
Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds

 ,
,  , , ,
, , ,
Abstract
1. Introduction
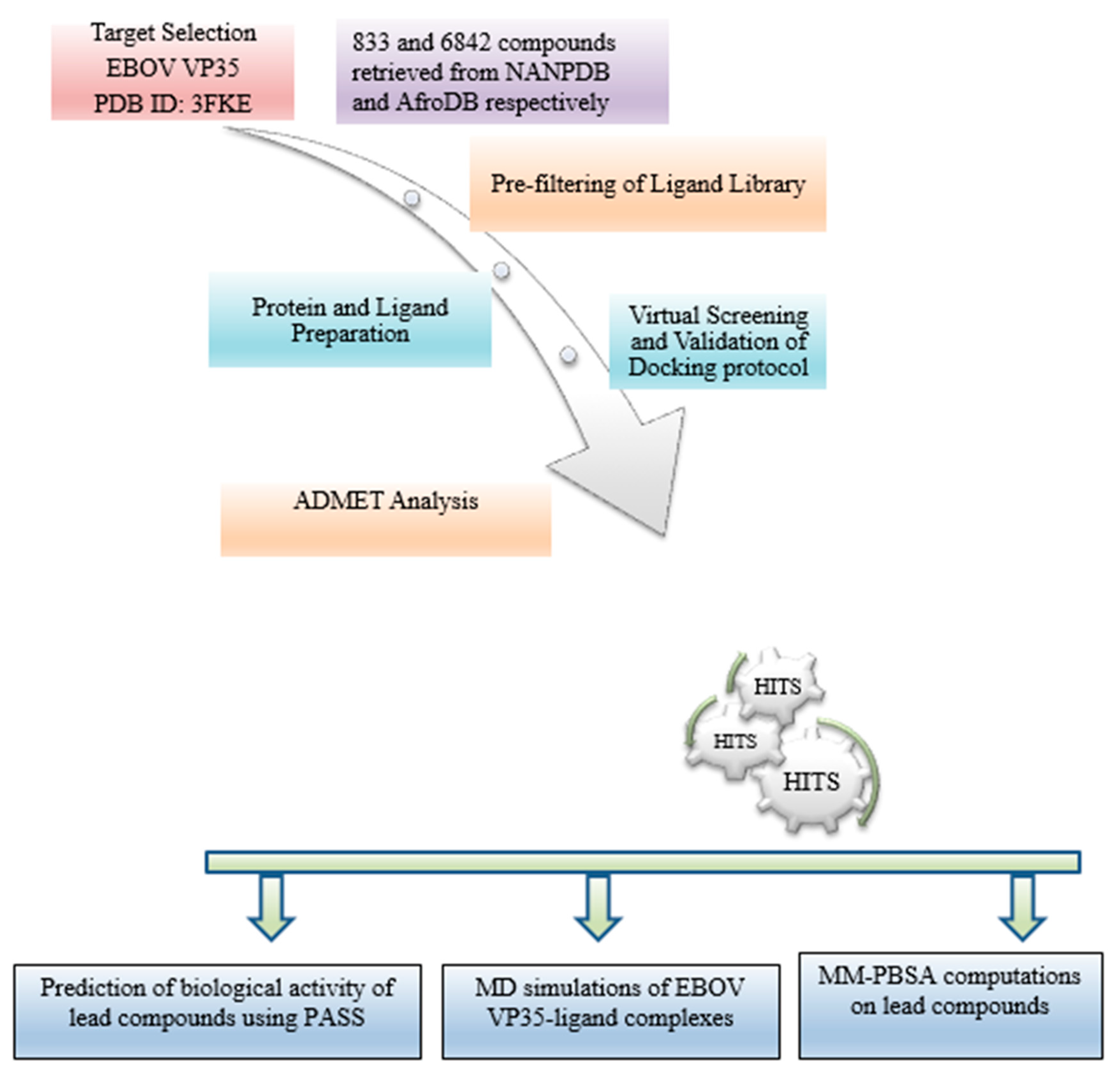
2. Materials and Methods
2.1. Protein Retrieval
2.2. Retrieval of Compounds from Natural Products Databases
2.3. Protein Active Site Evaluation
2.4. Pre-Filtering of Ligand Library
2.5. Protein and Ligand Preparation
2.6. Virtual Screening and Validation of Docking Protocol
2.7. Molecular Interaction Profiling
2.8. Pharmacokinetic Profiling
2.9. Prediction of Anti-Viral Activity of Lead Compounds
2.10. Quality and Efficiency of Evaluation of Potential Lead Compounds
2.11. Molecular Dynamic Simulations of Protein-Ligand Complexes
2.12. Binding Free Energy Calculations of Protein-Ligand Complexes by MM-PBSA
3. Results
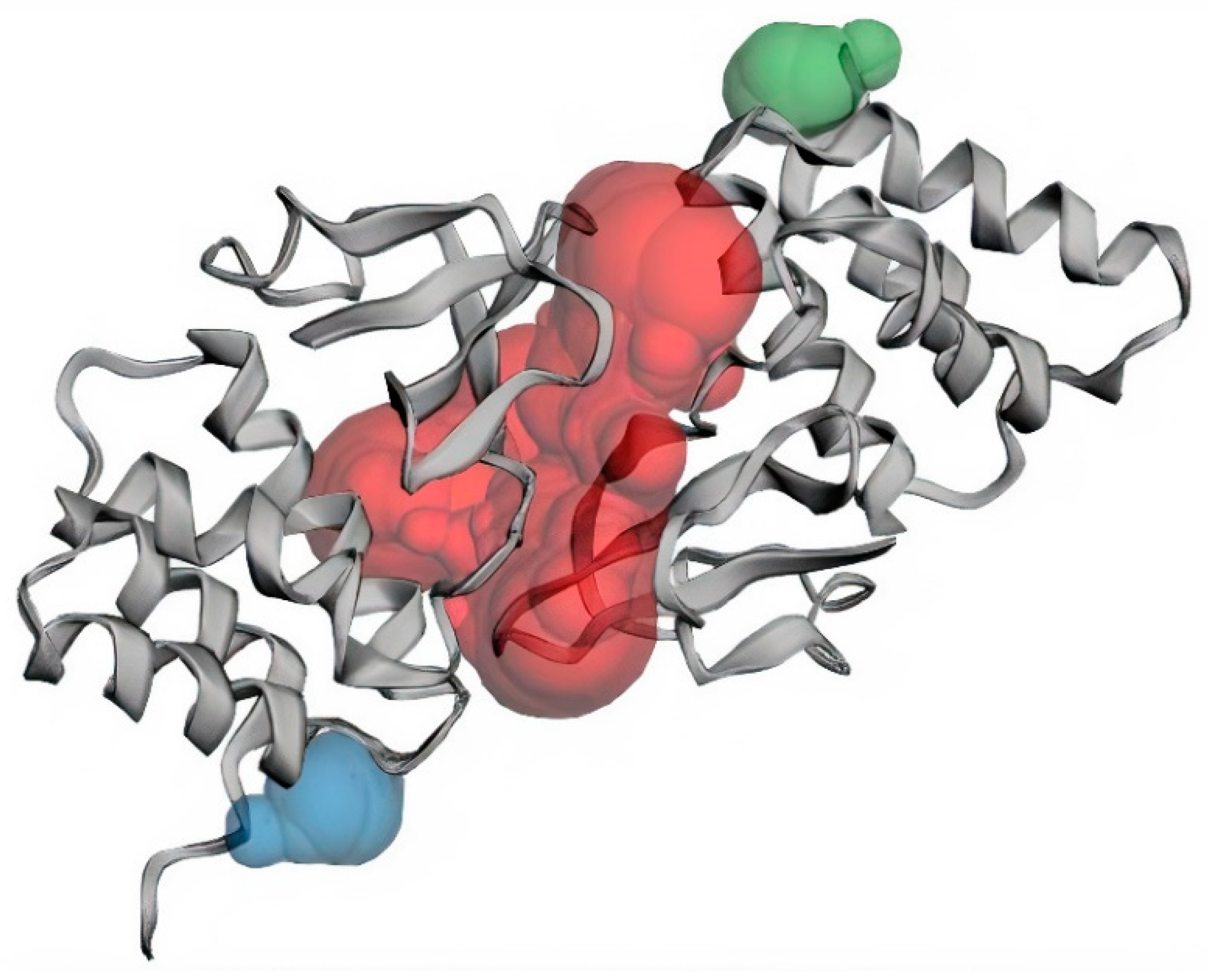
3.1. Structural and Binding Site Analysis
3.2. Molecular Docking Studies
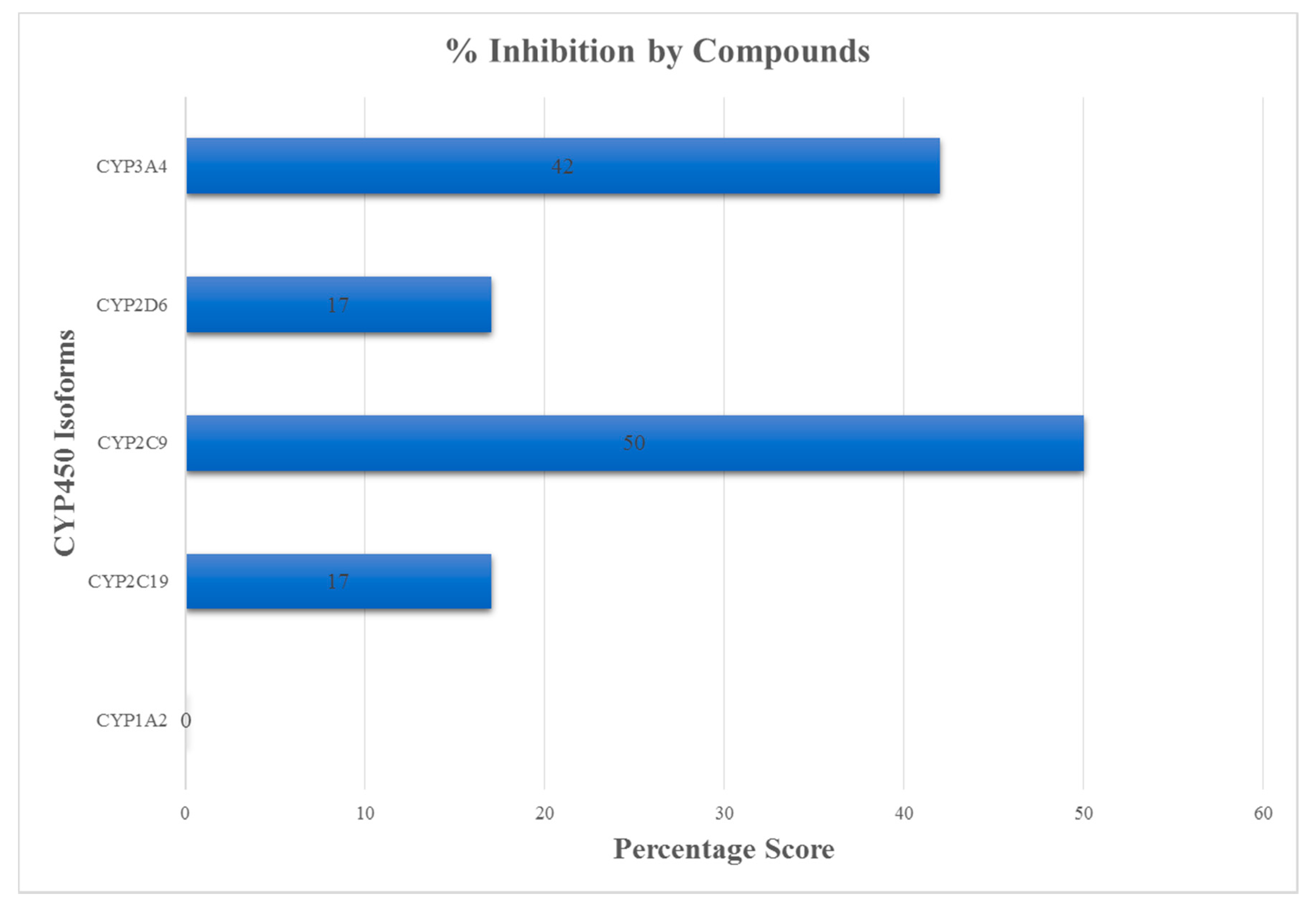
3.3. ADMET Profiling for Identification of Drug-Likeliness
3.4. Molecular Interactions of Protein-Ligand Complexes
3.5. Biological Activity Predictions for Ligands
3.6. Assessment of Quality of Ligands
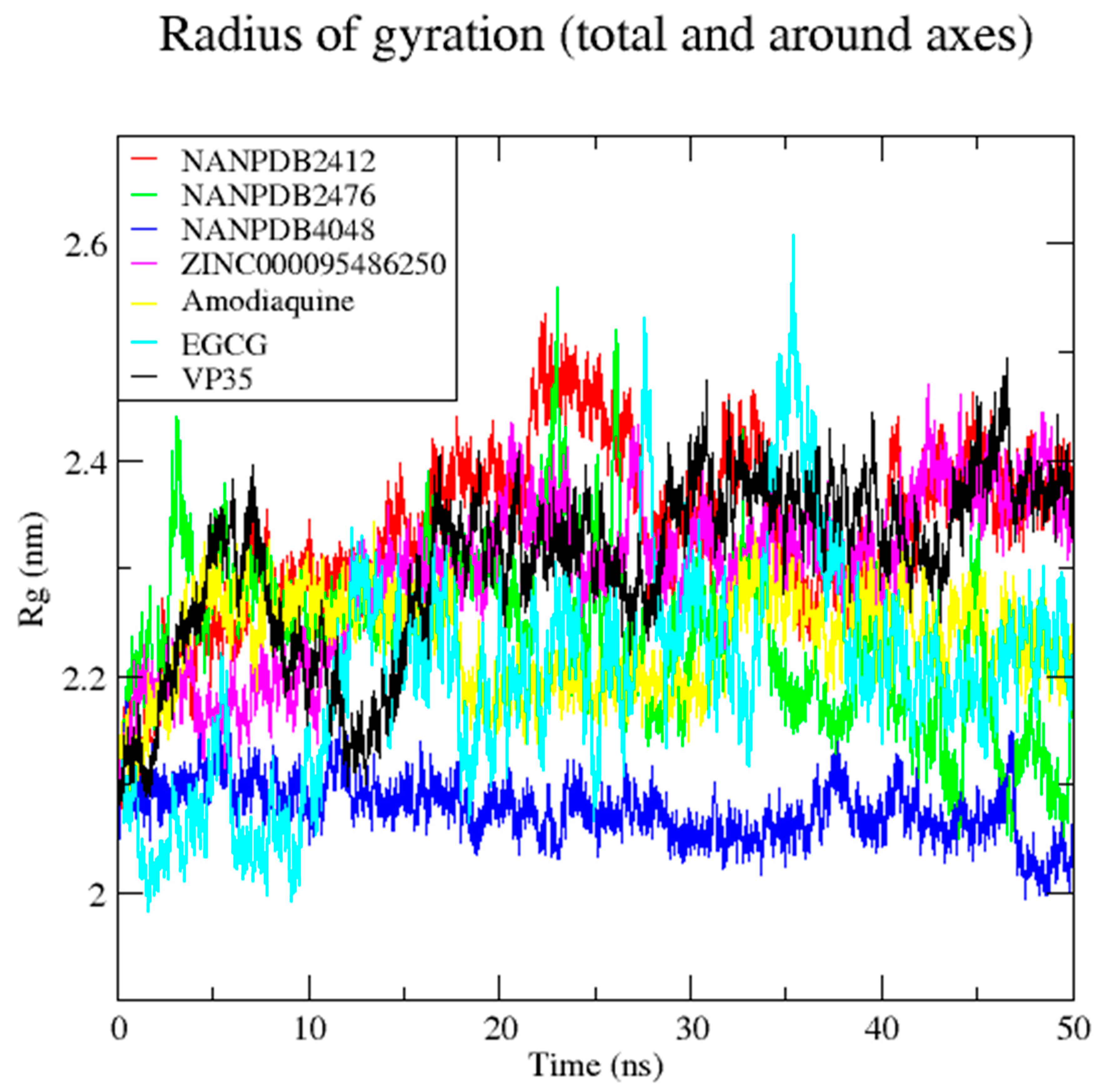
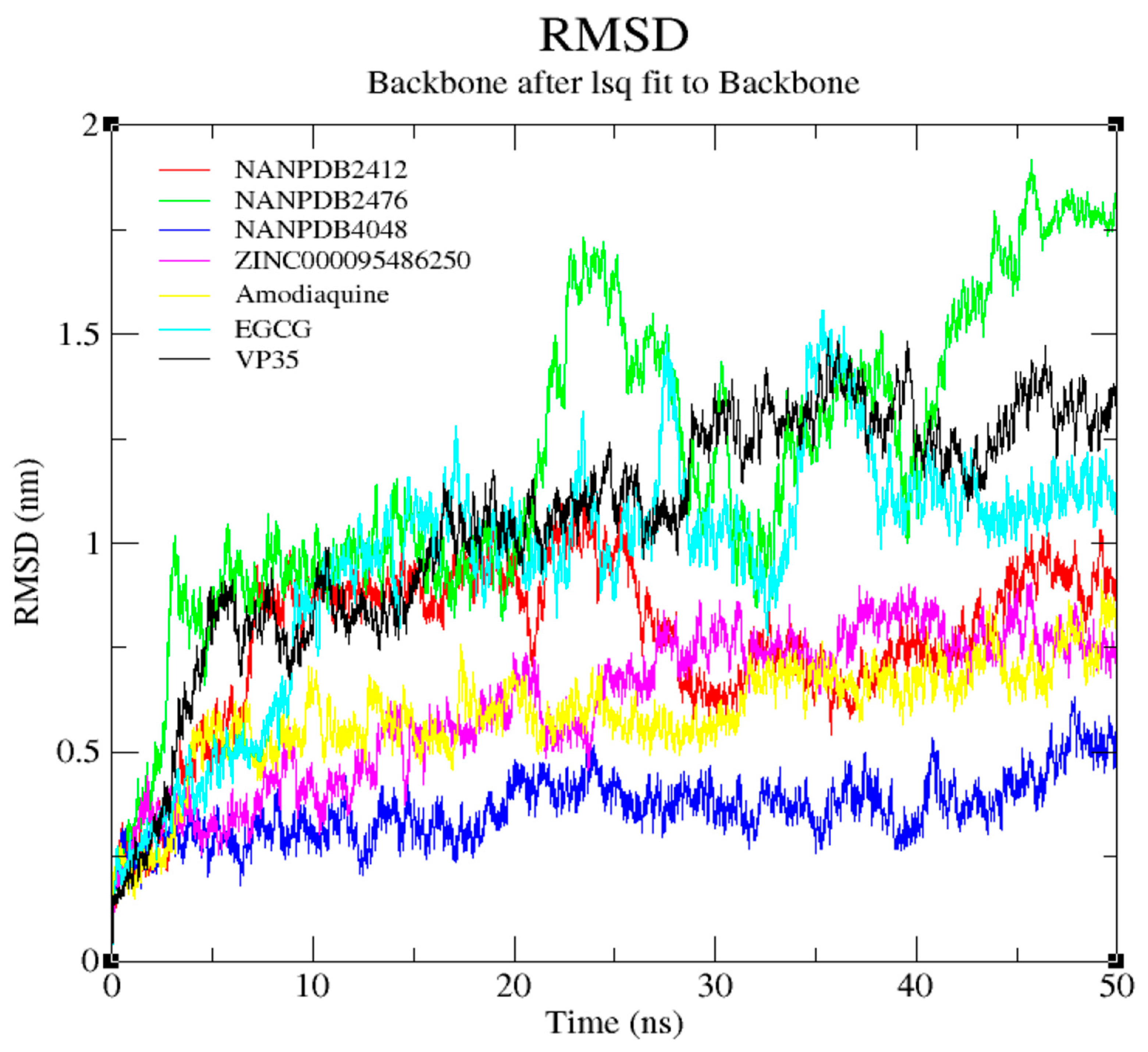
3.7. Molecular Dynamics Simulation of VP35-Ligand Complexes
3.8. MM-PBSA Computations on Potential Lead Compounds
3.9. Structural Similarity Search of Hits

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | IUPAC Names | Two-Dimensional Structure |
|---|---|---|
| NANPDB2412 | (1R,2R,5S,7S,8S,13R,14R,17R)-2,7,14-trimethyl-16-oxapentacyclo[9.7.0.02,8.05,7.013,17]octadeca-3,10-diene-12,15-dione |  |
| NANPDB2476 | (1S,3R,10S,11R,14R,16R)-5,11,14-trimethyl-2,7-dioxapentacyclo[8.8.0.0¹,³.0⁴,⁸.011,16]octadeca-4,8-dien-6-one |  |
| NANPDB4048 | (1Z,2S,3aR,3bS,9aR,9bS,11aS)-1-ethylidene-2-hydroxy-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,5H,7H,8H,9H,9aH,9bH,11aH-cyclopenta[a]phenanthren-7-one |  |
| ZINC000095486250 | (6aR,12aS)-6a,9,9,12a-tetramethyl-3H,4H,5H,6aH,7H,9H,10H,11H,12H,12aH-naphtho[2,1-b]oxocin-3-one |  |
4. Implications and Future Prospects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajak, H.; Jain, D.K.; Singh, A.; Sharma, A.K.; Dixit, A. Ebola virus disease: Past, present and future. Asian Pac. J. Trop. Biomed. 2015, 5, 337–343. [Google Scholar] [CrossRef]
- Breman, J.G.; Heymann, D.L.; Lloyd, G.; McCormick, J.B.; Miatudila, M.; Murphy, F.A.; Muyembé-Tamfun, J.J.; Piot, P.; Ruppol, J.F.; Sureau, P.; et al. Discovery and Description of Ebola Zaire Virus in 1976 and Relevance to the West African Epidemic during 2013–2016. J. Infect. Dis. 2016, 214, S93–S101. [Google Scholar] [CrossRef]
- Schwartz, D.A. Maternal and Infant Death and the rVSV-ZEBOV Vaccine Through Three Recent Ebola Virus Epidemics-West Africa, DRC Équateur and DRC Kivu: 4 Years of Excluding Pregnant and Lactating Women and Their Infants from Immunization. Curr. Trop. Med. Rep. 2019, 6, 213–222. [Google Scholar] [CrossRef]
- Chowell, G.; Nishiura, H. Transmission dynamics and control of Ebola virus disease (EVD): A review. BMC Med. 2014, 12, 196. [Google Scholar] [CrossRef]
- Safari, S.; Baratloo, A.; Rouhipour, A.; Ghelichkhani, P.; Yousefifard, M. Ebola Hemorrhagic Fever as a Public Health Emergency of International Concern; a Review Article. Emergency 2015, 3, 3–7. [Google Scholar] [CrossRef]
- Vetter, P.; Fischer, W.A.; Schibler, M.; Jacobs, M.; Bausch, D.G.; Kaiser, L. Ebola Virus Shedding and Transmission: Review of Current Evidence. J. Infect. Dis. 2016, 214, S177–S184. [Google Scholar] [CrossRef]
- Dixit, D.; Masumbuko Claude, K.; Kjaldgaard, L.; Hawkes, M.T. Review of Ebola virus disease in children—How far have we come? Paediatr. Int. Child Health 2021, 41, 12–27. [Google Scholar] [CrossRef]
- Judson, S.; Prescott, J.; Munster, V. Understanding Ebola Virus Transmission. Viruses 2015, 7, 511–521. [Google Scholar] [CrossRef]
- To, K.K.W.; Chan, J.F.W.; Tsang, A.K.L.; Cheng, V.C.C.; Yuen, K.-Y. Ebola virus disease: A highly fatal infectious disease reemerging in West Africa. Microbes Infect. 2015, 17, 84–97. [Google Scholar] [CrossRef]
- Alexander, K.A.; Sanderson, C.E.; Marathe, M.; Lewis, B.L.; Rivers, C.M.; Shaman, J.; Drake, J.M.; Lofgren, E.; Dato, V.M.; Eisenberg, M.C.; et al. What Factors Might Have Led to the Emergence of Ebola in West Africa? PLoS Negl. Trop. Dis. 2015, 9, e0003652. [Google Scholar] [CrossRef]
- Goeijenbier, M.; van Kampen, J.J.A.; Reusken, C.B.E.M.; Koopmans, M.P.G.; van Gorp, E.C.M. Ebola virus disease: A review on epidemiology, symptoms, treatment and pathogenesis. Neth. J. Med. 2014, 72, 442–448. [Google Scholar]
- Maras, M.H.; Miranda, M.D. The weaponization of Ebola: A new risk in the wake of an outbreak? Comp. Strategy 2016, 35, 72–79. [Google Scholar] [CrossRef]
- St Claire, M.C.; Ragland, D.R.; Bollinger, L.; Jahrling, P.B. Animal Models of Ebolavirus Infection. Comp. Med. 2017, 67, 253–262. [Google Scholar]
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: Focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef]
- Weyer, J.; Grobbelaar, A.; Blumberg, L. Ebola Virus Disease: History, Epidemiology and Outbreaks. Curr. Infect. Dis. Rep. 2015, 17, 21. [Google Scholar] [CrossRef]
- Hewlett, A.; Vasa, A.M.; Cieslak, T.J.; Lowe, J.J.; Schwedhelm, S. Viral Hemorrhagic Fever Preparedness. In Infection Prevention; Springer International Publishing: Cham, Switzerland, 2018; pp. 197–211. ISBN 9783319609805. [Google Scholar]
- Takamatsu, Y.; Kolesnikova, L.; Becker, S. Ebola virus proteins NP, VP35, and VP24 are essential and sufficient to mediate nucleocapsid transport. Proc. Natl. Acad. Sci. USA 2018, 115, 1075–1080. [Google Scholar] [CrossRef]
- Hume, A.J.; Mühlberger, E. Distinct Genome Replication and Transcription Strategies within the Growing Filovirus Family. J. Mol. Biol. 2019, 431, 4290–4320. [Google Scholar] [CrossRef]
- Balmith, M.; Soliman, M.E.S. Potential Ebola drug targets-filling the gap: A critical step forward towards the design and discovery of potential drugs. Biologia 2017, 72, 1–13. [Google Scholar] [CrossRef]
- Takada, A.; Kawaoka, Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 2001, 9, 506–511. [Google Scholar] [CrossRef]
- Olukitibi, T.A.; Ao, Z.; Mahmoudi, M.; Kobinger, G.A.; Yao, X. Dendritic Cells/Macrophages-Targeting Feature of Ebola Glycoprotein and its Potential as Immunological Facilitator for Antiviral Vaccine Approach. Microorganisms 2019, 7, 402. [Google Scholar] [CrossRef]
- Jasenosky, L.D.; Cadena, C.; Mire, C.E.; Borisevich, V.; Haridas, V.; Ranjbar, S.; Nambu, A.; Bavari, S.; Soloveva, V.; Sadukhan, S.; et al. The FDA-Approved Oral Drug Nitazoxanide Amplifies Host Antiviral Responses and Inhibits Ebola Virus. iScience 2019, 19, 1279–1290. [Google Scholar] [CrossRef]
- Kimberlin, C.R.; Bornholdt, Z.A.; Li, S.; Woods, V.L.; MacRae, I.J.; Saphire, E.O. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 314–319. [Google Scholar] [CrossRef]
- Cárdenas, W.B.; Loo, Y.-M.; Gale, M.; Hartman, A.L.; Kimberlin, C.R.; Martínez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola Virus VP35 Protein Binds Double-Stranded RNA and Inhibits Alpha/Beta Interferon Production Induced by RIG-I Signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef]
- Leung, D.W.; Basler, C.F.; Amarasinghe, G.K. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol. 2012, 20, 139–146. [Google Scholar] [CrossRef]
- Seesuay, W.; Jittavisutthikul, S.; Sae-lim, N.; Sookrung, N.; Sakolvaree, Y.; Chaicumpa, W. Human transbodies that interfere with the functions of Ebola virus VP35 protein in genome replication and transcription and innate immune antagonism. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Freundlich, J.S.; Coffee, M. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research 2014, 3, 277. [Google Scholar] [CrossRef] [PubMed]
- Pleško, S. In Silico Study of Plant Polyphenols’ Interactions with VP24–Ebola Virus Matrix Protein. Acta Chim. Slov. 2015, 62, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Raj, U.; Varadwaj, P.K. Flavonoids as Multi-target Inhibitors for Proteins Associated with Ebola Virus: In Silico Discovery Using Virtual Screening and Molecular Docking Studies. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 132–141. [Google Scholar] [CrossRef]
- Saxena, D.; Kaul, G.; Dasgupta, A.; Chopra, S. Atoltivimab/maftivimab/odesivimab (Inmazeb) combination to treat infection caused by Zaire ebolavirus. Drugs Today 2021, 57, 483. [Google Scholar] [CrossRef]
- Lane, T.; Anantpadma, M.; Freundlich, J.S.; Davey, R.A.; Madrid, P.B.; Ekins, S. The Natural Product Eugenol Is an Inhibitor of the Ebola Virus In Vitro. Pharm. Res. 2019, 36, 104. [Google Scholar] [CrossRef]
- Catarino, L.; Romeiras, M.M. Biodiversity of Vegetation and Flora in Tropical Africa. Diversity 2020, 12, 369. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Byrne, B.M.; McCoy, A.M.; James, B.J.; Frank, J.D.; Oakley, G.G. In silico and in vitro methods to identify ebola virus VP35-dsRNA inhibitors. Bioorg. Med. Chem. 2016, 24, 5388–5392. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Koulouridi, E.; Valli, M.; Ntie-Kang, F.; Bolzani, V.D.S. A primer on natural product-based virtual screening. Phys. Sci. Rev. 2019, 4, 251–290. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Reynolds, R.C. Fusing Dual-Event Data Sets for Mycobacterium tuberculosis Machine Learning Models and Their Evaluation. J. Chem. Inf. Model. 2013, 53, 3054–3063. [Google Scholar] [CrossRef]
- Serafim, M.S.M.; Kronenberger, T.; Oliveira, P.R.; Poso, A.; Honório, K.M.; Mota, B.E.F.; Maltarollo, V.G. The application of machine learning techniques to innovative antibacterial discovery and development. Expert Opin. Drug Discov. 2020, 15, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzym. Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Alam, S.; Khan, F. 3D-QSAR studies on Maslinic acid analogs for Anticancer activity against Breast Cancer cell line MCF-7. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Hussain, W.; Qaddir, I.; Mahmood, S.; Rasool, N. In silico targeting of non-structural 4B protein from dengue virus 4 with spiropyrazolopyridone: Study of molecular dynamics simulation, ADMET and virtual screening. VirusDisease 2018, 29, 147–156. [Google Scholar] [CrossRef]
- Haghighi, O.; Davaeifar, S.; Zahiri, H.S.; Maleki, H.; Noghabi, K.A. Homology Modeling and Molecular Docking Studies of Glutamate Dehydrogenase (GDH) from Cyanobacterium Synechocystis sp. PCC 6803. Int. J. Pept. Res. Ther. 2020, 26, 783–793. [Google Scholar] [CrossRef]
- Konidala, K.K.; Bommu, U.D.; Yeguvapalli, S.; Pabbaraju, N. In silico insights into prediction and analysis of potential novel pyrrolopyridine analogs against human MAPKAPK-2: A new SAR-based hierarchical clustering approach. 3 Biotech 2018, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput.-Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Réau, M.; Langenfeld, F.; Zagury, J.F.; Lagarde, N.; Montes, M. Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef]
- Goksuluk, D.; Korkmaz, S.; Zararsiz, G.; Karaagaoglu, A.E. easyROC: An Interactive Web-tool for ROC Curve Analysis Using R Language Environment. R J. 2016, 8, 213. [Google Scholar] [CrossRef]
- Shamsara, J. Correlation between Virtual Screening Performance and Binding Site Descriptors of Protein Targets. Int. J. Med. Chem. 2018, 2018, 3829307. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D. Discovery Studio Modeling Environment, Release 2017, San Diego: DassaultSystèmes, 2016. Adres. 2016. Available online: http//accelrys.com/products/collaborative-science/biovia-discoverystudio/visualizationdownload.php (accessed on 6 May 2020).
- Kumavath, R.; Azad, M.; Devarapalli, P.; Tiwari, S.; Kar, S.; Barh, D.; Azevedo, V.; Kumar, A.P. Novel aromatase inhibitors selection using induced fit docking and extra precision methods: Potential clinical use in ER-alpha-positive breast cancer. Bioinformation 2016, 12, 324–331. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Al Wasidi, A.S.; Hassan, A.S.; Naglah, A.M. In vitro cytotoxicity and druglikeness of pyrazolines and pyridines bearing benzofuran moiety. J. Appl. Pharm. Sci. 2020, 10, 142–148. [Google Scholar] [CrossRef]
- Zafar, F.; Gupta, A.; Thangavel, K.; Khatana, K.; Sani, A.A.; Ghosal, A.; Tandon, P.; Nishat, N. Physicochemical and Pharmacokinetic Analysis of Anacardic Acid Derivatives. ACS Omega 2020, 5, 6021–6030. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Druzhilovskiy, D.S.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Dmitriev, A.V.; Pogodin, P.V.; Poroikov, V.V. Computer-aided prediction of biological activity spectra for chemical compounds: Opportunities and limitation. Biomed. Chem. Res. Methods 2018, 1, e00004. [Google Scholar] [CrossRef]
- Tarasova, O.; Biziukova, N.; Kireev, D.; Lagunin, A.; Ivanov, S.; Filimonov, D.; Poroikov, V. A computational approach for the prediction of treatment history and the effectiveness or failure of antiretroviral therapy. Int. J. Mol. Sci. 2020, 21, 748. [Google Scholar] [CrossRef]
- Rajput, A.; Kumar, M. Anti-Ebola: An initiative to predict Ebola virus inhibitors through machine learning. Mol. Divers. 2021, 1, 1–10. [Google Scholar] [CrossRef]
- Kenny, P.W.; Leitão, A.; Montanari, C.A. Ligand efficiency metrics considered harmful. J. Comput.-Aided Mol. Des. 2014, 28, 699–710. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Reynolds, R.C. Group Additivity in Ligand Binding Affinity: An Alternative Approach to Ligand Efficiency. J. Chem. Inf. Model. 2017, 57, 3086–3093. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Islam, M.A.; Pillay, T.S. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2019, 38, 1798–1809. [Google Scholar] [CrossRef]
- Selvaraj, C.; Sakkiah, S.; Tong, W.; Hong, H. Molecular dynamics simulations and applications in computational toxicology and nanotoxicology. Food Chem. Toxicol. 2018, 112, 495–506. [Google Scholar] [CrossRef]
- Zhu, S. Validation of the Generalized Force Fields GAFF, CGenFF, OPLS-AA, and PRODRGFF by Testing against Experimental Osmotic Coefficient Data for Small Drug-Like Molecules. J. Chem. Inf. Model. 2019, 59, 4239–4247. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Viet, M.H.; Li, M.S. Effects of water models on binding affinity: Evidence from all-atom simulation of binding of tamiflu to A/H5N1 neuraminidase. Sci. World J. 2014, 2014, 536084. [Google Scholar] [CrossRef]
- Kumar, D.; Kumari, K.; Jayaraj, A.; Kumar, V.; Kumar, R.V.; Dass, S.K.; Chandra, R.; Singh, P. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct. Dyn. 2020, 39, 2659–2672. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles. J. Phys. Chem. B 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
- Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Ul Hasnain, M.J.; Shoaib, M.; Qadri, S.; Afzal, B.; Anwar, T.; Abbas, S.H.; Sarwar, A.; Malik, H.M.T.; Pervez, M.T. Computational analysis of functional single nucleotide polymorphisms associated with SLC26A4 gene. PLoS ONE 2020, 15, e0225368. [Google Scholar] [CrossRef]
- Paissoni, C.; Spiliotopoulos, D.; Musco, G.; Spitaleri, A. GMXPBSA 2.1: A GROMACS tool to perform MM/PBSA and computational alanine scanning. Comput. Phys. Commun. 2015, 186, 105–107. [Google Scholar] [CrossRef]
- Alkarkhi, A.F.M.; Alqaraghuli, W.A.A. R Statistical Software. In Applied Statistics for Environmental Science with R; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Leung, D.W.; Ginder, N.D.; Fulton, D.B.; Nix, J.; Basler, C.F.; Honzatko, R.B.; Amarasinghe, G.K. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA 2009, 106, 411–416. [Google Scholar] [CrossRef]
- Dilley, K.A.; Voorhies, A.A.; Luthra, P.; Puri, V.; Stockwell, T.B.; Lorenzi, H.; Basler, C.F.; Shabman, R.S. The Ebola virus VP35 protein binds viral immunostimulatory and host RNAs identified through deep sequencing. PLoS ONE 2017, 12, e0178717. [Google Scholar] [CrossRef]
- Prins, K.C.; Binning, J.M.; Shabman, R.S.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. Basic Residues within the Ebolavirus VP35 Protein Are Required for Its Viral Polymerase Cofactor Function. J. Virol. 2010, 84, 10581–10591. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitra, P. Ebola Virus VP35 Protein: Modeling of the Tetrameric Structure and an Analysis of Its Interaction with Human PKR. J. Proteome Res. 2020, 19, 4533–4542. [Google Scholar] [CrossRef]
- Brown, C.S.; Lee, M.S.; Leung, D.W.; Wang, T.; Xu, W.; Luthra, P.; Anantpadma, M.; Shabman, R.S.; Melito, L.M.; MacMillan, K.S.; et al. In Silico Derived Small Molecules Bind the Filovirus VP35 Protein and Inhibit Its Polymerase Cofactor Activity. J. Mol. Biol. 2014, 426, 2045–2058. [Google Scholar] [CrossRef]
- Leung, D.W.; Prins, K.C.; Borek, D.M.; Farahbakhsh, M.; Tufariello, J.M.; Ramanan, P.; Nix, J.C.; Helgeson, L.A.; Otwinowski, Z.; Honzatko, R.B.; et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat. Struct. Mol. Biol. 2010, 17, 165–172. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ikram, N. Integrated computational approach for virtual hit identification against ebola viral proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef]
- Kashyap, S. Comparative Insillico Studies on Phytochemicals of Ocimum as Natural Inhibitors of Ebola Vp-35 Protein. Indo Am. J. Pharm. Sci. 2019, 10, 489–511. [Google Scholar] [CrossRef]
- Empereur-mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.-F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef]
- Li, F.; He, H. Assessing the Accuracy of Diagnostic Tests. Shanghai Arch. Psychiatry 2018, 30, 207–212. [Google Scholar] [CrossRef]
- Mohan, R.R.; Wilson, M.; Gorham, R.D.; Harrison, R.E.S.; Morikis, V.A.; Kieslich, C.A.; Orr, A.A.; Coley, A.V.; Tamamis, P.; Morikis, D. Virtual Screening of Chemical Compounds for Discovery of Complement C3 Ligands. ACS Omega 2018, 3, 6427–6438. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, K.O.; Kolapo, T.U.; Onawole, A.T.; Islam, M.A.; Adegoke, R.O.; Badmus, S.O. Molecular dynamics and combined docking studies for the identification of Zaire ebola virus inhibitors. J. Biomol. Struct. Dyn. 2019, 9, 5142. [Google Scholar] [CrossRef]
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ.-Sci. 2019, 31, 595–601. [Google Scholar] [CrossRef]
- El-Kattan, A.; Varm, M. Oral Absorption, Intestinal Metabolism and Human Oral Bioavailability. In Topics on Drug Metabolism; BoD—Books on Demand: Norderstedt, Germany, 2012. [Google Scholar]
- Bowen, L.; Smith, B.; Steinbach, S.; Billioux, B.; Summers, A.; Azodi, S.; Ohayon, J.; Schindler, M.; Nath, A. Survivors of Ebola Virus Disease Have Persistent Neurological Deficits (Abstract S53.003). In Proceedings of the American Academy of Neurology Annual Meeting, Vancouver, BC, Canada, 15–21 April 2016. [Google Scholar]
- Billioux, B.J.; Smith, B.; Nath, A. Neurological Complications of Ebola Virus Infection. Neurotherapeutics 2016, 13, 461–470. [Google Scholar] [CrossRef]
- Sagui, E.; Janvier, F.; Baize, S.; Foissaud, V.; Koulibaly, F.; Savini, H.; Maugey, N.; Aletti, M.; Granier, H.; Carmoi, T. Severe Ebola Virus Infection with Encephalopathy: Evidence for Direct Virus Involvement. Clin. Infect. Dis. 2015, 61, 1627–1628. [Google Scholar] [CrossRef]
- de Greslan, T.; Billhot, M.; Rousseau, C.; Mac Nab, C.; Karkowski, L.; Cournac, J.-M.; Bordes, J.; Gagnon, N.; Dubrous, P.; Duron, S.; et al. Ebola Virus–Related Encephalitis: Table 1. Clin. Infect. Dis. 2016, 63, 1076–1078. [Google Scholar] [CrossRef][Green Version]
- Wong, G.; Qiu, X.; Bi, Y.; Formenty, P.; Sprecher, A.; Jacobs, M.; Gao, G.F.; Kobinger, G. More Challenges from Ebola: Infection of the Central Nervous System. J. Infect. Dis. 2016, 214, S294–S296. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef]
- Karthika, C.; Sureshkumar, R. P-Glycoprotein Efflux Transporters and Its Resistance Its Inhibitors and Therapeutic Aspects. In Creatinine—A Comprehensive Update [Working Title]; IntechOpen: London, UK, 2020. [Google Scholar]
- Ma, J.D.; Tsunoda, S.M.; Bertino, J.S.; Trivedi, M.; Beale, K.K.; Nafziger, A.N. Evaluation of in vivo P-glycoprotein phenotyping probes: A need for validation. Clin. Pharmacokinet. 2010, 49, 223–237. [Google Scholar] [CrossRef]
- Dutkiewicz, Z.; Mikstacka, R. Structure-Based Drug Design for Cytochrome P450 Family 1 Inhibitors. Bioinorg. Chem. Appl. 2018, 2018, 3924608. [Google Scholar] [CrossRef]
- Egieyeh, S.A.; Syce, J.; Malan, S.F.; Christoffels, A. Prioritization of anti-malarial hits from nature: Chemo-informatic profiling of natural products with in vitro antiplasmodial activities and currently registered anti-malarial drugs. Malar. J. 2016, 15, 50. [Google Scholar] [CrossRef]
- Ren, J.X.; Zhang, R.T.; Zhang, H.; Cao, X.S.; Liu, L.K.; Xie, Y. Identification of novel VP35 inhibitors: Virtual screening driven new scaffolds. Biomed. Pharmacother. 2016, 84, 199–207. [Google Scholar] [CrossRef]
- Baikerikar, S. Curcumin and natural derivatives inhibit Ebola viral proteins: An in silico approach. Pharmacognosy Res. 2017, 9, 15. [Google Scholar] [CrossRef]
- Parasuraman, S. Prediction of activity spectra for substances. J. Pharmacol. Pharmacother. 2011, 2, 52–53. [Google Scholar] [CrossRef]
- Tarasova, O.; Filimonov, D.; Poroikov, V. PASS-based approach to predict HIV-1 reverse transcriptase resistance. J. Bioinform. Comput. Biol. 2017, 15, 1650040. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Duplantier, A.J.; Bavari, S. Discovering Drugs for the Treatment of Ebola Virus. Curr. Treat. Options Infect. Dis. 2017, 9, 299–317. [Google Scholar] [CrossRef]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against Ebola virus. J. Med. Virol. 2019, 91, 2029–2048. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Elvati, P.; Violi, A. Antiviral Drug-Membrane Permeability: The Viral Envelope and Cellular Organelles. arXiv 2020, arXiv:2007.14965. [Google Scholar]
- Mazzon, M.; Marsh, M. Targeting viral entry as a strategy for broad-spectrum antivirals [version 1; peer review: 3 approved]. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Jamkhande, P.G.; Pathan, S.K.; Wadher, S.J. In silico PASS analysis and determination of antimycobacterial, antifungal, and antioxidant efficacies of maslinic acid in an extract rich in pentacyclic triterpenoids. Int. J. Mycobacteriol. 2016, 5, 417–425. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Teye, J.; Quansah, E.; Issah, I.; Wilson, M.D.; Miller, W.A.; Tiburu, E.K.; Bonney, J.H.K. Pharmacoinformatics-based identification of potential bioactive compounds against Ebola virus protein VP24. Comput. Biol. Med. 2019, 113, 103414. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H. Ligand efficiency metrics: Why all the fuss? Future Med. Chem. 2015, 7, 1363–1365. [Google Scholar] [CrossRef]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting Protein-Protein Interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.Y.; Coumar, M.S.; Shiao, H.Y.; Wang, W.C.; Chen, C.W.; Song, J.S.; Chen, C.H.; Lin, W.H.; Wu, S.H.; Hsu, J.T.A.; et al. Ligand efficiency based approach for efficient virtual screening of compound libraries. Eur. J. Med. Chem. 2014, 83, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Kumar, R.; Planey, S.L. Lipophilicity Indices for Drug Development. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 31–36. [Google Scholar]
- Xue, X.; Bao, G.; Zhang, H.Q.; Zhao, N.Y.; Sun, Y.; Zhang, Y.; Wang, X.L. An application of fit quality to screen MDM2/p53 protein-protein interaction inhibitors. Molecules 2018, 23, 3174. [Google Scholar] [CrossRef]
- Bembenek, S.D.; Tounge, B.A.; Reynolds, C.H. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today 2009, 14, 278–283. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Al Mamun, A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 39, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Calero-Rubio, C.; Paik, B.; Jia, X.; Kiick, K.L.; Roberts, C.J. Predicting unfolding thermodynamics and stable intermediates for alanine-rich helical peptides with the aid of coarse-grained molecular simulation. Biophys. Chem. 2016, 217, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.H.; Chen, K.B.; Lee, W.Y.; Sun, M.F.; Lee, C.C.; Chen, C.Y.C. Ligand-based and structure-based investigation for Alzheimer’s disease from traditional Chinese medicine. Evid.-Based Complement. Altern. Med. 2014, 2014, 364819. [Google Scholar] [CrossRef]
- Karthick, V.; Nagasundaram, N.; Doss, C.G.P.; Chakraborty, C.; Siva, R.; Lu, A.; Zhang, G.; Zhu, H. Virtual screening of the inhibitors targeting at the viral protein 40 of Ebola virus. Infect. Dis. Poverty 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Goyal, S.; Binnington, B.; McCarthy, S.D.S.; Desmaële, D.; Férrié, L.; Figadère, B.; Loiseau, P.M.; Branch, D.R. Inhibition of in vitro Ebola infection by anti-parasitic quinoline derivatives. F1000Research 2020, 9, 268. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Steinmetz, N.F.; Ching, W.Y. Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys. Chem. Chem. Phys. 2019, 21, 3877–3893. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Shen, C.; Liu, H.; Wang, X.; Lei, T.; Wang, E.; Xu, L.; Yu, H.; Li, D.; Yao, X. Importance of incorporating protein flexibility in molecule modeling: A theoretical study on type I1/2 NIK inhibitors. Front. Pharmacol. 2019, 10, 345. [Google Scholar] [CrossRef]
- Asiedu, S.O.; Kwofie, S.K.; Broni, E.; Wilson, M.D. Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm. Biomolecules 2021, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Chang, F.R.; Chen, S.R.; Wu, Y.H.Y.C.; Hu, H.C.; Wu, Y.H.Y.C.; Backlund, A.; Cheng, Y. Bin Anti-dengue virus constituents from Formosan zoanthid Palythoa mutuki. Mar. Drugs 2016, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Jomori, T.; Tanaka, J.; Senba, M.; Mori, N. Peridinin, a carotenoid, inhibits proliferation and survival of HTLV-1-infected T-cell lines. Int. J. Oncol. 2016, 49, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; He, L.; Xie, H.; Wu, S.; Li, Y.; Zhang, P.; Yang, Z.; Huang, X. Antiviral activity of Carbenoxolone disodium against dengue virus infection. J. Med. Virol. 2017, 89, 571–581. [Google Scholar] [CrossRef]
- Haga, I.R.; Simpson, J.L.; Hawes, P.C.; Beard, P.M. Carbenoxolone-mediated cytotoxicity inhibits Vaccinia virus replication in a human keratinocyte cell line. Sci. Rep. 2018, 8, 16956. [Google Scholar] [CrossRef]
- Dargan, D.J.; Subak-Sharpe, J.H. The antiviral activity against herpes simplex virus of the triterpenoid compounds carbenoxolone sodium and cicloxolone sodium. J. Antimicrob. Chemother. 1986, 18, 185–200. [Google Scholar] [CrossRef]
- Kim, J.; Park, K.E.; Jeong, Y.S.; Kim, Y.M.; Park, H.; Nam, J.H.; Jung, K.; Son, W.S.; Jung, H.S.; Lee, J.H.; et al. Therapeutic efficacy of ABN401, a highly potent and selective MET inhibitor, based on diagnostic biomarker test in MET-addicted cancer. Cancers 2020, 12, 1575. [Google Scholar] [CrossRef]
- Awadh Ali, N.; Al Sokari, S.; Gushash, A.; Anwar, S.; Al-Karani, K.; Al-Khulaidi, A. Ethnopharmacological survey of medicinal plants in Albaha Region, Saudi Arabia. Pharmacognosy Res. 2017, 9, 401–407. [Google Scholar] [CrossRef]
- Islam, M.A.; Pillay, T.S. Pharmacoinformatics-based identification of chemically active molecules against Ebola virus. J. Biomol. Struct. Dyn. 2018, 37, 4104–4119. [Google Scholar] [CrossRef]
- Setlur, A.S.; Naik, S.Y.; Skariyachan, S. Herbal Lead as Ideal Bioactive Compounds Against Probable Drug Targets of Ebola Virus in Comparison with Known Chemical Analogue: A Computational Drug Discovery Perspective. Interdiscip. Sci. Comput. Life Sci. 2017, 9, 254–277. [Google Scholar] [CrossRef] [PubMed]
- Tambunan, U.S.F.; Alkaff, A.H.; Nasution, M.A.F. Bioinformatics Approach to Screening and Developing Drug against Ebola. In Advances in Ebola Control; BoD—Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]








| Binding Sites | Chain | Amino Acid Residues | Surface Area (SA)/Å2 | Volume/Å3 |
|---|---|---|---|---|
| Pocket 1 | A | Val245, Lys248, Leu249, Asp252, Ser253, Ile286, Phe287, Gln288, Asp289, Ala290, Ala291, Pro292, Pro293, Val294, Ile295, His296, Ile297, Arg298, Val314, Pro315, Pro316, Ser317, Pro318, Lys319, Val327, Gln329, Leu330, Gln331, Gly333, Thr335. | 1155.05 | 1078.689 |
| B | Gln241, Gln244, Val245, Lys248, Leu249, Asp252, Ser253, Ile286, Gln288, Asp289, Ala290, Ala291, Pro292, Pro293, Val294, Ile295, His296, Val314, Pro315, Pro316, Ser317, Pro318, Lys319, Val327, Gln329, Leu330, Gln331, Gly333, Lys334, Thr335. | |||
| Pocket 2 | B | Asp218, Ile219, Asn254, Leu256, Asp257 | 48.092 | 35.5916 |
| Pocket 3 | A | Asp218, Ile219, Asn254, Leu256, Asp257 | 52.040 | 34.782 |
| Compound ID | Binding Energy (kcal/mol) | Number of Hydrogen Bonds | Hydrogen Bond Residues | Hydrogen Bond Length (Å) | Hydrophobic Contacts |
|---|---|---|---|---|---|
| NANPDB86 | −8.5 | 1 | Gln329 | 2.0 | Val245, Leu249, Pro293, Val294, Ile295 |
| NANPDB95 | −8.1 | 0 | - | - | Pro316, Ala291, Pro292, Leu249, Pro293, Val294, Val327, Ile286, Ala290, Pro315, Pro318, Val314 |
| NANPDB142 | −8.0 | 0 | - | - | Pro318, Ala291, Pro315, Pro316, Ala290, Val294, Val327, Val314, Leu249 |
| NANPDB205 | −8.3 | 0 | - | - | Leu249, Pro293, Val245, Ile295 |
| NANPDB397 | −8.1 | 0 | - | - | Pro318, Val314, Ala291, Pro292, Pro293, Val327, Val294 |
| NANPDB2412 | −8.2 | 0 | - | - | Pro318, Pro316, Ala290, Pro315, Ala291, Val314, Pro292, Val294, Pro293, Val327 |
| NANPDB2476 | −8.0 | 0 | - | - | Pro316, Ala291, Pro315, Pro318, Pro292, Val314, Val327, Val294 |
| NANPDB3355 | −8.2 | 0 | - | - | Pro316, Ala290, Ala291, Pro292, Val314, Pro318, Val294, Val327 |
| NANPDB4048 | −8.2 | 0 | - | - | Pro318, Ala291, Val314, Pro292, Pro293, Leu249, Val294, Val327 |
| ZINC000014612849 | −8.1 | 0 | - | - | Val314, Pro292, Ala291, Pro318, Pro315, Val327, Val294 |
| ZINC000033831303 | −8.0 | 0 | - | - | Pro293, Leu249, Ile295, Val245, Val294 |
| ZINC000095486250 | −8.1 | 0 | - | - | Ala291, Pro318, Pro292, Val314, Pro293, Val327, Val294 |
| Amodiaquine | −7.0 | 0 | - | - | Ala291, Pro318, Ala291, Pro315, Val327, Val294, Pro292, Val314 |
| Chloroquine | −5.9 | 0 | - | - | Pro318, Val314, Val327, Pro292, Ala291, Val294, Pro293 |
| EGCG | −8.1 | 1 | Gln244 | 2.01 | Val294, Pro293, Leu249, Val245, Cys247, Ile297, Leu330 |
| Gossypetin | −7.5 | 1 | Leu330 | 1.97 | Ile295, Val294, Pro293, Leu249, Val245 |
| Taxifolin | −7.4 | 0 | - | - | Val314, Ala290, Ala291, Pro318, Val294, Val327, Pro292, Leu249 |
| Compound ID | Estimated Solubility Log S | Estimated Solubility Class | GI Absorption | BBB Permeant | P-glycoprotein Substrate |
|---|---|---|---|---|---|
| NANPDB86 | −3.79 | Soluble | High | Yes | No |
| NANPDB95 | −3.57 | Soluble | High | Yes | No |
| NANPDB142 | −3.77 | Soluble | High | Yes | No |
| NANPDB205 | −2.61 | Soluble | High | Yes | No |
| NANPDB397 | −3.09 | Soluble | High | Yes | No |
| NANPDB2412 | −3.99 | Soluble | High | Yes | No |
| NANPDB2476 | −3.89 | Soluble | High | Yes | No |
| NANPDB3355 | −3.25 | Soluble | High | Yes | No |
| NANPDB4048 | −3.73 | Soluble | High | Yes | No |
| ZINC000014612849 | −3.00 | Soluble | High | Yes | No |
| ZINC000033831303 | −3.89 | Soluble | High | Yes | No |
| ZINC000095486250 | −3.41 | Soluble | High | Yes | No |
| Amodiaquine | −5.9 | Moderately soluble | High | Yes | No |
| Chloroquine | −4.55 | Moderately soluble | High | Yes | No |
| EGCG | −3.56 | Soluble | Low | No | No |
| Gossypetin | −3.40 | Soluble | Low | No | No |
| Taxifolin | −2.66 | Soluble | High | No | No |
| Compound ID | Mutagenic | Tumorigenic | Reproductive Effect | Irritant |
|---|---|---|---|---|
| NANPDB86 | None | None | None | None |
| NANPDB95 | None | None | None | None |
| NANPDB142 | None | None | None | None |
| NANPDB205 | None | None | High | None |
| NANPDB397 | None | None | None | None |
| NANPDB2412 | None | None | None | None |
| NANPDB2476 | None | None | None | High |
| NANPDB3355 | None | High | None | High |
| NANPDB4048 | None | None | High | None |
| ZINC000014612849 | Low | None | None | None |
| ZINC000033831303 | High | High | None | High |
| ZINC000095486250 | None | None | None | None |
| Amodiaquine | High | None | High | High |
| Chloroquine | High | None | None | High |
| EGCG | None | None | None | None |
| Gossypetin | High | None | None | None |
| Taxifolin | None | None | None | None |
| Compound ID | Biological Activity | Pa | Pi |
|---|---|---|---|
| NANPDB86 | Rhinovirus | 0.444 | 0.052 |
| Herpes | 0.334 | 0.069 | |
| Protein synthesis inhibitor | 0.467 | 0.008 | |
| Transcription factor inhibitor | 0.39 | 0.026 | |
| RNA synthesis inhibitor | 0.287 | 0..63 | |
| NANPDB95 | Herpes | 0.394 | 0.038 |
| Picornavirus | 0.337 | 0.173 | |
| Transcription factor inhibitor | 0.557 | 0.008 | |
| Protein synthesis inhibitor | 0.493 | 0.007 | |
| RNA synthesis inhibitor | 0.331 | 0.038 | |
| NANPDB142 | Rhinovirus | 0.413 | 0.078 |
| Herpes | 0.332 | 0.071 | |
| Picornavirus | 0.352 | 0.156 | |
| DNA polymerase 1 inhibitor | 0.625 | 0.003 | |
| RNA synthesis inhibitor | 0.285 | 0.065 | |
| NANPDB205 | Adenovirus | 0.222 | 0.176 |
| Protein synthesis inhibitor | 0.238 | 0.041 | |
| RNA synthesis inhibitor | 0.251 | 0.100 | |
| DNA synthesis inhibitor | 0.207 | 0.141 | |
| NANPDB397 | - | - | - |
| NANPDB2412 | Herpes | 0.410 | 0.031 |
| Rhinovirus | 0.345 | 0.167 | |
| Transcription factor inhibitor | 0.283 | 0.013 | |
| DNA synthesis inhibitor | 0.232 | 0.101 | |
| RNA synthesis inhibitor | 0.231 | 0.125 | |
| NANPDB2476 | Influenza | 0.476 | 0.027 |
| Rhinovirus | 0.381 | 0.114 | |
| Protein synthesis inhibitor | 0.376 | 0.019 | |
| RNA synthesis inhibitor | 0.277 | 0.072 | |
| NANPDB3355 | Rhinovirus | 0.552 | 0.012 |
| Protein synthesis inhibitor | 0.353 | 0.022 | |
| Transcription factor inhibitor | 0.240 | 0.093 | |
| RNA synthesis inhibitor | 0.241 | 0.111 | |
| NANPDB4048 | Influenza | 0.621 | 0.011 |
| Rhinovirus | 0.362 | 0.140 | |
| Membrane permeability inhibitor | 0.753 | 0.020 | |
| RNA synthesis inhibitor | 0.484 | 0.009 | |
| ZINC000014612849 | - | - | - |
| ZINC000033831303 | RNA synthesis inhibitor | 0.281 | 0.069 |
| ZINC000095486250 | Influenza | 0.399 | 0.047 |
| Herpes | 0.273 | 0.111 | |
| RNA synthesis inhibitor | 0.298 | 0.056 | |
| DNA polymerase I inhibitor | 0.275 | 0.098 | |
| Amodiaquine | - | - | - |
| Chloroquine | - | - | - |
| EGCG | Influenza | 0.771 | 0.003 |
| Rhinovirus | 0.514 | 0.020 | |
| Herpes | 0.480 | 0.012 | |
| HIV | 0.300 | 0.008 | |
| Hepatitis B | 0.316 | 0.029 | |
| Transcription factor inhibitor | 0.404 | 0.007 | |
| RNA synthesis inhibitor | 0.318 | 0.044 | |
| DNA polymerase I inhibitor | 0.294 | 0.070 | |
| Gossypetin | Hepatitis B | 0.498 | 0.005 |
| Influenza | 0.415 | 0.042 | |
| Membrane permeability inhibitor | 0.953 | 0.002 | |
| RNA synthesis inhibitor | 0.358 | 0.029 | |
| DNA polymerase I inhibitor | 0.331 | 0.040 | |
| Taxifolin | Influenza | 0.620 | 0.011 |
| Herpes | 0.492 | 0.010 | |
| Rhinovirus | 0.503 | 0.023 | |
| Hepatitis B | 0.399 | 0.015 | |
| Membrane permeability inhibitor | 0.850 | 0.005 | |
| Transcription factor inhibitor | 0.413 | 0.022 | |
| DNA polymerase I inhibitor | 0.329 | 0.041 | |
| RNA synthesis inhibitor | 0.394 | 0.021 |
| Compound ID | Number of Heavy Atoms | Log P | Ki | LE | LE_Scale | FQ | LELP |
|---|---|---|---|---|---|---|---|
| NANPDB86 | 24 | 2.79 | 5.87 × 10−7 | 0.354 | 0.404 | 0.876 | 7.88 |
| NANPDB95 | 24 | 2.94 | 1.15 × 10−6 | 0.338 | 0.404 | 0.837 | 8.70 |
| NANPDB95 | 24 | 2.94 | 1.15 × 10−6 | 0.338 | 0.404 | 0.837 | 8.70 |
| NANPDB142 | 25 | 2.93 | 1.37 × 10−6 | 0.320 | 0.391 | 0.818 | 9.16 |
| NANPDB205 | 20 | 1.81 | 8.23 × 10−7 | 0.415 | 0.467 | 0.889 | 4.36 |
| NANPDB397 | 24 | 2.66 | 1.15 × 10−6 | 0.338 | 0.404 | 0.837 | 7.87 |
| NANPDB2412 | 23 | 3.25 | 9.74 × 10−7 | 0.357 | 0.418 | 0.854 | 9.10 |
| NANPDB2476 | 22 | 3.55 | 1.37 × 10−6 | 0.364 | 0.433 | 0.841 | 9.75 |
| NANPDB3355 | 24 | 2.43 | 9.74 × 10−7 | 0.342 | 0.404 | 0.847 | 7.11 |
| NANPDB4048 | 23 | 3.61 | 9.74 × 10−7 | 0.357 | 0.418 | 0.854 | 10.11 |
| ZINC000014612849 | 25 | 2.22 | 1.15 × 10−6 | 0.324 | 0.391 | 0.829 | 6.85 |
| ZINC000033831303 | 23 | 3.37 | 1.37 × 10−6 | 0.348 | 0.418 | 0.833 | 9.68 |
| ZINC000095486250 | 21 | 3.68 | 1.15 × 10−6 | 0.386 | 0.449 | 0.860 | 9.53 |
| Compound ID | Binding Affinity from Docking [kcal/mol (kJ/mol)] | van der Waal Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|---|
| NANPDB2412 | −8.2 (−34.3088) | −112.794 ± 31.343 | −4.338 ± 7.888 | 63.305 ± 25.933 | −13.955 ± 3.243 | −67.782 ± 17.041 |
| NANPDB2476 | −8.0 (−33.472) | −72.353 ± 15.702 | −8.393 ± 9.299 | 46.887 ± 21.330 | −10.531 ± 2.288 | −44.390 ± 19.503 |
| NANPDB4048 | −8.2 (−34.3088) | −122.063 ± 24.789 | −3.170 ± 8.186 | 68.675 ± 23.656 | −15.854 ± 2.967 | −72.413 ± 15.915 |
| ZINC000095486250 | −8.1 (−33.8904) | −133.848 ± 15.162 | −6.489 ± 7.863 | 62.413 ± 10.653 | −16.289 ± 1.014 | −94.213 ± 12.755 |
| Amodiaquine | −7.0 (−29.288) | −150.934 ± 19.558 | −6.282 ± 8.679 | 83.311 ± 13.703 | −18.495 ± 1.647 | −92.400 ± 15.855 |
| EGCG | −8.1 (−33.8904) | −110.393 ± 27.459 | −46.227 ± 20.847 | 126.216 ± 35.236 | −14.160 ± 3.019 | −44.564 ± 23.104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darko, L.K.S.; Broni, E.; Amuzu, D.S.Y.; Wilson, M.D.; Parry, C.S.; Kwofie, S.K. Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds. Biomedicines 2021, 9, 1796. https://doi.org/10.3390/biomedicines9121796
Darko LKS, Broni E, Amuzu DSY, Wilson MD, Parry CS, Kwofie SK. Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds. Biomedicines. 2021; 9(12):1796. https://doi.org/10.3390/biomedicines9121796
Chicago/Turabian StyleDarko, Louis K. S., Emmanuel Broni, Dominic S. Y. Amuzu, Michael D. Wilson, Christian S. Parry, and Samuel K. Kwofie. 2021. "Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds" Biomedicines 9, no. 12: 1796. https://doi.org/10.3390/biomedicines9121796
APA StyleDarko, L. K. S., Broni, E., Amuzu, D. S. Y., Wilson, M. D., Parry, C. S., & Kwofie, S. K. (2021). Computational Study on Potential Novel Anti-Ebola Virus Protein VP35 Natural Compounds. Biomedicines, 9(12), 1796. https://doi.org/10.3390/biomedicines9121796