
Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems

 , , ,
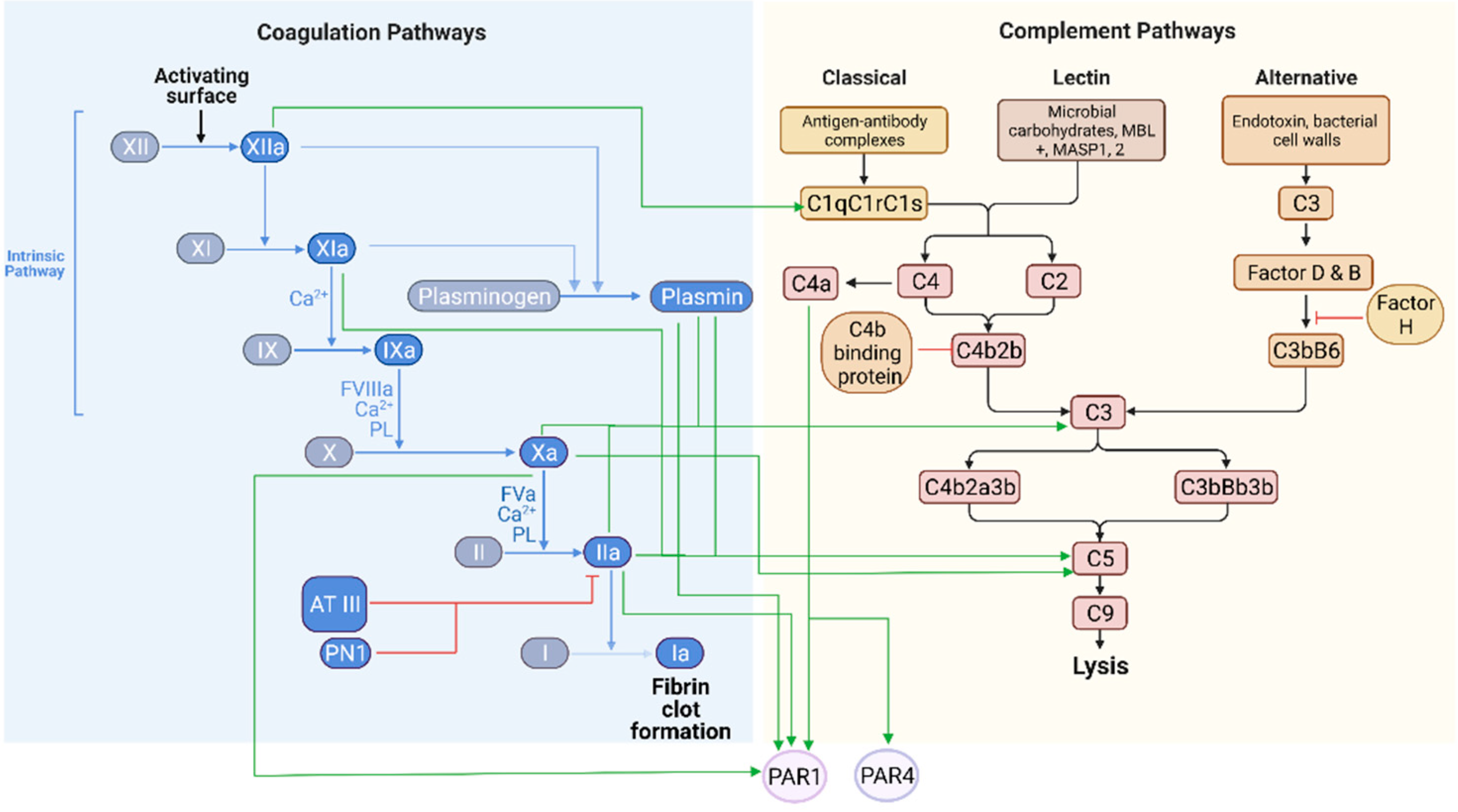
, , , 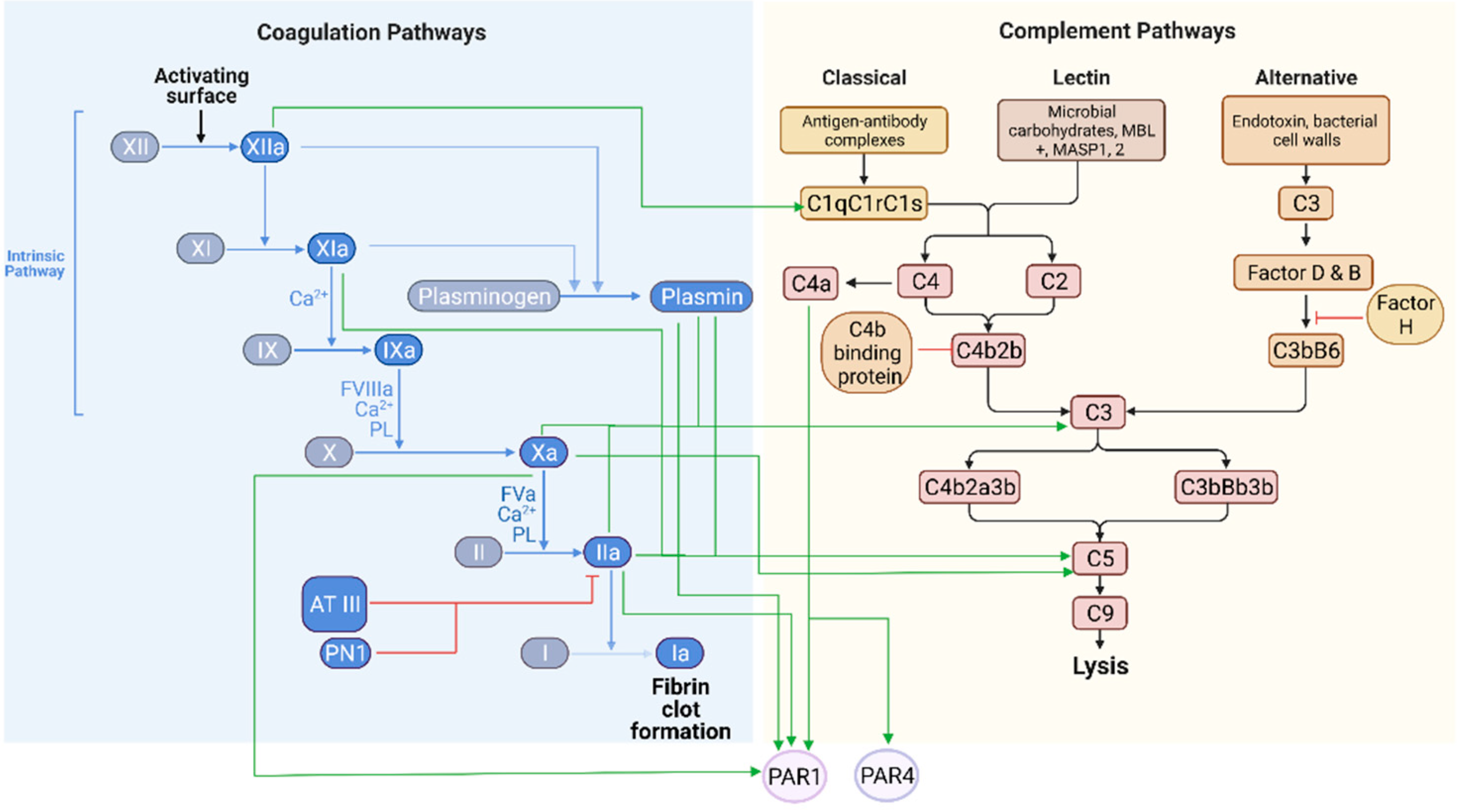{kind=link}
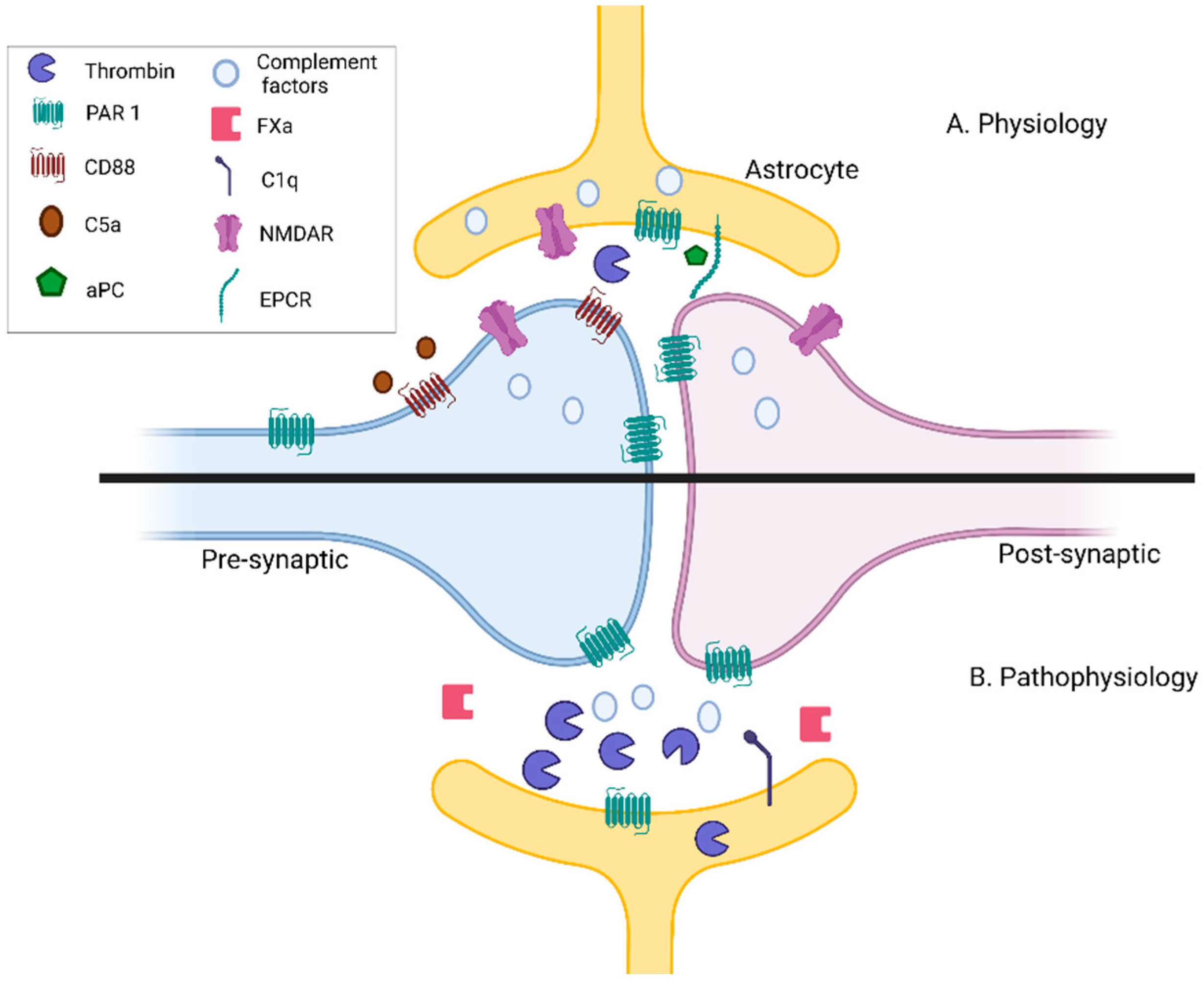{kind=link}
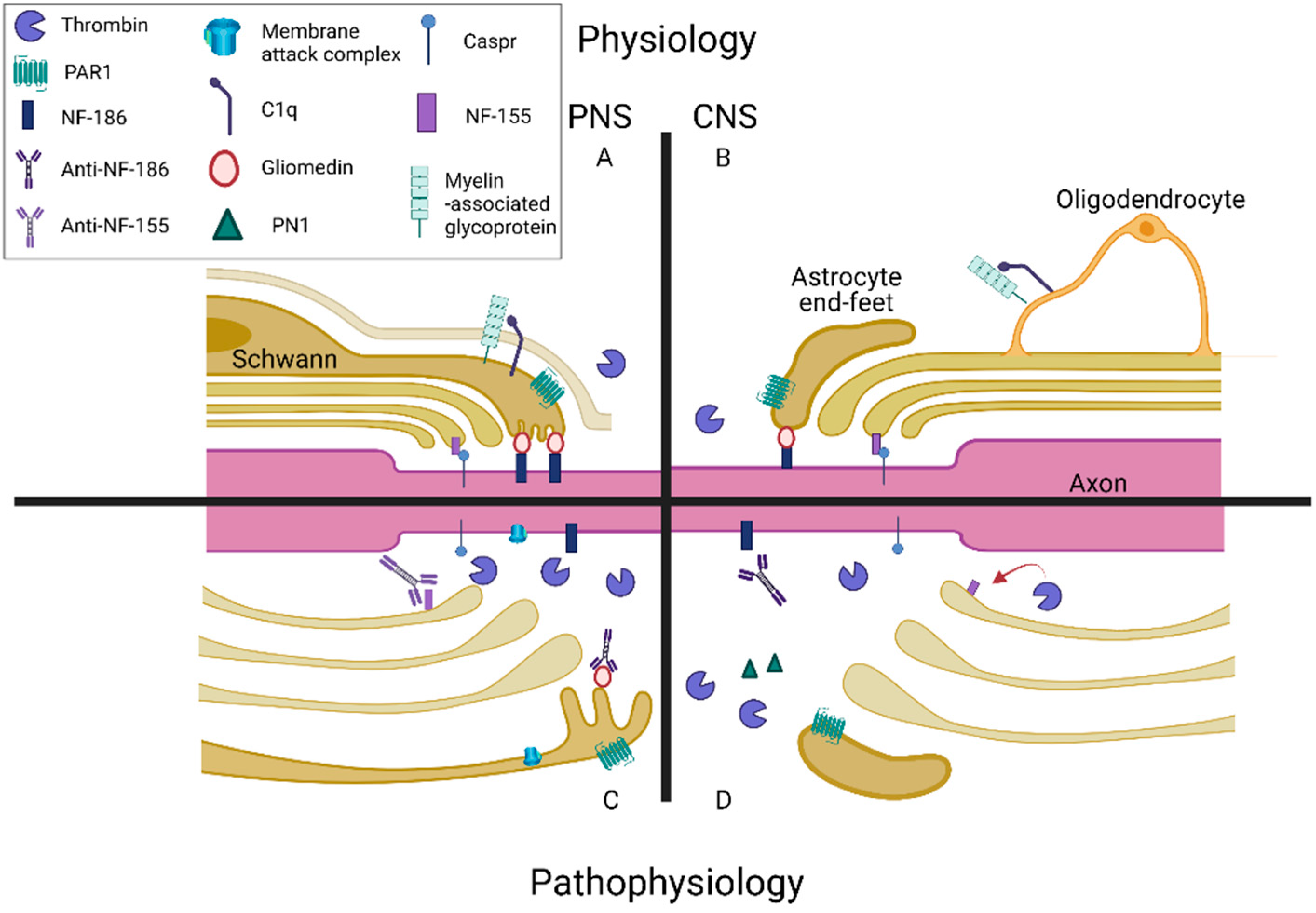{kind=link}
Abstract
:1. Introduction
2. The Complement and Coagulation Systems in Physiological States
3. The Complement and Coagulation Systems in Pathophysiology
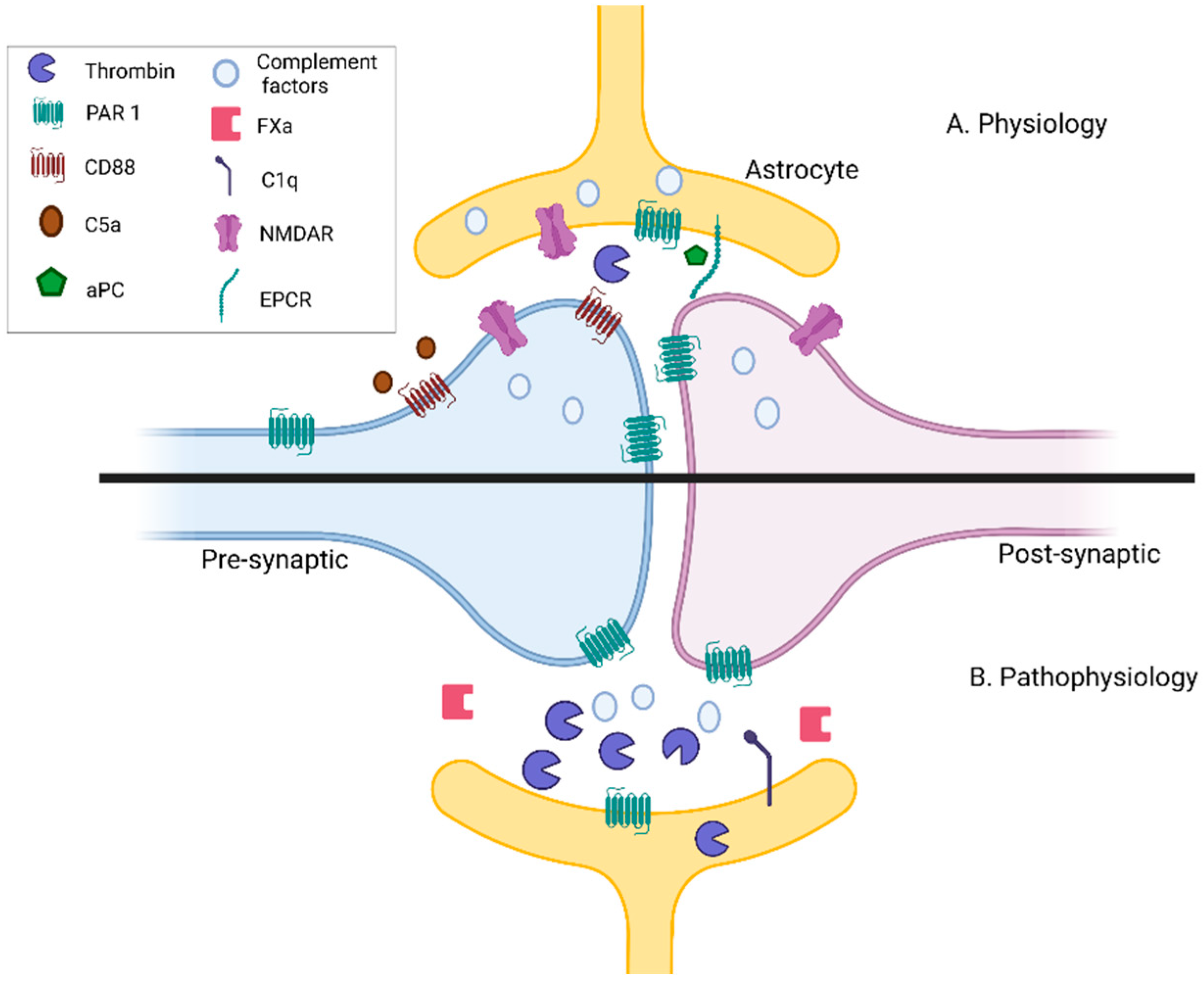
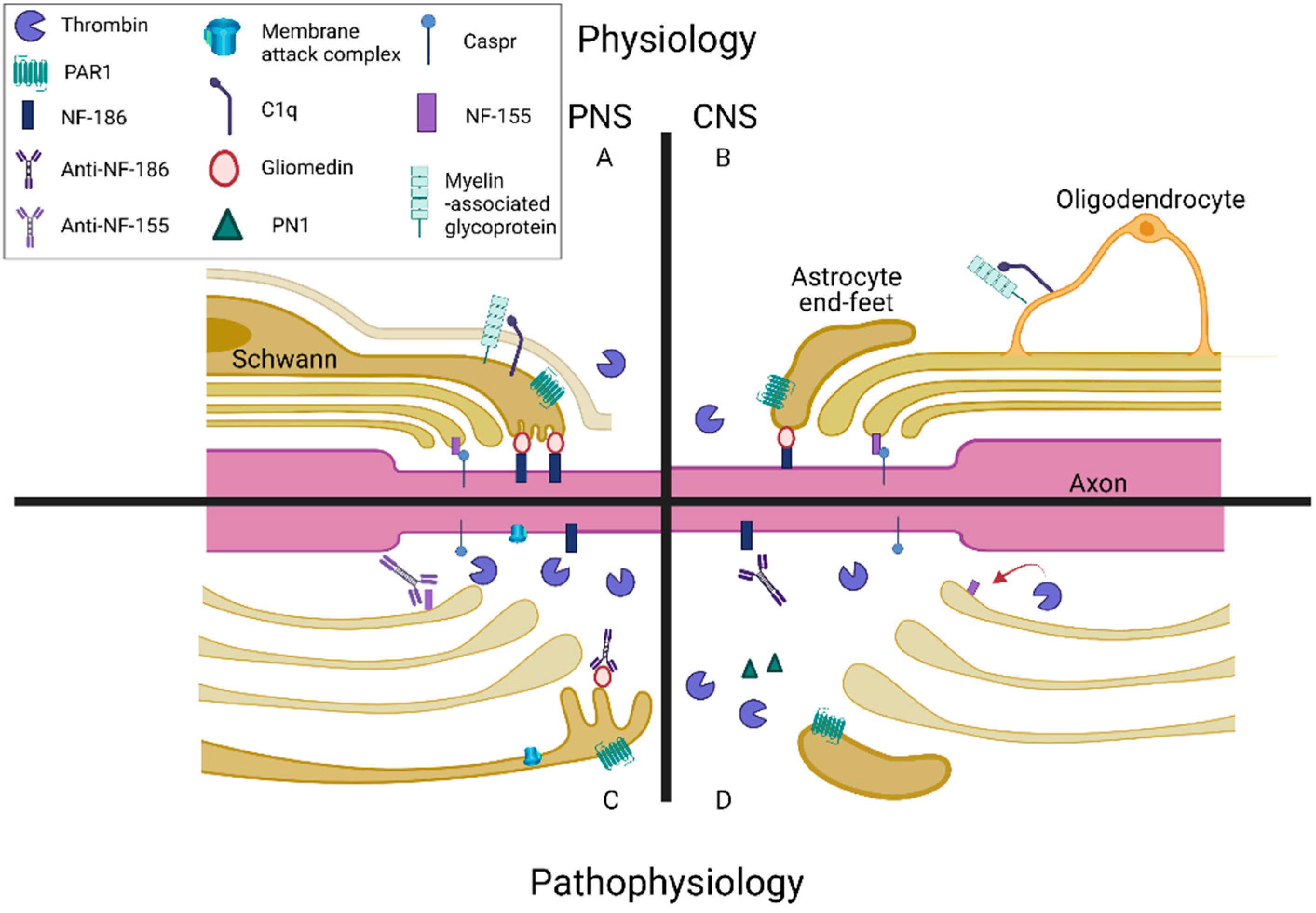
4. Neuronal Conduction Is Affected by the Complement and Coagulation Systems
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Douglas, S. Coagulation history, Oxford 1951–53. Br. J. Haematol. 1999, 107, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Matsushita, T.; Kojima, T. Historical perspective and future direction of coagulation research. J. Thromb. Haemost. 2011, 9, 352–363. [Google Scholar] [CrossRef]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.H. Examining coagulation-complement crosstalk: Complement activation and thrombosis. Thromb. Res. 2016, 141, S50–S54. [Google Scholar] [CrossRef]
- Wiegner, R.; Chakraborty, S.; Huber-Lang, M. Complement-coagulation crosstalk on cellular and artificial surfaces. Immunobiology 2016, 221, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Canobbio, I. Blood platelets: Circulating mirrors of neurons? Res. Pract. Thromb. Haemost. 2019, 3, 564–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beura, S.K.; Panigrahi, A.R.; Yadav, P.; Agrawal, S.; Singh, S.K. Role of Neurons and Glia Cells in Wound Healing as a Novel Perspective Considering Platelet as a Conventional Player. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Yin, W.; Ghebrehiwet, B.; Peerschke, E.I.B. Expression of complement components and inhibitors on platelet microparticles. Platelets 2008, 19, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Peerschke, E.I.B.; Yin, W.; Grigg, S.E.; Ghebrehiwet, B. Blood platelets activate the classical pathway of human complement. J. Thromb. Haemost. 2006, 4, 2035–2042. [Google Scholar] [CrossRef]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Mietto, B.S.; Mostacada, K.; Martinez, A.M.B. Neurotrauma and inflammation: CNS and PNS responses. Mediators Inflamm. 2015, 1–14. [Google Scholar] [CrossRef]
- Ziabska, K.; Ziemka-Nalecz, M.; Pawelec, P.; Sypecka, J.; Zalewska, T. Aberrant Complement System Activation in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4675. [Google Scholar] [CrossRef] [PubMed]
- Festoff, B.W.; Citron, B.A. Thrombin and the coag-inflammatory nexus in neurotrauma, ALS, and other neurodegenerative disorders. Front. Neurol. 2019, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflammation 2018, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.J.; Anderson, A.J.; Barnum, S.R.; Stevens, B.; Tenner, A.J. The complement cascade: Yin-Yang in neuroinflammation —Neuro-protection and degeneration. J. Neurochem. 2008, 107, 1169–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniak, S. The coagulation system in host defense. Res. Pract. Thromb. Haemost. 2018, 2, 549–557. [Google Scholar] [CrossRef]
- Tegla, C.A.; Cudrici, C.; Patel, S.; Trippe, R.; Rus, V.; Niculescu, F.; Rus, H. Membrane attack by complement: The assembly and biology of terminal complement complexes. Immunol. Res. 2011, 51, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Brennan, F.H.; Anderson, A.J.; Taylor, S.M.; Woodruff, T.M.; Ruitenberg, M.J. Complement activation in the injured central nervous system: Another dual-edged sword? J. Neuroinflam. 2012, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Grand, R.J.A.; Turnell, A.S.; Grabham, P.W. Cellular consequences of thrombin-receptor activation. Biochem. J. 1996, 313, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S. Molecular mechanisms of thrombin signaling. Semin Hematol. 1994, 31, 270–277. [Google Scholar]
- Coughlin, S. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. USA. 1994, 91, 9200–9202. [Google Scholar] [CrossRef] [Green Version]
- Willis Fox, O.; Preston, R.J.S. Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 2020, 18, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.-K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garred, P.; Tenner, A.J.; Mollnes, T.E. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol. Rev. 2021, 73, 792–827. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, J.W. Thrombina. Ann. N. Y. Acad. Sci. 1986, 485, 5–15. [Google Scholar] [CrossRef]
- Kenawy, H.I.; Boral, I.; Bevington, A. Complement-coagulation cross-talk: A potential mediator of the physiological activation of complement by low pH. Front. Immunol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Carson, S.D.; Johnson, D.R. Consecutive Enzyme Cascades: Complement Activation at the Cell Surface Triggers Increased Tissue Factor Activity. Blood 1990, 76, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Muhlfelder, T.W.; Niemetz, J.; Kreutzer, D.; Beebe, D.; Ward, P.A.; Rosenfeld, S.I. C5 chemotactic fragment induces leukocyte production of tissue factor activity: A link between complement and coagulation. J. Clin. Investig. 1979, 63, 147–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a Induces Tissue Factor Activity on Endothelial Cells. Thromb. Haemost. 2018, 77, 394–398. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular Intercommunication between the Complement and Coagulation Systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [Green Version]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef]
- Sallah, S. Inhibitors to clotting factors. Ann. Hematol. 1997, 75, 1–7. [Google Scholar] [CrossRef]
- Blom, A.M. The role of complement inhibitors beyond controlling inflammation. J. Intern. Med. 2017, 282, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.E.; Mejia, P.; Lu, F. Biological activities of C1 inhibitor. Mol. Immunol. 2008, 45, 4057–4063. [Google Scholar] [CrossRef] [Green Version]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.G.; Conway, E.M. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshari, R.S.; Silasi, R.; Lupu, C.; Taylor, F.B.; Lupu, F. In vivo–generated thrombin and plasmin do not activate the complement system in baboons. Blood 2017, 130, 2678–2681. [Google Scholar] [CrossRef] [Green Version]
- Mosnier, L.O.; Griffin, J.H. Protein C anticoagulant activity in relation to anti-inflammatory and anti-apoptotic activities. Front. Biosci. 2006, 11, 2381–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Reiser, G.; Keep, R.F. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem. 2003, 84, 3–9. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-coagulopathy: Blood coagulation factors in central nervous system diseases. Int. J. Mol. Sci. 2017, 18, 2128. [Google Scholar] [CrossRef]
- Gofrit, S.; Shavit-Stein, E. The neuro-glial coagulonome: The thrombin receptor and coagulation pathways as major players in neurological diseases. Neural Regen. Res. 2019, 14, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Deschepper, C.F.; Bigornia, V.; Berens, M.E.; Lapointe, M.C. Production of thrombin and antithrombin III by brain and astroglial cell cultures. Mol. Brain Res. 1991, 11, 355–358. [Google Scholar] [CrossRef]
- Pompili, E.; Fabrizi, C.; Fornai, F.; Fumagalli, L. Role of the protease-activated receptor 1 in regulating the function of glial cells within central and peripheral nervous system. J. Neural Transm. 2019, 126, 1259–1271. [Google Scholar] [CrossRef]
- Shavit, E.; Beilin, O.; Korczyn, A.D.; Sylantiev, C.; Aronovich, R.; Drory, V.E.; Gurwitz, D.; Horresh, I.; Bar-Shavit, R.; Peles, E.; et al. Thrombin receptor PAR-1 on myelin at the node of Ranvier: A new anatomy and physiology of conduction block. Brain 2008, 131, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Han, K.S.; Mannaioni, G.; Hamill, C.E.; Lee, J.; Junge, C.E.; Lee, C.J.; Traynelis, S.F. Activation of protease activated receptor 1 increases the excitability of the dentate granule neurons of hippocampus. Mol. Brain 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Reiser, G. Thrombin signaling in the brain: The role of protease-activated receptors. Biol. Chem. 2003, 384, 193–202. [Google Scholar] [CrossRef]
- Wang, H.; Ubl, J.J.; Stricker, R.; Reiser, G. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 2002, 283, 1351–1364. [Google Scholar] [CrossRef]
- Ellis, C.A.; Malik, A.B.; Gilchrist, A.; Hamm, H.; Sandoval, R.; Voyno-Yasenetskaya, T.; Tiruppathi, C. Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J. Biol. Chem. 1999, 274, 13718–13727. [Google Scholar] [CrossRef] [Green Version]
- De Ceunynck, K.; Peters, C.G.; Jain, A.; Higgins, S.J.; Aisiku, O.; Fitch-Tewfik, J.L.; Chaudhry, S.A.; Dockendorff, C.; Parikh, S.M.; Ingber, D.E.; et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc. Natl. Acad. Sci. USA. 2018, 115, E982–E991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Trejo, J.A. Protease-activated receptor signaling: New roles and regulatory mechanisms. Curr. Opin. Hematol. 2007, 14, 230–235. [Google Scholar] [CrossRef]
- Wang, H.; Ricklin, D.; Lambris, J.D. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc. Natl. Acad. Sci. USA 2017, 114, 10948–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; de la Fuente, M.; Nieman, M.T. Complement factor C4a does not activate protease-activated receptor 1 (PAR1) or PAR4 on human platelets. Res. Pract. Thromb. Haemost. 2021, 5, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Luchena, C.; Zuazo-Ibarra, J.; Alberdi, E.; Matute, C.; Capetillo-Zarate, E. Contribution of neurons and glial cells to complement-mediated synapse removal during development, aging and in Alzheimer’s disease. Mediat. Inflamm. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement System in Neural Synapse Elimination in Development and Disease. Adv. Immunol. 2017, 135, 53–79. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisalyaput, K.; Tenner, A.J. Complement component C1q inhibits β-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J. Neurochem. 2008, 104, 696–707. [Google Scholar] [CrossRef]
- Cong, Q.; Soteros, B.M.; Wollet, M.; Kim, J.H.; Sia, G.M. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci. 2020, 23, 1067–1078. [Google Scholar] [CrossRef]
- Ben Shimon, M.; Lenz, M.; Ikenberg, B.; Becker, D.; Shavit-Stein, E.; Chapman, J.; Tanne, D.; Pick, C.G.; Blatt, I.; Neufeld, M.; et al. Thrombin regulation of synaptic transmission and plasticity: Implications for health and disease. Front. Cell. Neurosci. 2015, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000, 20, 4582–4595. [Google Scholar] [CrossRef]
- Maggio, N.; Shavit, E.; Chapman, J.; Segal, M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: Toward understanding the functional consequences of cerebrovascular insults. J. Neurosci. 2008, 28, 732–736. [Google Scholar] [CrossRef]
- Maggio, N.; Itsekson, Z.; Dominissini, D.; Blatt, I.; Amariglio, N.; Rechavi, G.; Tanne, D.; Chapman, J. Thrombin regulation of synaptic plasticity: Implications for physiology and pathology. Exp. Neurol. 2013, 247, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Krenzlin, H.; Lorenz, V.; Danckwardt, S.; Kempski, O.; Alessandri, B. The importance of thrombin in cerebral injury and disease. Int. J. Mol. Sci. 2016, 17, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef]
- Pompili, E.; Fabrizi, C. Thrombin in peripheral nerves: Friend or foe? Neural Regen. Res. 2021, 16, 1223–1224. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Yaffe, K.; Peltz, C.B.; Ledreux, A.; Gorgens, K.; Davidson, B.; Granholm, A.C.; Mustapic, M.; Kapogiannis, D.; Tweedie, D.; et al. Traumatic brain injury increases plasma astrocyte-derived exosome levels of neurotoxic complement proteins. FASEB J. 2020, 34, 3359–3366. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziliotto, N.; Bernardi, F.; Jakimovski, D.; Zivadinov, R. Coagulation pathways in neurological diseases: Multiple sclerosis. Front. Neurol. 2019, 10, 1–21. [Google Scholar] [CrossRef]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef]
- Beilin, O.; Karussis, D.M.; Korczyn, A.D.; Gurwitz, D.; Aronovich, R.; Hantai, D.; Grigoriadis, N.; Mizrachi-Kol, R.; Chapman, J. Increased thrombin inhibition in experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2005, 79, 351–359. [Google Scholar] [CrossRef]
- Hammond, J.W.; Bellizzi, M.J.; Ware, C.; Qiu, W.Q.; Saminathan, P.; Li, H.; Luo, S.; Ma, S.A.; Li, Y.; Gelbard, H.A. Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain Behav. Immun. 2020, 87, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Yalcin, E.; Presumey, J.; Aw, E.; Ma, M.; Whelan, C.W.; Stevens, B.; McCarroll, S.A.; Carroll, M.C. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 2021, 24, 214–224. [Google Scholar] [CrossRef]
- Abe, T.; Kubo, K.; Izumoto, S.; Shimazu, S.; Goan, A.; Tanaka, T.; Koroki, T.; Saito, K.; Kawana, R.; Ochiai, H. Complement Activation in Human Sepsis is Related to Sepsis-Induced Disseminated Intravascular Coagulation. Shock 2020, 54, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.J.C.; Nobre, V.; Barichello, T.; Teixeira, A.L. Post-sepsis cognitive impairment and associated risk factors: A systematic review. Aust. Crit. Care 2018, 31, 242–253. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA J. Am. Med. Assoc. 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Orhun, G.; Tüzün, E.; Özcan, P.E.; Ulusoy, C.; Yildirim, E.; Küçükerden, M.; Gürvit, H.; Ali, A.; Esen, F. Association between inflammatory markers and cognitive outcome in patients with acute brain dysfunction due to sepsis. Noropsikiyatri Ars. 2019, 56, 63–70. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Schwartz, J.B.; Elahi, F.M.; Rissman, R.A. Complement protein levels in plasma astrocyte-derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer’s disease dementia. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Levy, M. Single-Cell Transcriptomics of the Microglial Complement Pathway in the Progression of Alzheimer’s Dementia (3009). Neurology 2021, 96. [Google Scholar]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Van Nostrand, W.E.; Schmaier, A.H.; Farrow, J.S.; Cunningham, D.D. Protease Nexin-II(amyloid β-protein Precursor): A Platelet α-Granule Protein. Science 1990, 248, 745–748. [Google Scholar] [CrossRef]
- Jenkins, D.R.; Craner, M.J.; Esiri, M.M.; Deluca, G.C. Contribution of Fibrinogen to Inflammation and Neuronal Density in Human Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2259–2271. [Google Scholar] [CrossRef] [PubMed]
- Itsekson-Hayosh, Z.; Shavit-Stein, E.; Katzav, A.; Rubovitch, V.; Maggio, N.; Chapman, J.; Harnof, S.; Pick, C.G. Minimal Traumatic Brain Injury in Mice: Protease-Activated Receptor 1 and Thrombin-Related Changes. J. Neurotrauma 2016, 33, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Uchino, K.; Fakhran, S.; Sattar, M.A.; Branstetter, B.F.; Au, K.; Navratil, J.S.; Paul, B.; Lee, M.; Gallagher, K.M.; et al. Platelet C4d is associated with acute ischemic stroke and stroke severity. Stroke 2008, 39, 3236–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, Z.G.; Campbell, R.A.; Weyrich, A.S.; Rondina, M.T. Platelet-leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, A.M.; Stevens, B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv. Exp. Med. Biol. 2010, 703, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Stasi, K.; Nagel, D.; Yang, X.; Wang, R.-F.; Ren, L.; Podos, S.M.; Mittag, T.; Danias, J. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1024–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Li, S.-M.; Li, B.; Zhang, L.; Zhang, G.-F.; Sun, J.; Ji, M.-H.; Yang, J.-J. A complement-microglial axis driving inhibitory synapse related protein loss might contribute to systemic inflammation-induced cognitive impairment. Int. Immunopharmacol. 2020, 87. [Google Scholar] [CrossRef]
- Shavit-Stein, E.; Itsekson-Hayosh, Z.; Aronovich, A.; Reisner, Y.; Bushi, D.; Pick, C.G.; Tanne, D.; Chapman, J.; Vlachos, A.; Maggio, N. Thrombin induces ischemic LTP (iLTP): Implications for synaptic plasticity in the acute phase of ischemic stroke. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.; Ikenberg, B.; Schiener, S.; Maggio, N.; Vlachos, A. NMDA-receptor inhibition restores Protease-Activated Receptor 1 (PAR1) mediated alterations in homeostatic synaptic plasticity of denervated mouse dentate granule cells. Neuropharmacology 2014, 86, 212–218. [Google Scholar] [CrossRef]
- Crane, J.W.; Baiquni, G.P.; Sullivan, R.K.P.; Lee, J.D.; Sah, P.; Taylor, S.M.; Noakes, P.G.; Woodruff, T.M. The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J. Neuroinflam. 2009, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shavit-Stein, E.; Mindel, E.; Gofrit, S.G.; Chapman, J.; Maggio, N. Ischemic stroke in PAR1 KO mice: Decreased brain plasmin and thrombin activity along with decreased infarct volume. PLoS ONE 2021, 16, e248431. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Xie, X.L.; Zhang, X.; Wang, Y.; Chu, K.; Brown, J.; Chen, L.; Hong, G. Inhibition of Complement Drives Increase in Early Growth Response Proteins and Neuroprotection Mediated by Salidroside After Cerebral Ischemia. Inflammation 2018, 41, 449–463. [Google Scholar] [CrossRef]
- Salcedo, R.M.; Festoff, B.W.; Citron, B.A. Quantitative reverse transcriptase PCR to gauge increased protease- activated receptor 1 (PAR-1) mRNA copy numbers in the Wobbler mutant mouse. J. Mol. Neurosci. 1998, 10, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Levin, S.C.; Willis, E.F.; Li, R.; Woodruff, T.M.; Noakes, P.G. Complement components are upregulated and correlate with disease progression in the TDP-43 Q331K mouse model of amyotrophic lateral sclerosis. J. Neuroinflam. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shavit-Stein, E.; Rahal, I.A.; Bushi, D.; Gera, O.; Sharon, R.; Gofrit, S.G.; Pollak, L.; Mindel, K.; Maggio, N.; Kloog, Y.; et al. Brain protease activated receptor 1 pathway: A therapeutic target in the superoxide dismutase 1 (SOD1) mouse model of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2020, 21, 3419. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.E.; Jin, J.G.; Ji, R.R.; Tong, J.; Pomonis, J.D.; Lavery, D.J.; Miller, S.W.; Chiang, L.W. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain 2008, 137, 182–201. [Google Scholar] [CrossRef]
- Anjum, A.; Yazid, M.D.; Daud, M.F.; Idris, J.; Hwei Ng, A.M.; Naicker, A.S.; Rashidah Ismail, O.H.; Kumar, R.K.A.; Lokanathan, Y. Spinal cord injury: Pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.L.; Anderson, A.J. Complement and spinal cord injury: Traditional and non-traditional aspects of complement cascade function in the injured spinal cord microenvironment. Exp. Neurol. 2014, 258, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, M.; Yoon, H.; Wu, J.; Mustafa, K.; Scarisbrick, I.A. Targeting the thrombin receptor modulates inflammation and astrogliosis to improve recovery after spinal cord injury. Neurobiol. Dis. 2016, 93, 226–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.L.; Nguyen, H.X.; Mendez, O.A.; Anderson, A.J. Complement Protein C3 Suppresses Axon Growth and Promotes Neuron Loss. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.L.; Nguyen, H.X.; Mendez, O.A.; Anderson, A.J. Complement protein C1q modulates neurite outgrowth in vitro and spinal cord axon regeneration in vivo. J. Neurosci. 2015, 35, 4332–4349. [Google Scholar] [CrossRef] [Green Version]
- Pompili, E.; Ciraci, V.; Leone, S.; De Franchis, V.; Familiari, P.; Matassa, R.; Familiari, G.; Tata, A.M.; Fumagalli, L.; Fabrizi, C. Thrombin regulates the ability of Schwann cells to support neuritogenesis and to maintain the integrity of the nodes of Ranvier. Eur. J. Histochem. 2020, 64, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Akassoglou, K.; Yu, W.M.; Akpinar, P.; Strickland, S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron 2002, 33, 861–875. [Google Scholar] [CrossRef] [Green Version]
- Gera, O.; Shavit-Stein, E.; Bushi, D.; Harnof, S.; Ben Shimon, M.; Weiss, R.; Golderman, V.; Dori, A.; Maggio, N.; Finegold, K.; et al. Thrombin and protein C pathway in peripheral nerve Schwann cells. Neuroscience 2016, 339, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Gera, O.; Bushi, D.; Shimon, M.B.; Artan-Furman, A.; Harnof, S.; Maggio, N.; Dori, A.; Chapman, J.; Shavit-Stein, E. Local Regulation of Thrombin Activity by Factor Xa in Peripheral Nerve Schwann Cells. Neuroscience 2018, 371, 445–454. [Google Scholar] [CrossRef]
- de Jonge, R.R.; van Schalik, I.N.; Vreijling, J.P.; Troost, D.; Baas, F. Expression of complement components in the peripheral nervous system. Hum. Mol. Genet. 2004, 13, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.F. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. NY Acad. Sci. 2018, 1412, 113–128. [Google Scholar] [CrossRef]
- Glazner, G.W.; Yadav, K.; Fitzgerald, S.; Coven, E.; Brenneman, D.E.; Nelson, P.G. Cholinergic stimulation increases thrombin activity and gene expression in cultured mouse muscle. Dev. Brain Res. 1997, 99, 148–154. [Google Scholar] [CrossRef]
- Salvany, S.; Casanovas, A.; Piedrafita, L.; Tarabal, O.; Hernández, S.; Calderó, J.; Esquerda, J.E. Microglial recruitment and mechanisms involved in the disruption of afferent synaptic terminals on spinal cord motor neurons after acute peripheral nerve injury. GLIA 2021, 69, 1216–1240. [Google Scholar] [CrossRef]
- Arancibia-Carcamo, I.L.; Attwell, D. The node of Ranvier in CNS pathology. Acta Neuropathol. 2014, 128, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasband, M.N.; Peles, E. The nodes of Ranvier: Molecular assembly and maintenance. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–16. [Google Scholar] [CrossRef]
- D’Este, E.; Kamin, D.; Balzarotti, F.; Hell, S.W. Ultrastructural anatomy of nodes of Ranvier in the peripheral nervous system as revealed by STED microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, E191–E199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.J.; Woo, D.H.; Lee, P.R.; Pajevic, S.; Bukalo, O.; Huffman, W.C.; Wake, H.; Basser, P.J.; SheikhBahaei, S.; Lazarevic, V.; et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 11832–11837. [Google Scholar] [CrossRef] [Green Version]
- Eshed, Y.; Feinberg, K.; Poliak, S.; Sabanay, H.; Sarig-Nadir, O.; Spiegel, I.; Bermingham, J.R.; Peles, E. Gliomedin Mediates Schwann Cell-Axon Interaction and the Molecular Assembly of the Nodes of Ranvier. Neuron 2005, 47, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzer, J.L.; Brophy, P.J.; Peles, E. Molecular Domains of Myelinated Axons in the Peripheral Nervous System. GLIA 2008, 56, 1532–1540. [Google Scholar] [CrossRef]
- Stathopoulos, P.; Alexopoulos, H.; Dalakas, M.C. Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat. Rev. Neurol. 2015, 11, 143–156. [Google Scholar] [CrossRef]
- Shavit-Stein, E.; Aronovich, R.; Sylantiev, C.; Gofrit, S.G.; Chapman, J.; Dori, A. The role of thrombin in the pathogenesis of diabetic neuropathy. PLoS ONE 2019, 14, e219453. [Google Scholar] [CrossRef]
- Koski, C.L.; Sanders, M.E.; Swoveland, P.T.; Lawley, T.J.; Shin, M.L.; Frank, M.M.; Joiner, K.A. Activation of terminal components of complement in patients with Guillain-Barre syndrome and other demyelinating neuropathies. J. Clin. Investig. 1987, 80, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Willison, H.J.; Halstead, S.K.; Beveridge, E.; Zitman, F.M.P.; Greenshields, K.N.; Morgan, B.P.; Plomp, J.J. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain-Barré syndrome. J. Neuroimmunol. 2008, 201–202, 172–182. [Google Scholar] [CrossRef]
- Yell, P.C.; Burns, D.K.; Dittmar, E.G.; White, C.L.; Cai, C. Diffuse microvascular C5b-9 deposition is a common feature in muscle and nerve biopsies from diabetic patients. Acta Neuropathol. Commun. 2018, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Quast, I.; Keller, C.W.; Hiepe, F.; Tackenberg, B.; Lünemann, J.D. Terminal complement activation is increased and associated with disease severity in CIDP. Ann. Clin. Transl. Neurol. 2016, 3, 730–735. [Google Scholar] [CrossRef]
- Devaux, J.J. Antibodies to gliomedin cause peripheral demyelinating neuropathy and the dismantling of the nodes of Ranvier. Am. J. Pathol. 2012, 181, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Susuki, K.; Rasband, M.N.; Tohyama, K.; Koibuchi, K.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Baba, H.; Yuki, N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J. Neurosci. 2007, 27, 3956–3967. [Google Scholar] [CrossRef] [PubMed]
- Phongsisay, V.; Susuki, K.; Matsuno, K.; Yamahashi, T.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Shinoda, M.; Yuki, N. Complement inhibitor prevents disruption of sodium channel clusters in a rabbit model of Guillain-Barré syndrome. J. Neuroimmunol. 2008, 205, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Kira, J.I.; Yamasaki, R.; Ogata, H. Anti-neurofascin autoantibody and demyelination. Neurochem. Int. 2019, 130, 104360. [Google Scholar] [CrossRef]
- Dutta, D.J.; Fields, R.D. Deletion of the Thrombin Proteolytic Site in Neurofascin 155 Causes Disruption of Nodal and Paranodal Organization. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Shavit-Stein, E.; Aronovich, R.; Sylantiev, C.; Gera, O.; Gofrit, S.G.; Chapman, J.; Dori, A. Blocking thrombin significantly ameliorates experimental autoimmune neuritis. Front. Neurol. 2019, 10, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Shavit-stein, E.; Gofrit, S.G.; Gayster, A.; Teldan, Y.; Ron, A.; Bandora, E.A.; Golderman, V.; Gera, O.; Harnof, S.; Chapman, J.; et al. Treatment of diabetic neuropathy with a novel PAR1-targeting molecule. Biomolecules 2020, 10, 1552. [Google Scholar] [CrossRef]
- Davidson, A.I.; Halstead, S.K.; Goodfellow, J.A.; Chavada, G.; Mallik, A.; Overell, J.; Lunn, M.P.; McConnachie, A.; van Doorn, P.; Willison, H.J. Inhibition of complement in Guillain-Barré syndrome: The ICA-GBS study. J. Peripher. Nerv. Syst. 2017, 22, 4–12. [Google Scholar] [CrossRef]
- Alawieh, A.; Farris Langley, E.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10, 6459. [Google Scholar] [CrossRef] [Green Version]
- Shlobin, N.A.; Har-Even, M.; Itsekson-Hayosh, Z.; Harnof, S.; Pick, C.G. Role of thrombin in central nervous system injury and disease. Biomolecules 2021, 11, 562. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J. Thrombin in inflammatory brain diseases. Autoimmun. Rev. 2006, 5, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.M.; Connell, N.T. Ravulizumab: A novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther. Adv. Hematol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.L.; Huda, S.; Whittam, D.H.; Matiello, M.; Morgan, B.P.; Jacob, A. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: A brief review. J. Neurol. 2021, 268, 1643–1664. [Google Scholar] [CrossRef]
- Shavit-Stein, E.; Ben Shimon, M.; Artan Furman, A.; Golderman, V.; Chapman, J.; Maggio, N. Thrombin Inhibition Reduces the Expression of Brain Inflammation Markers upon Systemic LPS Treatment. Neural Plast. 2018, 2018, 7692182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkowitz, S.; Chapman, J.; Dori, A.; Gofrit, S.G.; Maggio, N.; Shavit-Stein, E. Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems. Biomedicines 2021, 9, 1950. https://doi.org/10.3390/biomedicines9121950
Berkowitz S, Chapman J, Dori A, Gofrit SG, Maggio N, Shavit-Stein E. Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems. Biomedicines. 2021; 9(12):1950. https://doi.org/10.3390/biomedicines9121950
Chicago/Turabian StyleBerkowitz, Shani, Joab Chapman, Amir Dori, Shany Guly Gofrit, Nicola Maggio, and Efrat Shavit-Stein. 2021. "Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems" Biomedicines 9, no. 12: 1950. https://doi.org/10.3390/biomedicines9121950
APA StyleBerkowitz, S., Chapman, J., Dori, A., Gofrit, S. G., Maggio, N., & Shavit-Stein, E. (2021). Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems. Biomedicines, 9(12), 1950. https://doi.org/10.3390/biomedicines9121950