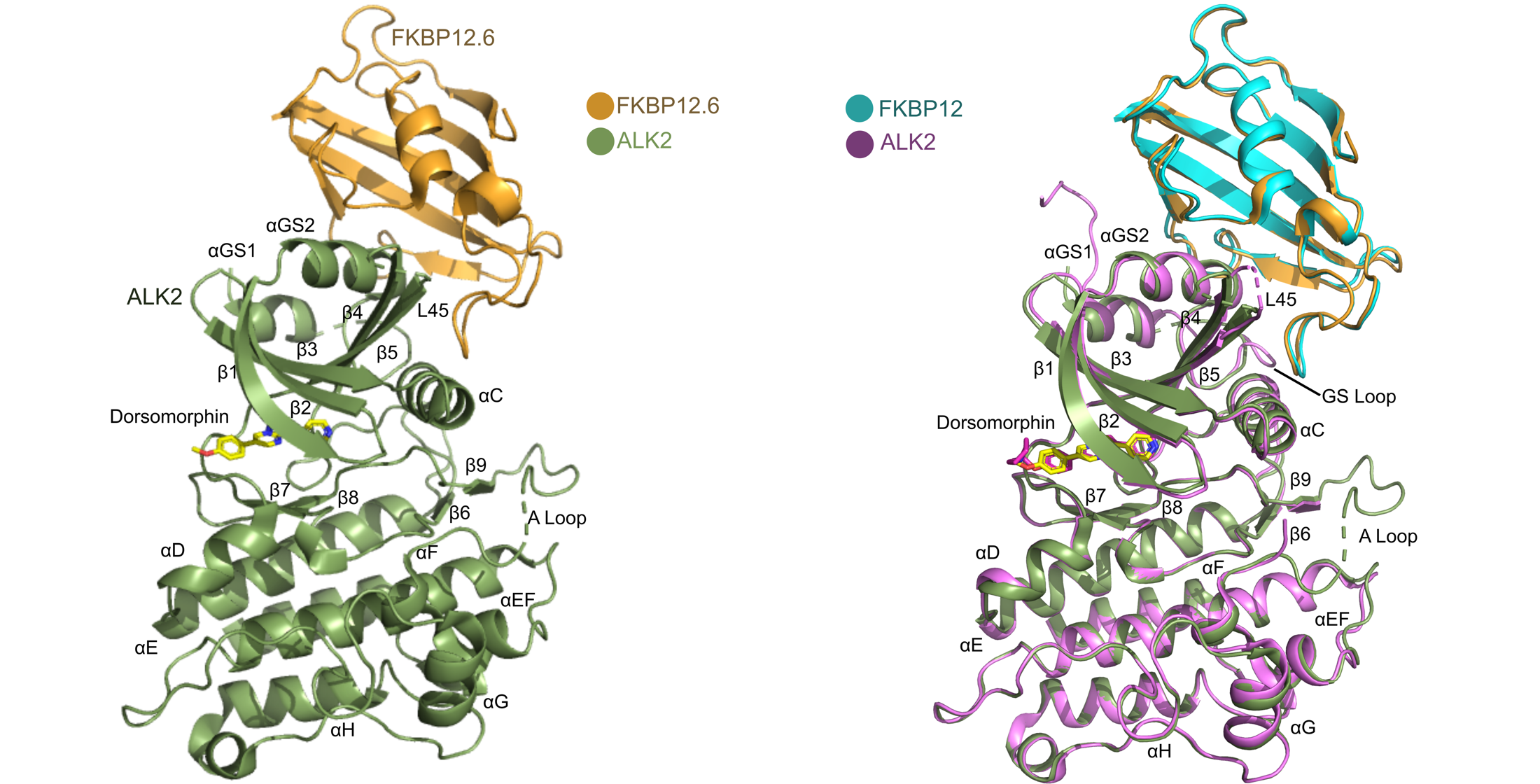
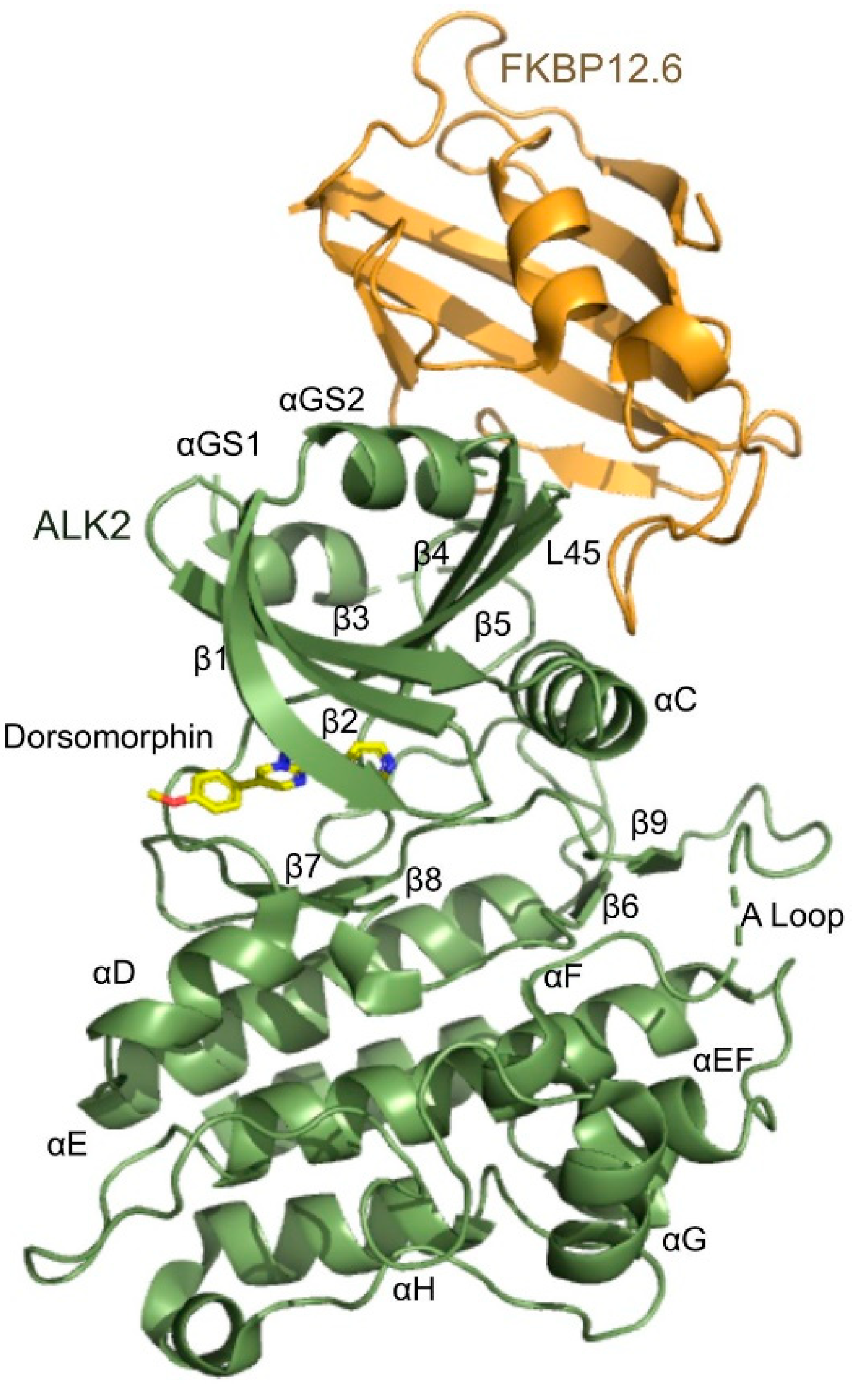
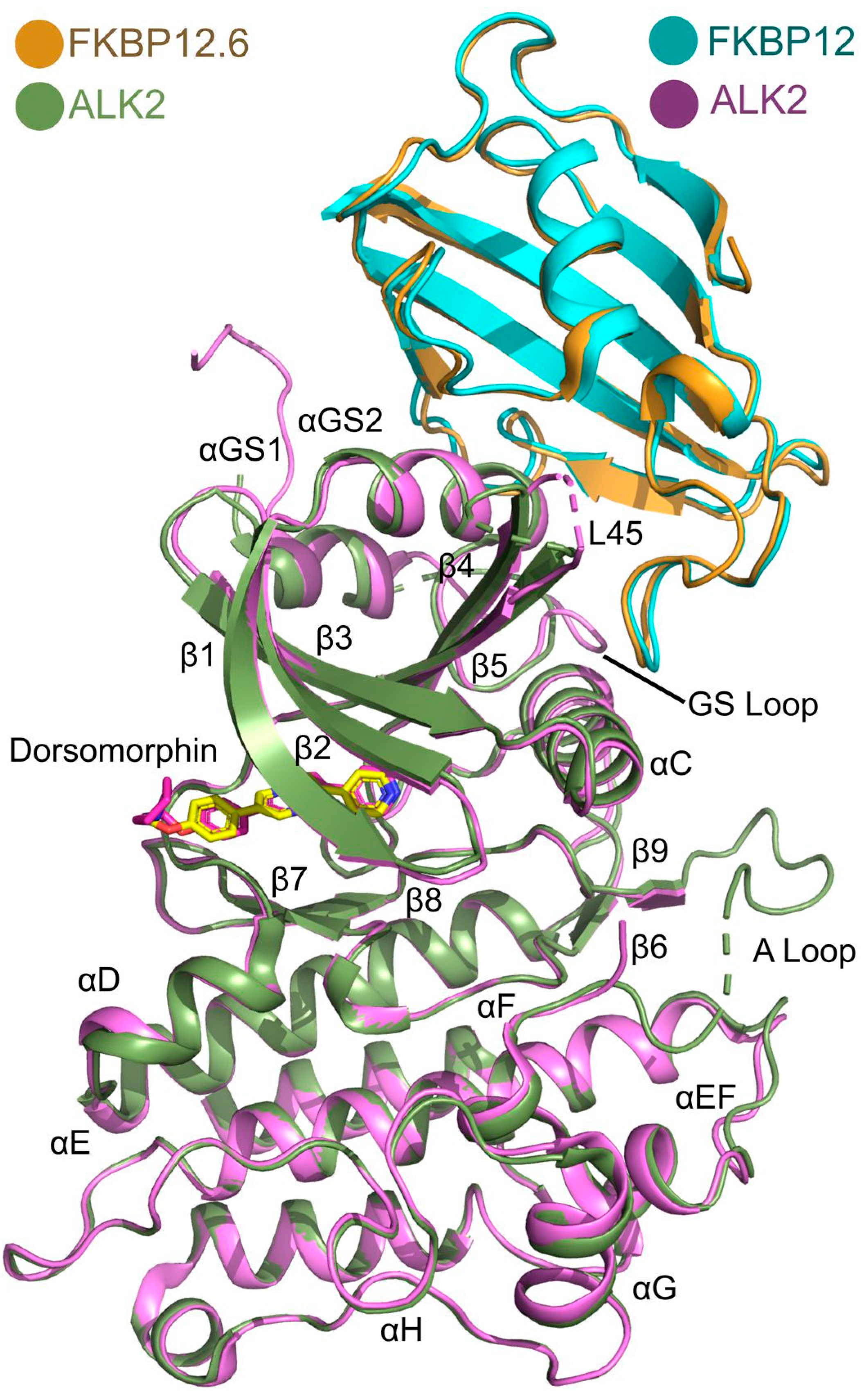
3.3. Overview of the Structure of the ALK2-FKBP12.6 Complex
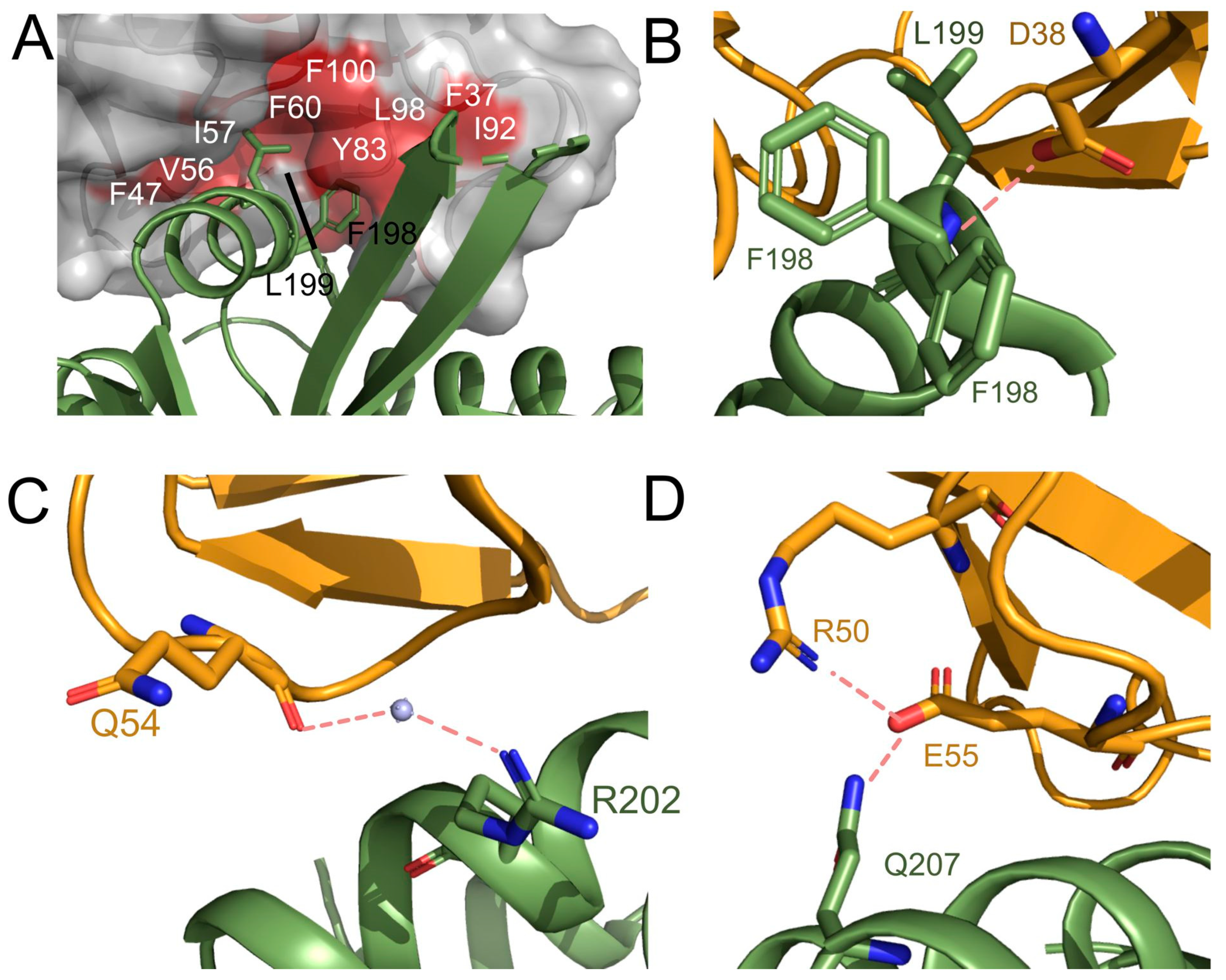
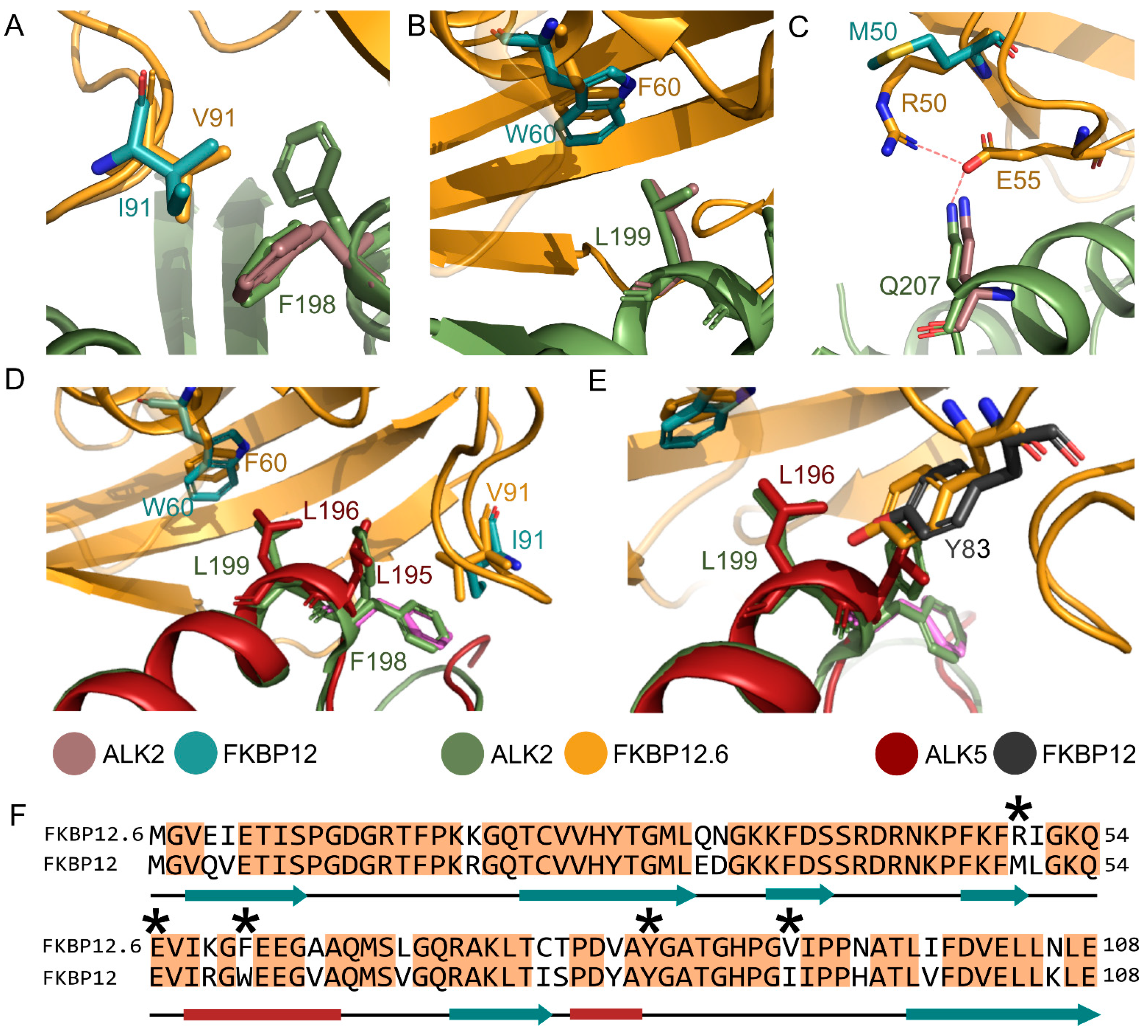
3.4. Comparison with the ALK2-FKBP12 Complex
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGF beta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hill, C.S. TGF-beta Superfamily Signaling in Embryonic Development and Homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef]
- Lowery, J.W.; Rosen, V. Bone Morphogenetic Protein-Based Therapeutic Approaches. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Taylor, K.R.; Vinci, M.; Bullock, A.N.; Jones, C. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. [Google Scholar] [CrossRef]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharm. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massague, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Chaikuad, A.; Bullock, A.N. Structural Basis of Intracellular TGF-beta Signaling: Receptors and Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia Ossificans Progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massague, J. The TGF beta receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2 rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef]
- van Dinther, M.; Visser, N.; de Gorter, D.J.J.; Doorn, J.; Goumans, M.-J.; de Boer, J.; ten Dijke, P. ALK2 R206H Mutation Linked to Fibrodysplasia Ossificans Progressiva Confers Constitutive Activity to the BMP Type I Receptor and Sensitizes Mesenchymal Cells to BMP-Induced Osteoblast Differentiation and Bone Formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef]
- Groppe, J.C.; Wu, J.; Shore, E.M.; Kaplan, F.S. In vitro Analyses of the Dysregulated R206H ALK2 Kinase-FKBP12 Interaction Associated with Heterotopic Ossification in FOP. Cells Tissues Organs 2011, 194, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, W.; Lacroix, J.J.; Luo, Y.; Hao, J. Insight into Molecular Mechanism for Activin A-Induced Bone Morphogenetic Protein Signaling. Int. J. Mol. Sci. 2020, 21, 6498. [Google Scholar] [CrossRef] [PubMed]
- Strain-Damerell, C.; Mahajan, P.; Fernandez-Cid, A.; Gileadi, O.; Burgess-Brown, N.A. Screening and Production of Recombinant Human Proteins: Ligation-Independent Cloning. Methods Mol. Biol. 2021, 2199, 23–43. [Google Scholar] [CrossRef]
- Gronroos, E.; Kingston, I.J.; Ramachandran, A.; Randall, R.A.; Vizan, P.; Hill, C.S. Transforming growth factor beta inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol. Cell Biol. 2012, 32, 2904–2916. [Google Scholar] [CrossRef] [PubMed]
- Burgess-Brown, N.A.; Mahajan, P.; Strain-Damerell, C.; Fernandez-Cid, A.; Gileadi, O.; Graslund, S. Screening and Production of Recombinant Human Proteins: Protein Production in E. coli. Methods Mol. Biol. 2021, 2199, 45–66. [Google Scholar] [CrossRef]
- Mahajan, P.; Strain-Damerell, C.; Mukhopadhyay, S.; Fernandez-Cid, A.; Gileadi, O.; Burgess-Brown, N.A. Screening and Production of Recombinant Human Proteins: Protein Production in Insect Cells. Methods Mol. Biol. 2021, 2199, 67–94. [Google Scholar] [CrossRef]
- Leslie, A.G. The integration of macromolecular diffraction data. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 48–57. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Kovalevskiy, O.; Nicholls, R.A.; Long, F.; Carlon, A.; Murshudov, G.N. Overview of refinement proceduRes. within REFMAC5: Utilizing data from different sources. Acta Cryst. D Struct. Biol. 2018, 74, 215–227. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef]
- Ng, J.T.; Dekker, C.; Reardon, P.; von Delft, F. Lessons from ten years of crystallization experiments at the SGC. Acta Cryst. D Struct. Biol. 2016, 72, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hahle, A.; Geiger, T.M.; Merz, S.; Meyners, C.; Tianqi, M.; Kolos, J.; Hausch, F. FKBP51 and FKBP12.6-Novel and tight interactors of Glomulin. PLoS ONE 2019, 14, e0221926. [Google Scholar] [CrossRef] [PubMed]
- Sinars, C.R.; Cheung-Flynn, J.; Rimerman, R.A.; Scammell, J.G.; Smith, D.F.; Clardy, J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 868–873. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y. FK506-Binding Proteins and Their Diverse Functions. Curr. Mol. Pharm. 2015, 9, 48–65. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Accession Code | PDB ID: 4C02 |
|---|---|
| Data collection | |
| Beamline | Diamond I04 |
| Wavelength (Å) | 0.9686 |
| Resolution (Å) | 39.79–2.17 |
| Spacegroup | P 41 3 2 |
| Cell dimensions | a = 182.33, b = 182.33, c = 182.33 Å |
| α = 90°, β = 90°, γ = 90.0° | |
| No. unique reflections | 54946 |
| Completeness 1 (%) | 99.8 (99.7) |
| I/σI 1 | 8.0 (2.0) |
| Rmerge 1 | 0.17 (1.07) |
| Redundancy 1 | 9.0 (9.3) |
| Refinement | |
| No. refinement atoms: | |
| Macromolecules | 3271 |
| Ligands | 179 |
| Waters | 231 |
| Rfact (%) | 17.73 |
| Rfree (%) | 19.75 |
| rms deviation bond 2 (Å) | 0.0069 |
| rms deviation angle 2 (°) | 1.5390 |
| Ramachandran favour | 97.4% |
| Ramachandran allowed | 99.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.; Riesebos, E.; Kerr, G.; Bullock, A.N. ALK2 Receptor Kinase Association with FKBP12.6 Is Structurally Conserved with the ALK2-FKBP12 Complex. Biomedicines 2021, 9, 129. https://doi.org/10.3390/biomedicines9020129
Williams E, Riesebos E, Kerr G, Bullock AN. ALK2 Receptor Kinase Association with FKBP12.6 Is Structurally Conserved with the ALK2-FKBP12 Complex. Biomedicines. 2021; 9(2):129. https://doi.org/10.3390/biomedicines9020129
Chicago/Turabian StyleWilliams, Eleanor, Elise Riesebos, Georgina Kerr, and Alex N. Bullock. 2021. "ALK2 Receptor Kinase Association with FKBP12.6 Is Structurally Conserved with the ALK2-FKBP12 Complex" Biomedicines 9, no. 2: 129. https://doi.org/10.3390/biomedicines9020129
APA StyleWilliams, E., Riesebos, E., Kerr, G., & Bullock, A. N. (2021). ALK2 Receptor Kinase Association with FKBP12.6 Is Structurally Conserved with the ALK2-FKBP12 Complex. Biomedicines, 9(2), 129. https://doi.org/10.3390/biomedicines9020129