
Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis


 and
and 
Abstract
:1. Sideroflexins: From Structure to Function
1.1. Sideroflexins from an Historical Point of View
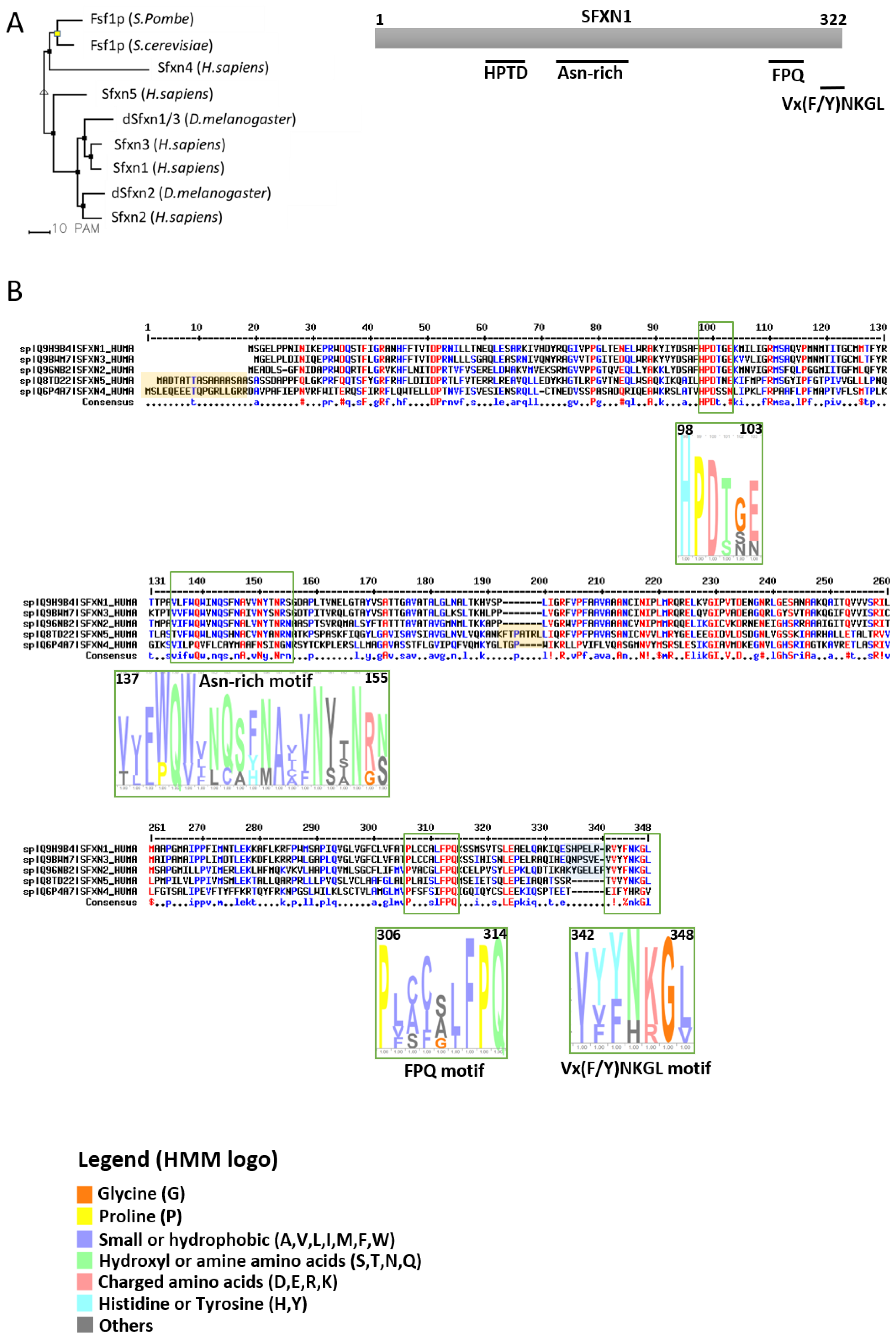
1.2. The Sideroflexin Family: From Genes to Proteins
1.3. Sideroflexins Are Mitochondrial Transporters Implicated in One-Carbon Metabolism
1.4. Sideroflexins in Disease
2. Sideroflexins and Mitochondrial Respiration
2.1. Overview of the Mitochondrial Respiratory Complexes and the Place of Iron in RC
2.2. Current Knowledge on the Regulation of Mitochondrial Respiration by SFXN Proteins
3. Which Place for Sideroflexins in the Regulation of Mitochondrial Metabolism?
3.1. Sideroflexins and One-Carbon Metabolism (OCM)
3.2. Sideroflexins in Central Carbon Metabolism
4. Sideroflexins, Iron Homeostasis and Heme Biosynthesis
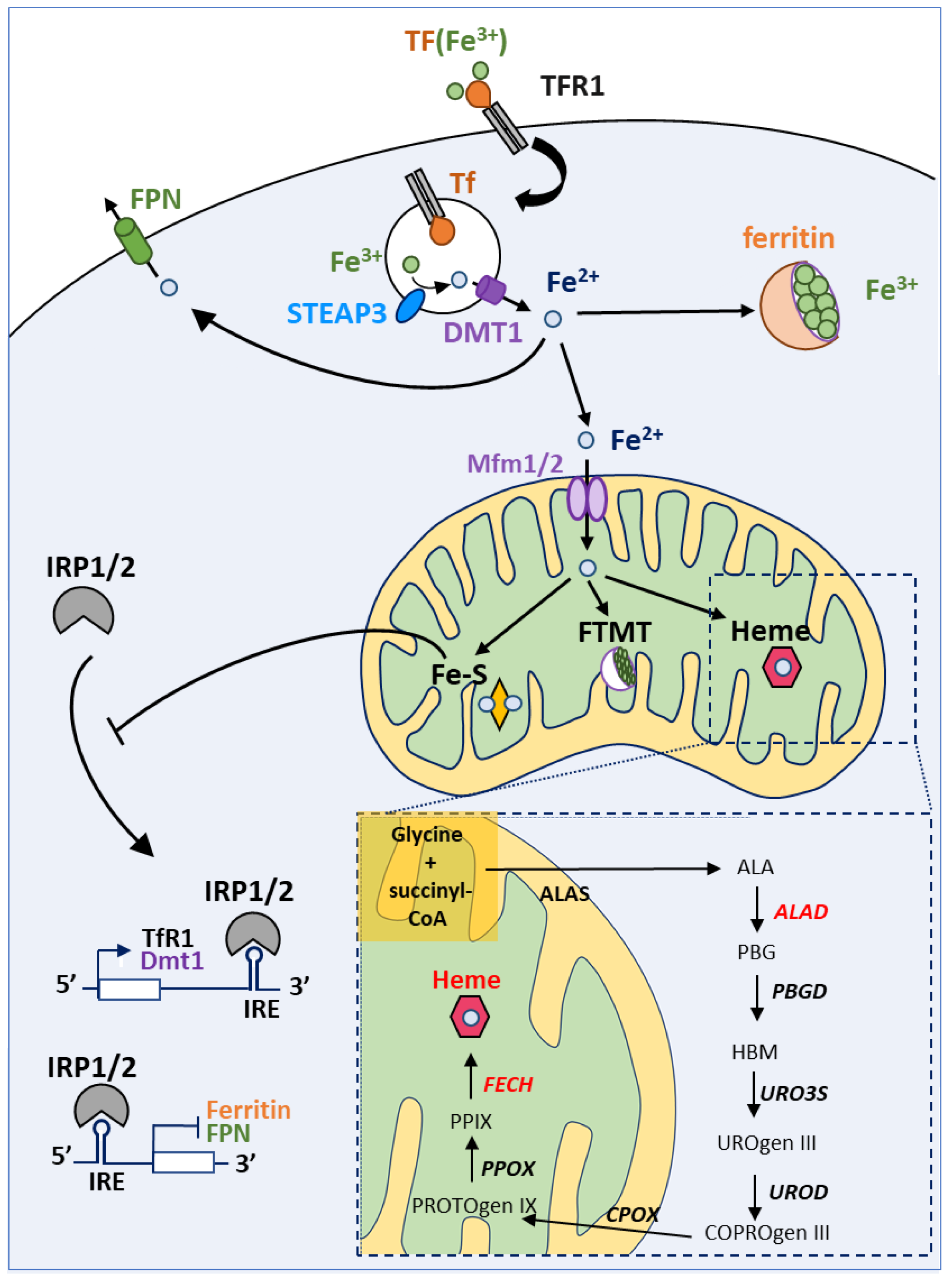
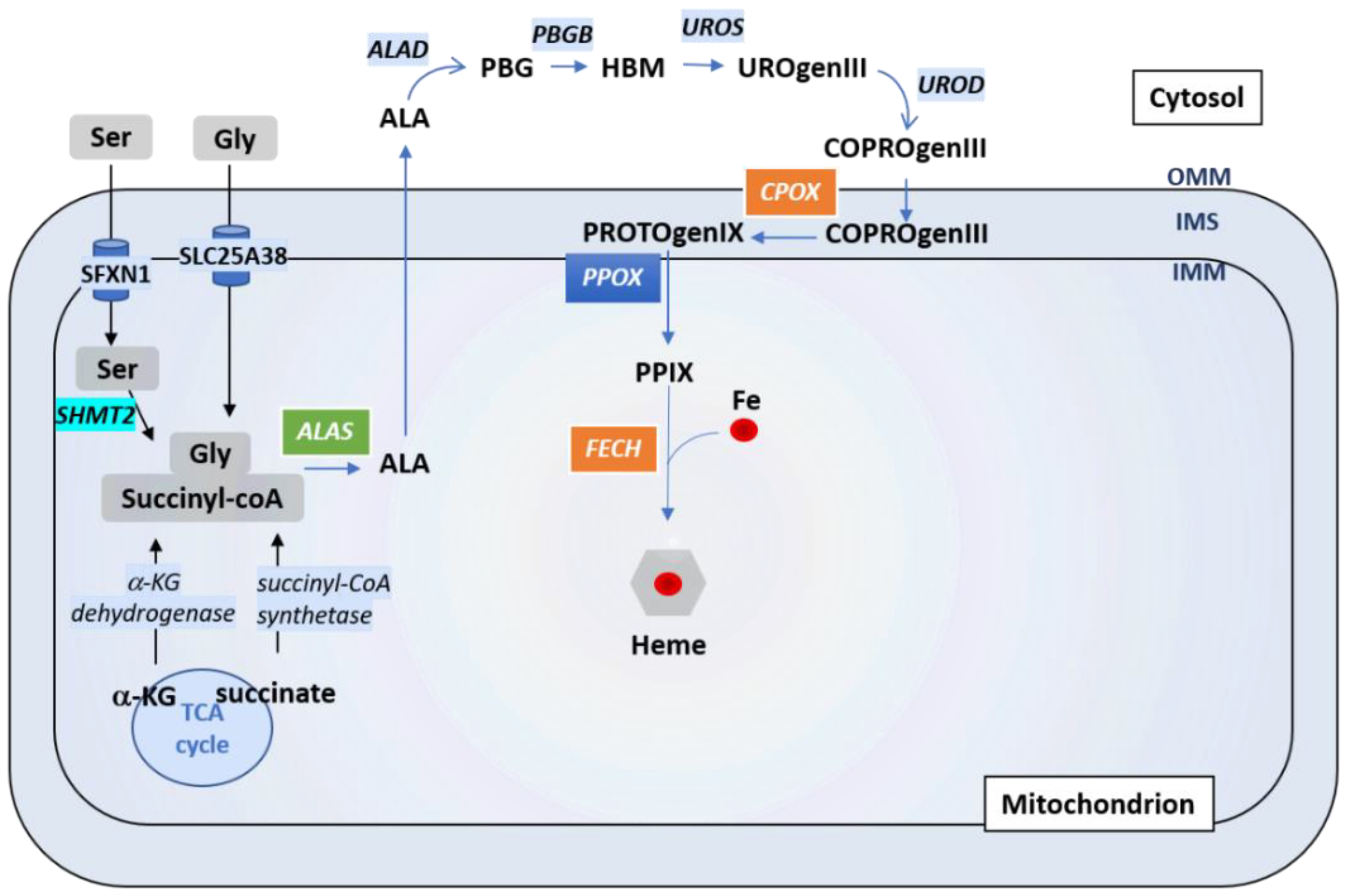
4.1. A Brief Overview of Iron Homeostasis, ISCs and Heme Biosynthesis
4.2. Can Sideroflexins Regulate Iron Homeostasis?
4.3. Which Role for Sideroflexins in Heme Biosynthesis and ISC Biosynthesis?
4.4. Which Role for Sideroflexins in ISC Biosynthesis?
5. Sideroflexins, Ferroptosis and Ferritinophagy
5.1. SFXN, Cell Death and Ferroptosis
5.2. SFXN1 and Ferritinophagy
6. Sideroflexins in Aging: May SFXN Regulate Neuronal Physiology and Retinal Function?
6.1. Sideroflexins and Biometals in Neuronal Physiopathology
6.2. Sfxn and Retinal Degeneration
7. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A
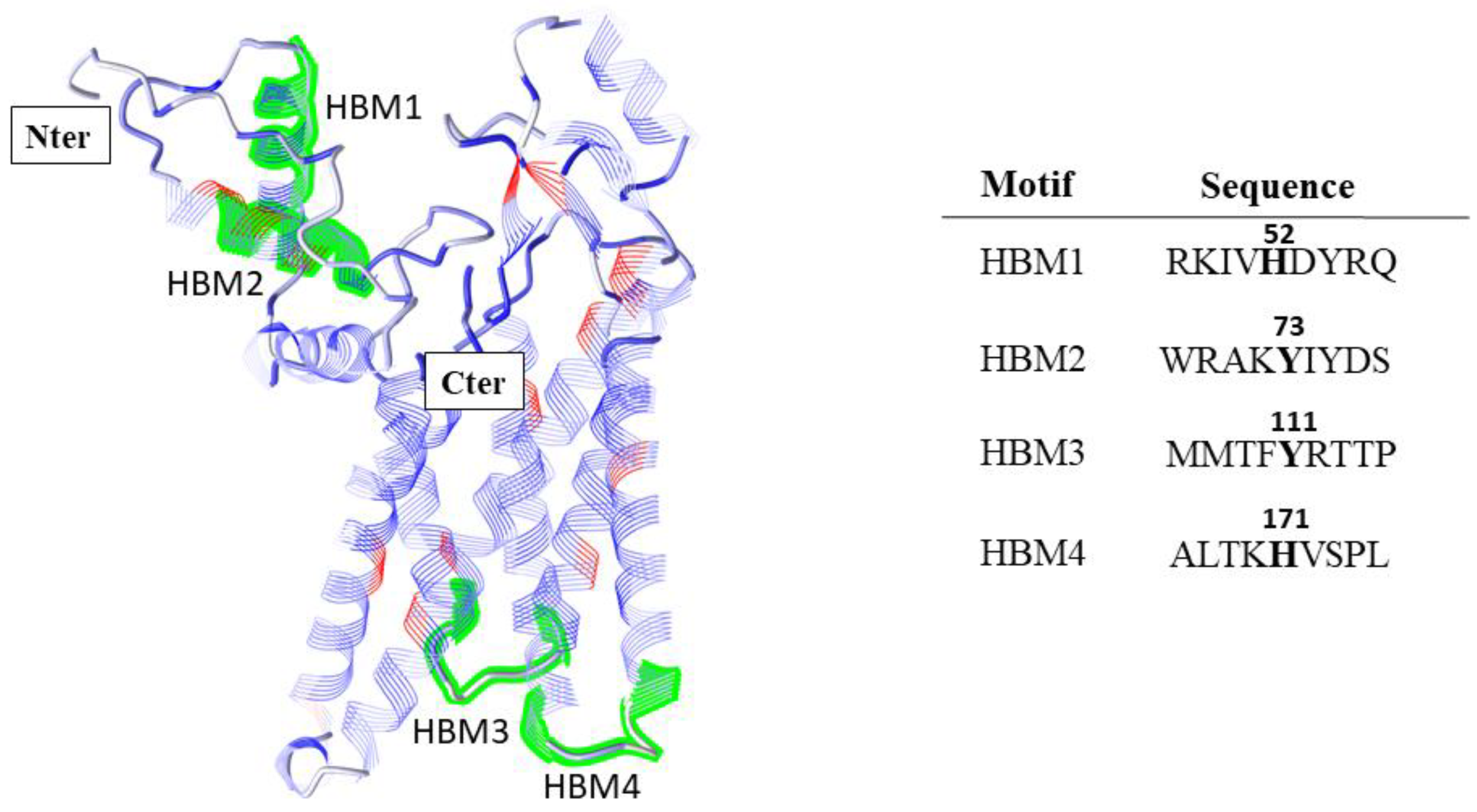
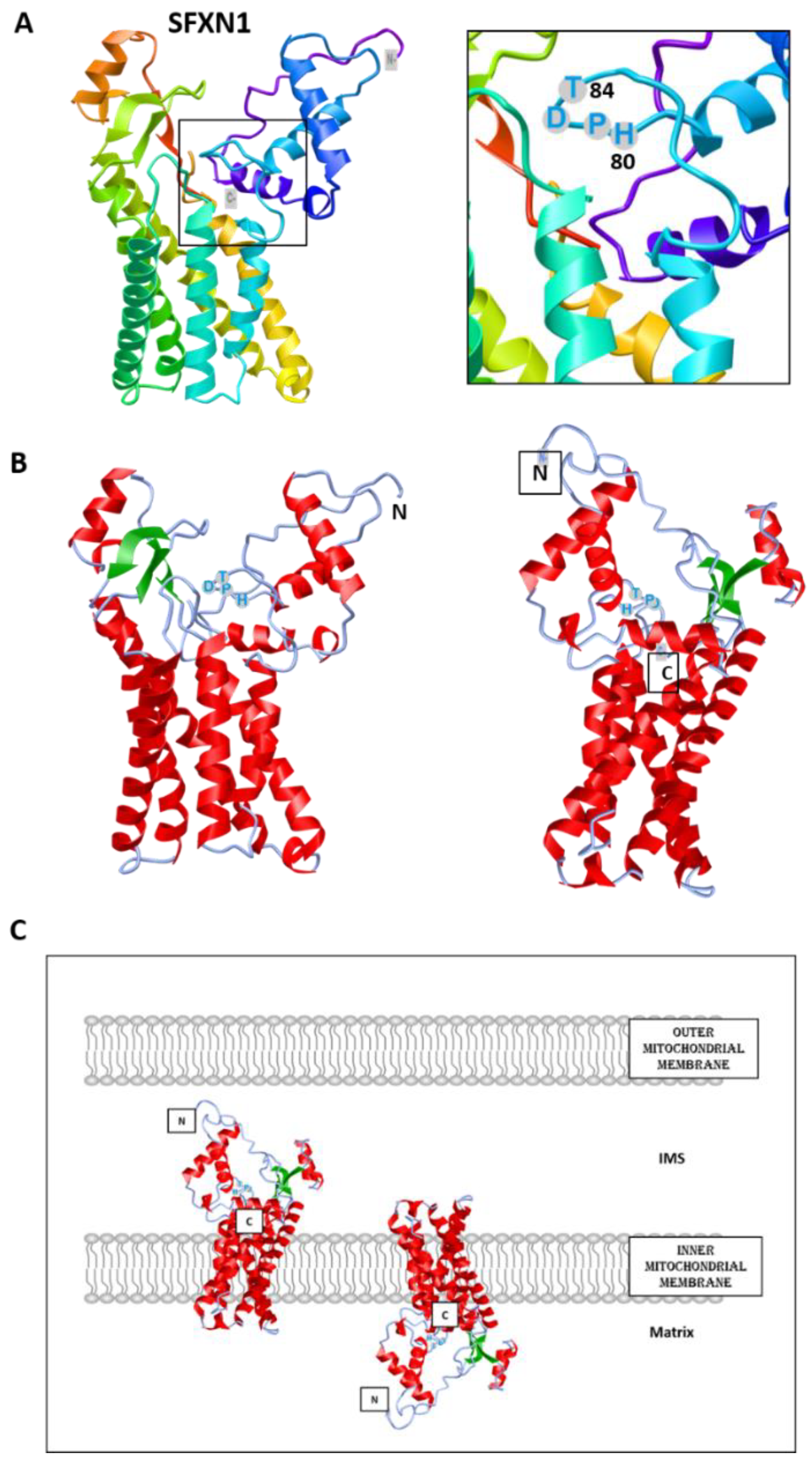
Appendix A.1. Prediction of Heme Binding Motifs in SFXN1
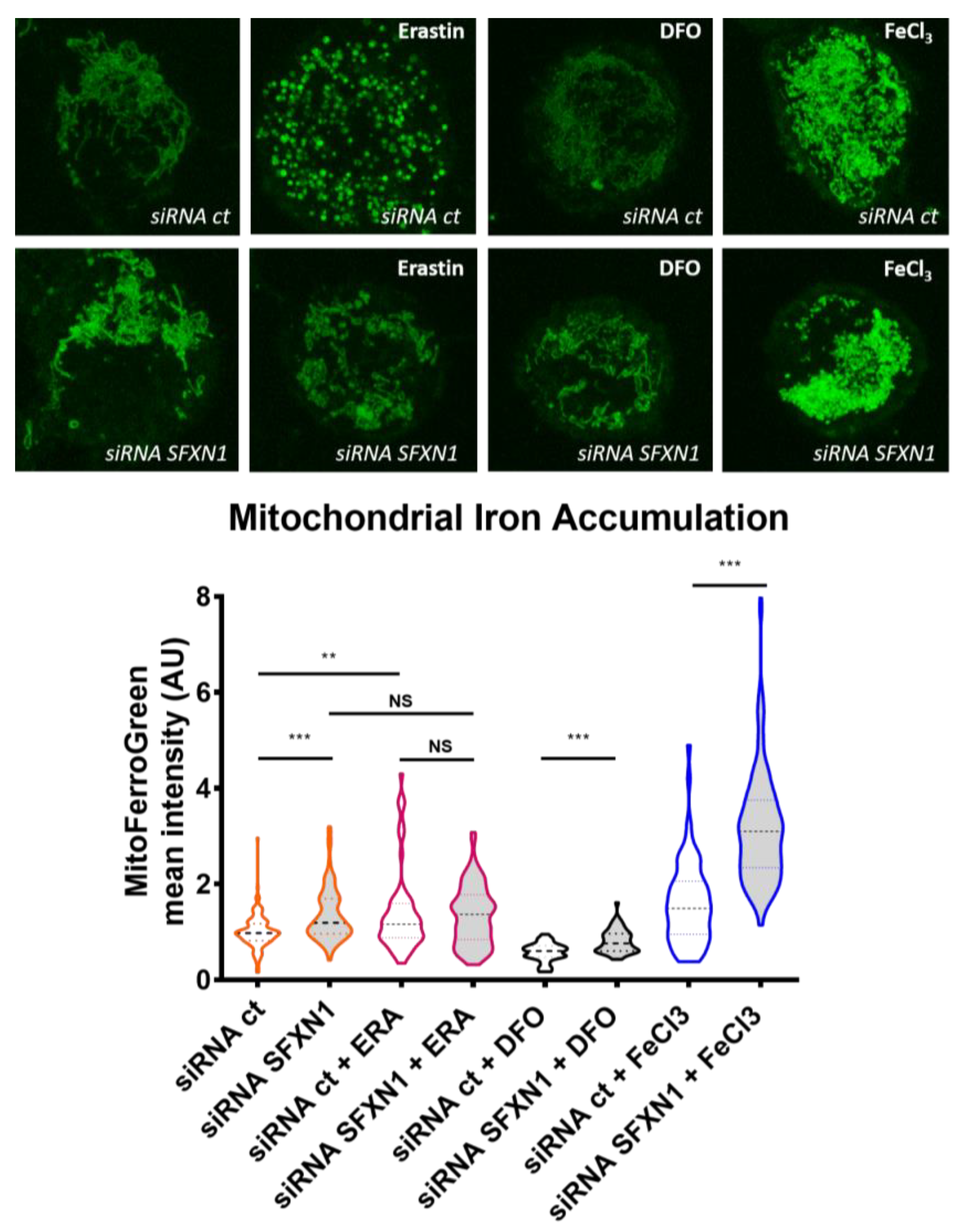
Appendix A.2. Mitochondrial Labile Iron Staining with the Mito-FerroGreen Fluorescent Probe
References
- Fleming, M.D. A Mutation in a Mitochondrial Transmembrane Protein Is Responsible for the Pleiotropic Hematological and Skeletal Phenotype of Flexed-Tail (f/f) Mice. Genes Dev. 2001, 15, 652–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenox, L.E.; Perry, J.M.; Paulson, R.F. BMP4 and Madh5 Regulate the Erythroid Response to Acute Anemia. Blood 2005, 105, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, S.; Lenox, L.E.; Lariviere, A.; Porayette, P.; Perry, J.M.; Yon, M.; Paulson, R.F. An Intronic Sequence Mutated in Flexed-Tail Mice Regulates Splicing of Smad5. Mamm. Genome 2007, 18, 852–860. [Google Scholar] [CrossRef] [PubMed]
- SLC56 Sideroflexins. IUPHAR/BPS Guide to PHARMACOLOGY. 2020. Available online: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1008 (accessed on 20 January 2021).
- Li, X.; Han, D.; Kin Ting Kam, R.; Guo, X.; Chen, M.; Yang, Y.; Zhao, H.; Chen, Y. Developmental Expression of Sideroflexin Family Genes in Xenopus Embryos. Dev. Dyn. 2010, 239, 2742–2747. [Google Scholar] [CrossRef]
- Lockhart, P.J.; Holtom, B.; Lincoln, S.; Hussey, J.; Zimprich, A.; Gasser, T.; Wszolek, Z.K.; Hardy, J.; Farrer, M.J. The Human Sideroflexin 5 (SFXN5) Gene: Sequence, Expression Analysis and Exclusion as a Candidate for PARK3. Gene 2002, 285, 229–237. [Google Scholar] [CrossRef]
- Miotto, G.; Tessaro, S.; Rotta, G.A.; Bonatto, D. In Silico Analyses of Fsf1 Sequences, a New Group of Fungal Proteins Orthologous to the Metazoan Sideroblastic Anemia-Related Sideroflexin Family. Fungal Genet. Biol. 2007, 44, 740–753. [Google Scholar] [CrossRef]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; Chan, S.H.; Lewis, C.A.; Wang, T.; Keys, H.R.; et al. SFXN1 Is a Mitochondrial Serine Transporter Required for One-Carbon Metabolism. Science 2018, 362, eaat9528. [Google Scholar] [CrossRef] [Green Version]
- Mon, E.E.; Wei, F.-Y.; Ahmad, R.N.R.; Yamamoto, T.; Moroishi, T.; Tomizawa, K. Regulation of Mitochondrial Iron Homeostasis by Sideroflexin 2. J. Physiol. Sci. 2019, 69, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Corpet, F. Multiple Sequence Alignment with Hierarchical Clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Gyimesi, G.; Hediger, M.A. Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules 2020, 10, 1611. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Youkharibache, P.; Zhang, D.; Lanczycki, C.J.; Geer, R.C.; Madej, T.; Phan, L.; Ward, M.; Lu, S.; Marchler, G.H.; et al. ICn3D, a Web-Based 3D Viewer for Sharing 1D/2D/3D Representations of Biomolecular Structures. Bioinformatics 2020, 36, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.-M.; Rhee, H.-W. APEX, a Master Key to Resolve Membrane Topology in Live Cells. Biochemistry 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kang, M.-G.; Park, J.-S.; Lee, G.; Ting, A.Y.; Rhee, H.-W. APEX Fingerprinting Reveals the Subcellular Localization of Proteins of Interest. Cell Rep. 2016, 15, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acoba, M.G.; Selen Alpergin, E.S.; Renuse, S.; Fernández-del-Río, L.; Lu, Y.-W.; Clarke, C.F.; Pandey, A.; Wolfgang, M.J.; Claypool, S.M. The Mitochondrial Carrier SFXN1 Is Critical for Complex III Integrity and Cellular Metabolism; Cell Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Jackson, T.D.; Hock, D.; Palmer, C.S.; Kang, Y.; Fujihara, K.M.; Clemons, N.J.; Thorburn, D.R.; Stroud, D.A.; Stojanovski, D. The TIM22 Complex Regulates Mitochondrial One-Carbon Metabolism by Mediating the Import of Sideroflexins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rivell, A.; Petralia, R.S.; Wang, Y.-X.; Mattson, M.P.; Yao, P.J. Sideroflexin 3 Is a Mitochondrial Protein Enriched in Neurons. Neuromol. Med. 2019, 21, 314–321. [Google Scholar] [CrossRef]
- Hildick-Smith, G.J.; Cooney, J.D.; Garone, C.; Kremer, L.S.; Haack, T.B.; Thon, J.N.; Miyata, N.; Lieber, D.S.; Calvo, S.E.; Akman, H.O.; et al. Macrocytic Anemia and Mitochondriopathy Resulting from a Defect in Sideroflexin 4. Am. J. Hum. Genet. 2013, 93, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-Type-Specific Profiling of Brain Mitochondria Reveals Functional and Molecular Diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Azzi, A.; Glerum, M.; Koller, R.; Mertens, W.; Spycher, S. The Mitochondrial Tricarboxylate Carrier. J. Bioenergy Biomembr. 1993, 25, 515–524. [Google Scholar] [CrossRef]
- Miyake, S.; Yamashita, T.; Taniguchi, M.; Tamatani, M.; Sato, K.; Tohyama, M. Identification and Characterization of a Novel Mitochondrial Tricarboxylate Carrier. Biochem. Biophys. Res. Commun. 2002, 295, 463–468. [Google Scholar] [CrossRef]
- Kovaleva, G.Y.; Bazykin, G.A.; Brudno, M.; Gelfand, M.S. Comparative genomics of transcriptional regulation in yeasts and ITS application to identification of a candidate alpha-isopropylmalate transporter. J. Bioinform. Comput. Biol. 2006, 4, 981–998. [Google Scholar] [CrossRef] [Green Version]
- Wood, V.; Harris, M.A.; McDowall, M.D.; Rutherford, K.; Vaughan, B.W.; Staines, D.M.; Aslett, M.; Lock, A.; Bahler, J.; Kersey, P.J.; et al. PomBase: A Comprehensive Online Resource for Fission Yeast. Nucleic Acids Res. 2012, 40, D695–D699. [Google Scholar] [CrossRef] [PubMed]
- Lock, A.; Rutherford, K.; Harris, M.A.; Hayles, J.; Oliver, S.G.; Bähler, J.; Wood, V. PomBase 2018: User-Driven Reimplementation of the Fission Yeast Database Provides Rapid and Intuitive Access to Diverse, Interconnected Information. Nucleic Acids Res. 2019, 47, D821–D827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Xu, J.; Cheng, C.; Yin, G.; Zeng, L.; Ji, C.; Gu, S.; Xie, Y.; Mao, Y. Isolation and Characterization of a Novel Human Putative Anemia-Related Gene Homologous to Mouse Sideroflexin. Biochem. Genet. 2003, 41, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, M.M.; Penmatsa, A.; Whittaker, J.W. The Mtm1p Carrier and Pyridoxal 5′-Phosphate Cofactor Trafficking in Yeast Mitochondria. Arch. Biochem. Biophys. 2015, 568, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, J.W. Intracellular Trafficking of the Pyridoxal Cofactor. Implications for Health and Metabolic Disease. Arch. Biochem. Biophys. 2016, 592, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, R.; Lunetti, P.; Zara, V.; Ferramosca, A.; Marra, F.; Fiermonte, G.; Cappello, A.R.; De Leonardis, F.; Capobianco, L.; Dolce, V. Drosophila Melanogaster Mitochondrial Carriers: Similarities and Differences with the Human Carriers. Int. J. Mol. Sci. 2020, 21, 6052. [Google Scholar] [CrossRef]
- Sofou, K.; Hedberg-Oldfors, C.; Kollberg, G.; Thomsen, C.; Wiksell, Å.; Oldfors, A.; Tulinius, M. Prenatal onset of mitochondrial disease is associated with sideroflexin 4 deficiency. Mitochondrion 2019, 47, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 Affects Fe-S Cluster Biogenesis, Iron Metabolism, Mitochondrial Respiration and Heme Biosynthetic Enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef]
- Gylfe, A.E.; Katainen, R.; Kondelin, J.; Tanskanen, T.; Cajuso, T.; Hänninen, U.; Taipale, J.; Taipale, M.; Renkonen-Sinisalo, L.; Järvinen, H.; et al. Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Weston, C.; Klobusicky, J.; Weston, J.; Connor, J.; Toms, S.A.; Marko, N.F. Aberrations in the Iron Regulatory Gene Signature Are Associated with Decreased Survival in Diffuse Infiltrating Gliomas. PLoS ONE 2016, 11, e0166593. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.D.; Coffman, L.G.; Chou, J.W.; Black, M.A.; Bergh, J.; D’Agostino, R.; Torti, S.V.; Torti, F.M. An Iron Regulatory Gene Signature Predicts Outcome in Breast Cancer. Cancer Res. 2011, 71, 6728–6737. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Gu, J.; Zong, S.; Guo, R.; Liu, T.; Yang, M. Research Journey of Respirasome. Protein Cell 2020, 11, 318–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and Mechanism of Mitochondrial Electron Transport Chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; So, M.; Kaguni, L.S. Iron-Sulfur Clusters in Mitochondrial Metabolism: Multifaceted Roles of a Simple Cofactor. Biochem. Mosc. 2016, 81, 1066–1080. [Google Scholar] [CrossRef]
- Barros, M.H.; McStay, G.P. Modular Biogenesis of Mitochondrial Respiratory Complexes. Mitochondrion 2020, 50, 94–114. [Google Scholar] [CrossRef]
- Letts, J.A.; Sazanov, L.A. Clarifying the Supercomplex: The Higher-Order Organization of the Mitochondrial Electron Transport Chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Ndi, M.; Marin-Buera, L.; Salvatori, R.; Singh, A.P.; Ott, M. Biogenesis of the Bc1 Complex of the Mitochondrial Respiratory Chain. J. Mol. Biol. 2018, 430, 3892–3905. [Google Scholar] [CrossRef]
- Sá, E.; Lobo, T.; Fox, J.L.; Zeviani, M.; Winge, D.R.; Fernández-Vizarra, E. LYRM7/MZM1L Is a UQCRFS1 Chaperone Involved in the Last Steps of Mitochondrial Complex III Assembly in Human Cells. Biochim. Biophys. Acta (BBA) Bioenergy 2013, 1827, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.K.; Borgnia, M.J.; Hsu, A.L.; Esser, L.; Fox, T.; de Val, N.; Xia, D. Structures of AAA Protein Translocase Bcs1 Suggest Translocation Mechanism of a Folded Protein. Nat. Struct. Mol. Biol. 2020, 27, 202–209. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE Syndrome, a Lethal Metabolic Disorder with Iron Overload, Is Caused by a Point Mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, I.S.; Graham, L.C.; Carter, R.N.; Morton, N.M.; Hammachi, F.; Kunath, T.; Pennetta, G.; Carpanini, S.M.; Manson, J.C.; Lamont, D.J.; et al. Sideroflexin 3 Is an α-Synuclein-Dependent Mitochondrial Protein That Regulates Synaptic Morphology. J. Cell. Sci. 2017, 130, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K.; et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-TRNAs. Mol. Cell 2018, 69, 610–621.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniewicz, M.R. A Guide to 13C Metabolic Flux Analysis for the Cancer Biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef] [Green Version]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.Q.; Li, C.; Stanley, C.A.; Smith, T.J. Glutamate Dehydrogenase, a Complex Enzyme at a Crucial Metabolic Branch Point. Neurochem. Res. 2019, 44, 117–132. [Google Scholar] [CrossRef]
- Kurmi, K.; Haigis, M.C. Nitrogen Metabolism in Cancer and Immunity. Trends Cell Biol. 2020, 30, 408–424. [Google Scholar] [CrossRef]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.-H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial Iron Trafficking and the Integration of Iron Metabolism between the Mitochondrion and Cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and Cancer: More Ore to Be Mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafina, M.D.; Paw, B.H. Intracellular Iron and Heme Trafficking and Metabolism in Developing Erythroblasts. Metallomics 2017, 9, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic Iron Homeostasis and the Iron-Responsive Element/Iron-Regulatory Protein (IRE/IRP) Regulatory Network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-H.; Rouault, T.A. Metabolic Regulation of Citrate and Iron by Aconitases: Role of Iron–Sulfur Cluster Biogenesis. Biometals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.-K. Iron Regulatory Protein (IRP)-Iron Responsive Element (IRE) Signaling Pathway in Human Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Mühlenhoff, U. Maturation of Iron-Sulfur Proteins in Eukaryotes: Mechanisms, Connected Processes, and Diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as Iron−Sulfur Protein, Enzyme, and Iron-Regulatory Protein. Chem. Rev. 1996, 96, 2335–2374. [Google Scholar] [CrossRef]
- Netz, D.J.A.; Stith, C.M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.J.; Pierik, A.J. Eukaryotic DNA Polymerases Require an Iron-Sulfur Cluster for the Formation of Active Complexes. Nat. Chem. Biol. 2012, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.R.; White, M.F. The DNA Repair Helicases XPD and FancJ Have Essential Iron-Sulfur Domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef]
- Rouault, T.A. Biogenesis of Iron-Sulfur Clusters in Mammalian Cells: New Insights and Relevance to Human Disease. Dis. Models Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guengerich, F.P. Cytochrome P450 Research and The Journal of Biological Chemistry. J. Biol. Chem. 2019, 294, 1671–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-W. Structure and Function of Heme Proteins Regulated by Diverse Post-Translational Modifications. Arch. Biochem. Biophys. 2018, 641, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of Heme in Mammals. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2006, 1763, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Stojanovski, B.M.; Hunter, G.A.; Na, I.; Uversky, V.N.; Jiang, R.H.Y.; Ferreira, G.C. 5-Aminolevulinate Synthase Catalysis: The Catcher in Heme Biosynthesis. Mol. Genet. Metab. 2019, 128, 178–189. [Google Scholar] [CrossRef]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, T.; Kadota, S.; Niwa, M.; Nagasawa, H. A Mitochondria-Targeted Fluorescent Probe for Selective Detection of Mitochondrial Labile Fe(II). Metallomics 2018, 10, 794–801. [Google Scholar] [CrossRef]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis Is Controlled by the Coordinated Transcriptional Regulation of Glutathione and Labile Iron Metabolism by the Transcription Factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Jairaj Naik, T.; Ardehali, H. Disruption of ATP-Binding Cassette B8 in Mice Leads to Cardiomyopathy through a Decrease in Mitochondrial Iron Export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.T.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, D.L.; Lynch, J.; Wang, Y.; Fukuda, Y.; Nachagari, D.; Du, G.; Sun, D.; Fan, Y.; Tsurkan, L.; Potter, P.M.; et al. ATP-Dependent Mitochondrial Porphyrin Importer ABCB6 Protects against Phenylhydrazine Toxicity. J. Biol. Chem. 2012, 287, 12679–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Arimura, H.; Fukushige, T.; Minami, K.; Nishizawa, Y.; Tanimoto, A.; Kanekura, T.; Nakagawa, M.; Akiyama, S.-I.; Furukawa, T. Abcb10 Role in Heme Biosynthesis In Vivo: Abcb10 Knockout in Mice Causes Anemia with Protoporphyrin IX and Iron Accumulation. Mol. Cell. Biol. 2014, 34, 1077–1084. [Google Scholar] [CrossRef] [Green Version]
- Wißbrock, A.; Paul George, A.A.; Brewitz, H.H.; Kühl, T.; Imhof, D. The Molecular Basis of Transient Heme-Protein Interactions: Analysis, Concept and Implementation. Biosci. Rep. 2019, 39, BSR20181940. [Google Scholar] [CrossRef]
- Paul George, A.A.; Lacerda, M.; Syllwasschy, B.F.; Hopp, M.-T.; Wißbrock, A.; Imhof, D. HeMoQuest: A Webserver for Qualitative Prediction of Transient Heme Binding to Protein Motifs. BMC Bioinform. 2020, 21, 124. [Google Scholar] [CrossRef]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel Mechanisms for Heme-Dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Adam-Vizi, V. Alpha-Ketoglutarate Dehydrogenase: A Target and Generator of Oxidative Stress. Phil. Trans. R. Soc. B 2005, 360, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.-L. Frataxin Acts as an Iron Chaperone Protein to Modulate Mitochondrial Aconitase Activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Mochel, F.; Knight, M.A.; Tong, W.-H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice Mutation in the Iron-Sulfur Cluster Scaffold Protein ISCU Causes Myopathy with Exercise Intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Taketani, S.; Kakimoto, K.; Ueta, H.; Masaki, R.; Furukawa, T. Involvement of ABC7 in the Biosynthesis of Heme in Erythroid Cells: Interaction of ABC7 with Ferrochelatase. Blood 2003, 101, 3274–3280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically Relevant Reconstitution of Iron-Sulfur Cluster Biosynthesis Uncovers Persulfide-Processing Functions of Ferredoxin-2 and Frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Ecker, D.; Hoffmann, M.; Müting, G.; Maglioni, S.; Herebian, D.; Mayatepek, E.; Ventura, N.; Distelmaier, F. Caenorhabditis Elegans ATAD-3 Modulates Mitochondrial Iron and Heme Homeostasis. Biochem. Biophys. Res. Commun. 2015, 467, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Harel, T.; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.E.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T.; et al. Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes. Am. J. Hum. Genet. 2016, 99, 831–845. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria Regulation in Ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, W.; Zhou, H.; Wu, Q.; Duan, M.; Liu, C.; Wu, H.; Deng, W.; Shen, D.; Tang, Q. Ferritinophagy-Mediated Ferroptosis Is Involved in Sepsis-Induced Cardiac Injury. Free Radic. Biol. Med. 2020, 160, 303–318. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Huang, Z.; Luo, X.; Liu, M.; Wang, L.; Qi, Z.; Huang, S.; Zhong, J.; Chen, J.-X.; Li, L.; et al. Ferritinophagy Activation and Sideroflexin1-Dependent Mitochondria Iron Overload Is Involved in Apelin-13-Induced Cardiomyocytes Hypertrophy. Free Radic. Biol. Med. 2019, 134, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Caughman, S.W.; Casey, J.L.; Kodier, D.M.; Rouault, T.A.; Harford, J.B.; Klausner, R.D. A Model for the Structure and Functions of Iron-Responsive Elements. Gene 1988, 72, 201–208. [Google Scholar] [CrossRef]
- Campillos, M.; Cases, I.; Hentze, M.W.; Sanchez, M. SIREs: Searching for Iron-Responsive Elements. Nucleic Acids Res. 2010, 38, W360–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, B.R.; Menotti, E.; Bonnard, C.; Kühn, L.C. Optimal Sequence and Structure of Iron-Responsive Elements. Selection of RNA Stem-Loops with High Affinity for Iron Regulatory Factor. J. Biol. Chem. 1994, 269, 17481–17489. [Google Scholar] [CrossRef]
- Henderson, B.R.; Menotti, E.; Kühn, L.C. Iron Regulatory Proteins 1 and 2 Bind Distinct Sets of RNA Target Sequences. J. Biol. Chem. 1996, 271, 4900–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, J.; Kim, H.Y.; Basilion, J.P.; Cohen, S.; Iwai, K.; Philpott, C.C.; Altschul, S.; Klausner, R.D.; Rouault, T.A. Differences in the RNA Binding Sites of Iron Regulatory Proteins and Potential Target Diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 4345–4349. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Lee, M.-S. Brain Iron Accumulation in Atypical Parkinsonian Syndromes: In Vivo MRI Evidences for Distinctive Patterns. Front. Neurol. 2019, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain Iron Is Associated with Accelerated Cognitive Decline in People with Alzheimer Pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron(II) Binding to Amyloid-β, the Alzheimer’s Peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef] [PubMed]
- Derry, P.J.; Kent, T.A. Correlating Quantitative Susceptibility Mapping with Cognitive Decline in Alzheimer’s Disease. Brain 2017, 140, 2069–2072. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kolandaivel, P. Fe2+ Binding on Amyloid β-Peptide Promotes Aggregation: Fe2+ Promotes Aβ Aggregation. Proteins 2016, 84, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Uranga, R.M.; Salvador, G.A. Unraveling the Burden of Iron in Neurodegeneration: Intersections with Amyloid Beta Peptide Pathology. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Ott, S.; Dziadulewicz, N.; Crowther, D.C. Iron Is a Specific Cofactor for Distinct Oxidation- and Aggregation-Dependent A Toxicity Mechanisms in a Drosophila Model. Dis. Models Mech. 2015, 8, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Minjarez, B.; Calderón-González, K.G.; Rustarazo, M.L.V.; Herrera-Aguirre, M.E.; Labra-Barrios, M.L.; Rincon-Limas, D.E.; Del Pino, M.M.S.; Mena, R.; Luna-Arias, J.P. Identification of Proteins That Are Differentially Expressed in Brains with Alzheimer’s Disease Using ITRAQ Labeling and Tandem Mass Spectrometry. J. Proteom. 2016, 139, 103–121. [Google Scholar] [CrossRef]
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene Expression Profiling of Substantia Nigra Dopamine Neurons: Further Insights into Parkinson’s Disease Pathology. Brain 2009, 132, 1795–1809. [Google Scholar] [CrossRef] [Green Version]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid Precursor Protein and Alpha Synuclein Translation, Implications for Iron and Inflammation in Neurodegenerative Diseases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.-Y.; Zeng, P.; Qu, N.; Ning, L.-N.; Chu, J.; Zhang, T.; Zhou, X.-W.; Tian, Q. Evidence of Altered Depression and Dementia-Related Proteins in the Brains of Young Rats after Ovariectomy. J. Neurochem. 2018, 146, 703–721. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, R.C.; Mukhopadhyay, S.; McBride, D.; Veevers, J.; Harrison, F.E.; Aschner, M.; Haynes, E.N.; Bowman, A.B. Brain Manganese and the Balance between Essential Roles and Neurotoxicity. J. Biol. Chem. 2020, 295, 6312–6329. [Google Scholar] [CrossRef] [Green Version]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson’s Disease That Is Regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Wang, D.-W.; Xu, S.-F.; Zhang, S.; Fan, Y.-G.; Yang, Y.-Y.; Guo, S.-Q.; Wang, S.; Guo, T.; Wang, Z.-Y.; et al. α-Lipoic Acid Improves Abnormal Behavior by Mitigation of Oxidative Stress, Inflammation, Ferroptosis, and Tauopathy in P301S Tau Transgenic Mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Country, M.W. Retinal Metabolism: A Comparative Look at Energetics in the Retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Daruich, A.; Youale, J.; Courtois, Y.; Behar-Cohen, F. From Rust to Quantum Biology: The Role of Iron in Retina Physiopathology. Cells 2020, 9, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigot, K.; Gondouin, P.; Bénard, R.; Montagne, P.; Youale, J.; Piazza, M.; Picard, E.; Bordet, T.; Behar-Cohen, F. Transferrin Non-Viral Gene Therapy for Treatment of Retinal Degeneration. Pharmaceutics 2020, 12, 836. [Google Scholar] [CrossRef]
- Chen, B.; Aredo, B.; Ding, Y.; Zhong, X.; Zhu, Y.; Zhao, C.X.; Kumar, A.; Xing, C.; Gautron, L.; Lyon, S.; et al. Forward Genetic Analysis Using OCT Screening Identifies Sfxn3 Mutations Leading to Progressive Outer Retinal Degeneration in Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 12931–12942. [Google Scholar] [CrossRef]
- Chen, H. Prediction of Solvent Accessibility and Sites of Deleterious Mutations from Protein Sequence. Nucleic Acids Res. 2005, 33, 3193–3199. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Wang, G.; Zhou, H.X. Fold Recognition and Accurate Query-Template Alignment by a Combination of PSI-BLAST and Threading. Proteins 2001, 42, 23–37. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SFXN | Model | Localization | Experiment | Reference |
|---|---|---|---|---|
| SFXN1 | Mouse | IMM | Co-fractionation | Fleming et al. 2001 [1] |
| Human cells (Jurkat, K562) | Immunoblot on affinity-purified mitochondria STED (co-localization of Flag-SFXN1 and COX4) | Kory et al. 2018 [8] | ||
| Human cells (MCF7, HT1080), Drosophila | Immunoblot on mitochondrial extracts (fractionation) Confocal microscopy, Proteomics (LC-MS/MS on SFXN1 IP) | Our unpublished data | ||
| Human cells (HEK) | SILAC-based proteomics coupled LC-MS/MS, carbonate extraction, digitonin fractionation | Acoba et al. 2020 [16] | ||
| SFXN2 | Human cells (HeLa) | OMM or IMM | Confocal microscopy (Tom20 co-localization) | Mon et al. 2018 [9] |
| Human cells (Jurkat, K562) | Immunoblot on affinity-purified mitochondria | Kory et al. 2018 [8] | ||
| Human cells (HEK) | SILAC-based proteomics coupled LC-MS/MS | Acoba et al. 2020 [16] | ||
| SFXN3 | Rat embryonic brain cells | IMM | Fractionation, Confocal microscopy (co-localization with COX4), TEM | Rivell et al. 2019 [18] |
| Human cells (Jurkat, K562) | Immunoblot on affinity-purified mitochondria | Kory et al. 2018 [8] | ||
| Human cells (HEK) | SILAC-based proteomics coupled LC-MS/MS | Acoba et al. 2020 [16] | ||
| SFXN4 | Human cells (HeLa) | IMM | Fractionation and protease protection assay | Hildick-Smith et al. 2013 [19] |
| Human cells (Jurkat, K562) | Immunoblot on affinity-purified mitochondria | Kory et al. 2018 [8] | ||
| Human cells (HEK) | SILAC-based proteomics coupled LC-MS/MS | Acoba et al. 2020 [16] | ||
| SFXN5 | Human cells (HEK) Mouse astrocytes, human cortex and spinal cord | SILAC-based proteomics coupled LC-MS/MS Immunocapture of GFP-OMM-tagged mitochondria (MitoTag mice), immunostaining | Acoba et al. 2020 [16] Fecher et al. 2019 [20] |
| SFXN | Model | Complex | Data | Reference |
|---|---|---|---|---|
| SFXN1 | HEK SFXN1 KO cells HeLa SFXN1 KO cells | CI | No significant loss of activity SDHB ↓ | Acoba et al. 2020 [16] |
| CII | No significant loss of activity UQCRC2 ↓↓ UQCRFS1 ↓↓ | |||
| CIII | Cytochrome b ↓↓↓ Significant loss of activity Reduced levels of CIII2 and CIII2-CIV respiratory complexes | |||
| SFXN2 | HEK SFXN2 KO cells | CI CII-CIII CIV | No significant loss of activity Significant loss of activity Significant loss of activity | Mon et al. 2019 [9] |
| SFXN3 | SFXN3 KO mouse | CI, CIV | No significant loss of activity | Amorim et al. 2017 [45] |
| SFXN4 | Primary fibroblasts from two individuals with SFXN4 mutations | CI + CIII | Decreased activity | Hildick-Smith et al. 2013 [19] |
| SFXN4 KD zebrafish | CI CI + CIII | Decreased activity | Sofou et al. 2019 [30] | |
| K562 SFXN4 KO cells | CI CII CIII CIV | NDUFB8 ↓ SDHB ↓ UQCRC2 ↓ COX2 ↓ | Paul et al. 2019 [31] | |
| SFXN5 | N.A. 1 |
| Protein | Model | Evidence | Methodology | Reference |
|---|---|---|---|---|
| SFXN1 | Mouse | Iron overload in mitochondria of erythrocytes in the flexed-tail mouse | Iron mitochondrial staining | Fleming et al. 2001 [1] |
| HEK SFXN1 KO cells | Increased mitochondrial iron | ICP-MS | Acoba et al. 2020 [15] | |
| SFXN2 | HEK SFXN2 KO cells | Increased mitochondrial iron levels | ICP-MS Mito-FerroGreen staining and confocal microscopy | Mon et al. 2019 [9] |
| SFXN3 | Mouse Sfnx3 KO | Decreased circulating iron levels in male transgenic mice homozygous for the Sfxn3tm1b(KOMP)Wtsi allele | Biochemical assay | The IMPC database 1 |
| SFXN4 | K562 SFXN4 KO cells | Decreased labile iron pool | Indirect biochemical measure based on the dequenching of calcein upon release of iron TEM-EDX | Paul et al. 2019 [31] |
| Increased mitochondrial iron levels | ||||
| SFXN5 | no data available | - | - |
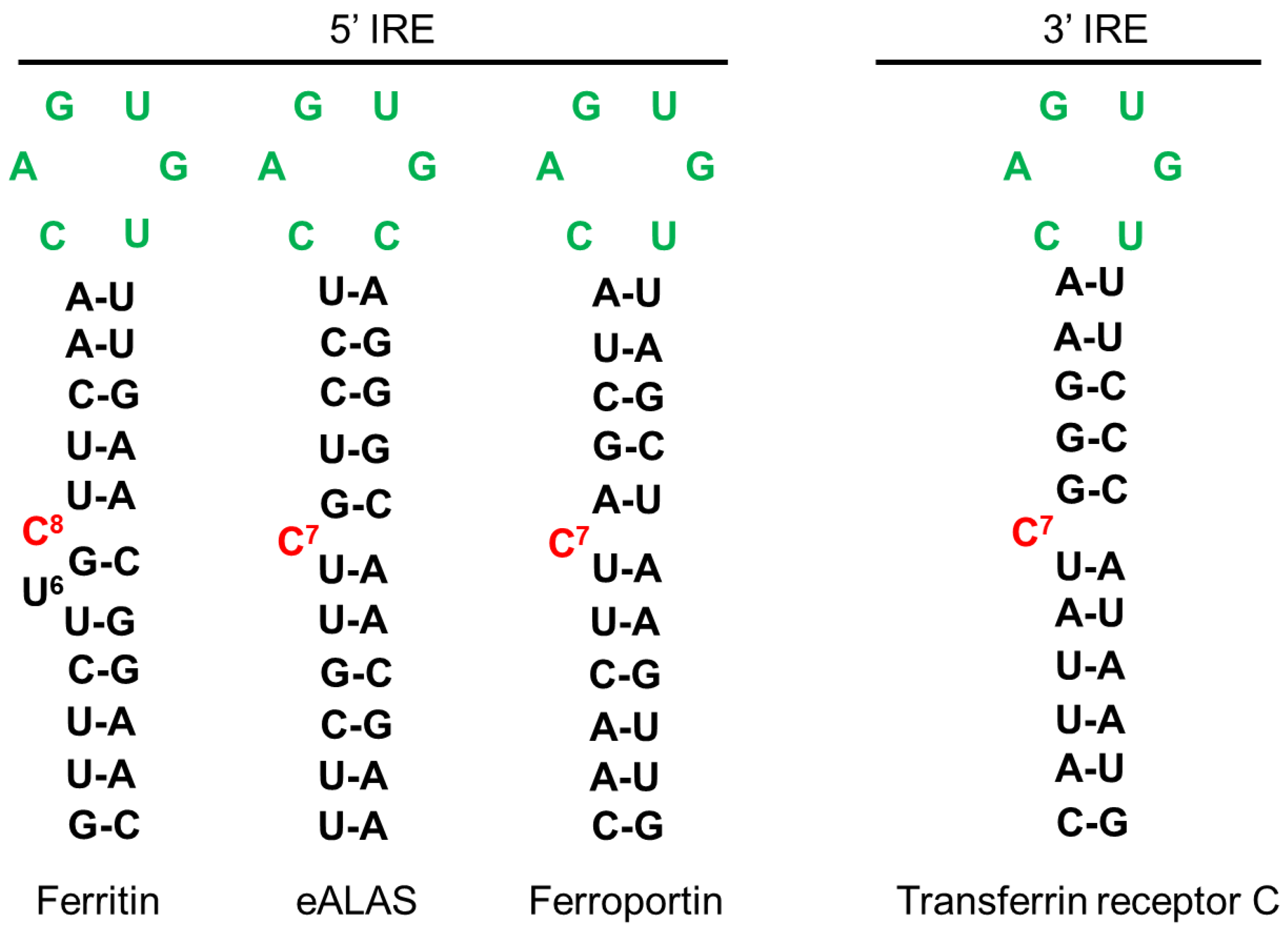
| Sequence ID | mRNA Length | CDS Position | Product | IRE Position |
|---|---|---|---|---|
| NM_022754.7 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 1, mRNA | 4066 | 90–1058 | sideroflexin-1 isoform 1 | 1000–1031 2698–2729 |
| NM_001322977.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 2, mRNA | 4094 | 118–1086 | sideroflexin-1 isoform 1 | 1028–1059 2726–2757 |
| NM_001322978.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 3, mRNA | 4037 | 244–1029 | sideroflexin-1 isoform 2 | 971–1002 2669–2700 |
| NM_001322980.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 4, mRNA | 3938 | 90–875 | sideroflexin-1 isoform 4 | 872–903 2570–2601 |
| NM_001322981.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 5, mRNA | 3966 | 118-903 | sideroflexin-1 isoform 4 | 900–931 2598–2629 |
| NM_001322982.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 6, mRNA | 4065 | 272–1057 | sideroflexin-1 isoform 2 | 999–1030 2697–2728 |
| NM_001322983.2 Homo sapiens sideroflexin 1 (SFXN1), transcript variant 7, mRNA | 959 | 90–818 | sideroflexin-1 isoform 3 | No IRE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tifoun, N.; De las Heras, J.M.; Guillaume, A.; Bouleau, S.; Mignotte, B.; Le Floch, N. Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis. Biomedicines 2021, 9, 103. https://doi.org/10.3390/biomedicines9020103
Tifoun N, De las Heras JM, Guillaume A, Bouleau S, Mignotte B, Le Floch N. Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis. Biomedicines. 2021; 9(2):103. https://doi.org/10.3390/biomedicines9020103
Chicago/Turabian StyleTifoun, Nesrine, José M. De las Heras, Arnaud Guillaume, Sylvina Bouleau, Bernard Mignotte, and Nathalie Le Floch. 2021. "Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis" Biomedicines 9, no. 2: 103. https://doi.org/10.3390/biomedicines9020103
APA StyleTifoun, N., De las Heras, J. M., Guillaume, A., Bouleau, S., Mignotte, B., & Le Floch, N. (2021). Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis. Biomedicines, 9(2), 103. https://doi.org/10.3390/biomedicines9020103