
Generation of a Novel High-Affinity Antibody Binding to PCSK9 Catalytic Domain with Slow Dissociation Rate by CDR-Grafting, Alanine Scanning and Saturated Site-Directed Mutagenesis for Favorably Treating Hypercholesterolemia

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Bacterial Strains and Cell Lines
2.3. Antigen Preparation
2.4. Generation of Murine Monoclonal Antibodies
2.5. Enzyme-Linked Immunosorbent Assay
2.6. Western Blot Analysis
2.7. LDL-C Uptake Assay
2.8. Cloning of VH and VL Gene from Hybridoma Cells
2.9. Computer Modeling of Single-Chain Variable-Fragment Antibodies
2.10. Design of Humanized scFvs
2.11. Construction, Expression, and Purification of scFvs
2.12. Saturated Site-Directed Mutagenesis of Humanized scFv
2.13. Generation of Full-Length Antibodies
2.14. Binding Affinity Measurement
2.15. Homology Modeling and Protein Contact Identification
2.16. Studies in Mice
2.17. Immunofluorescence Analysis
2.18. Statistical Analysis
3. Results
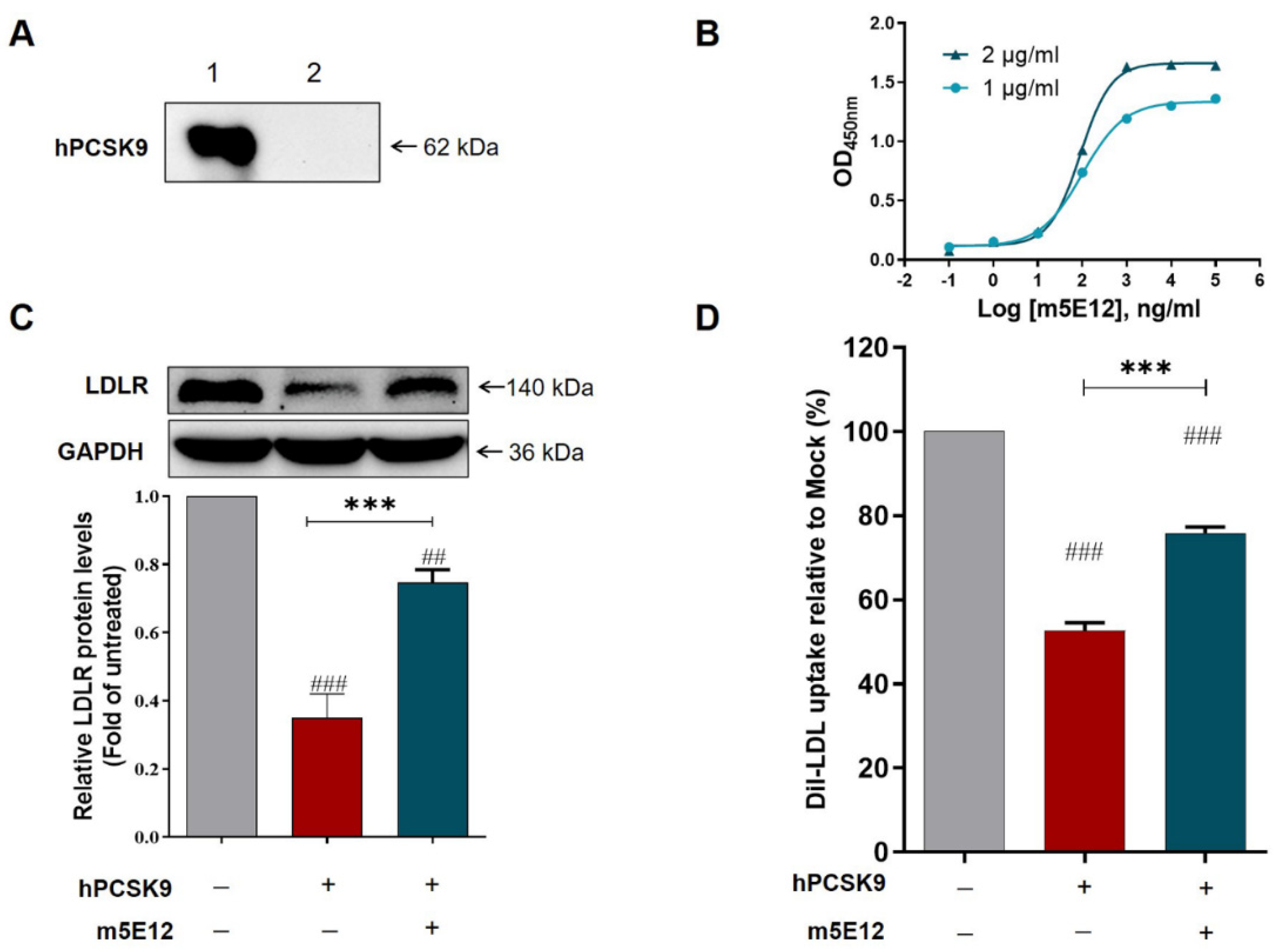
3.1. Generation of Murine Mab against Hpcsk9 by Hybridoma Technology
3.2. Characterization of Generated m5E12
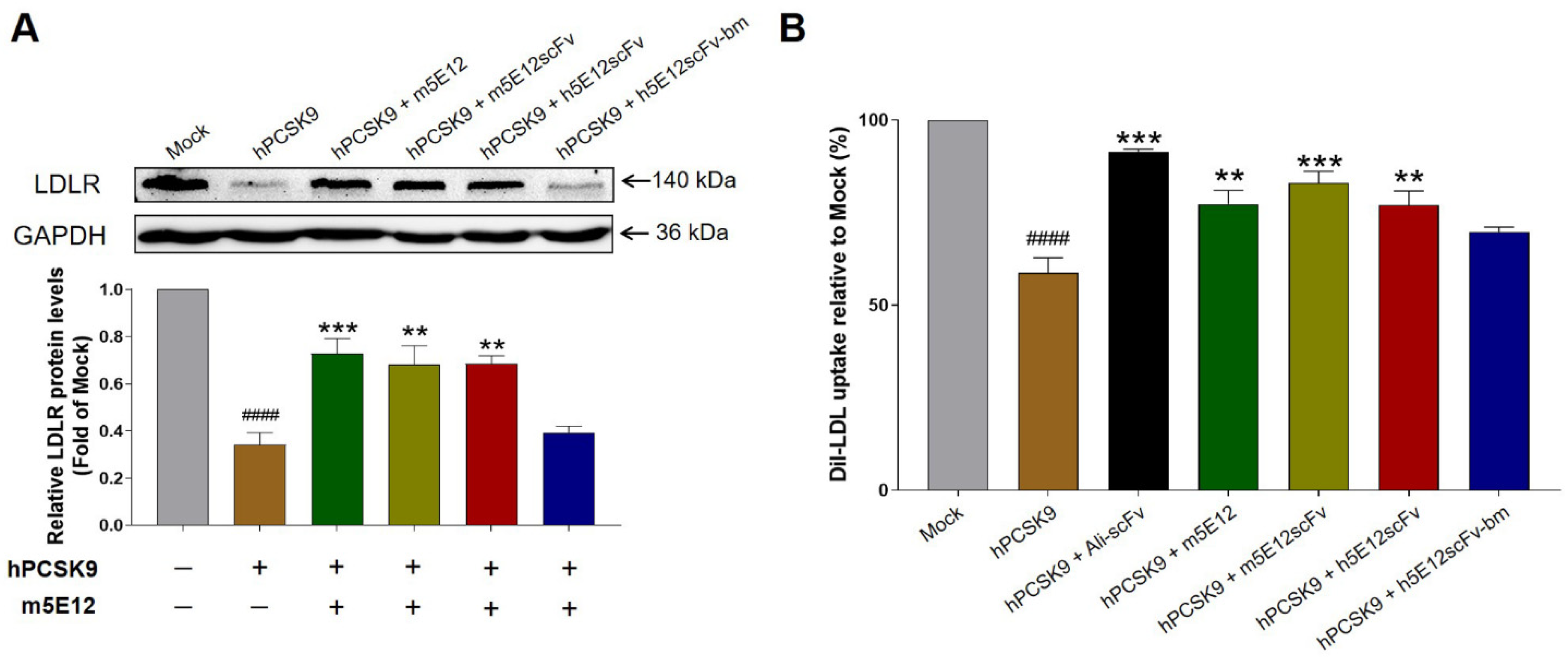
3.3. Humanization of Murine 5E12 scFv (m5E12scFv)
3.4. Preparation and Selection of Humanized 5E12 scFv
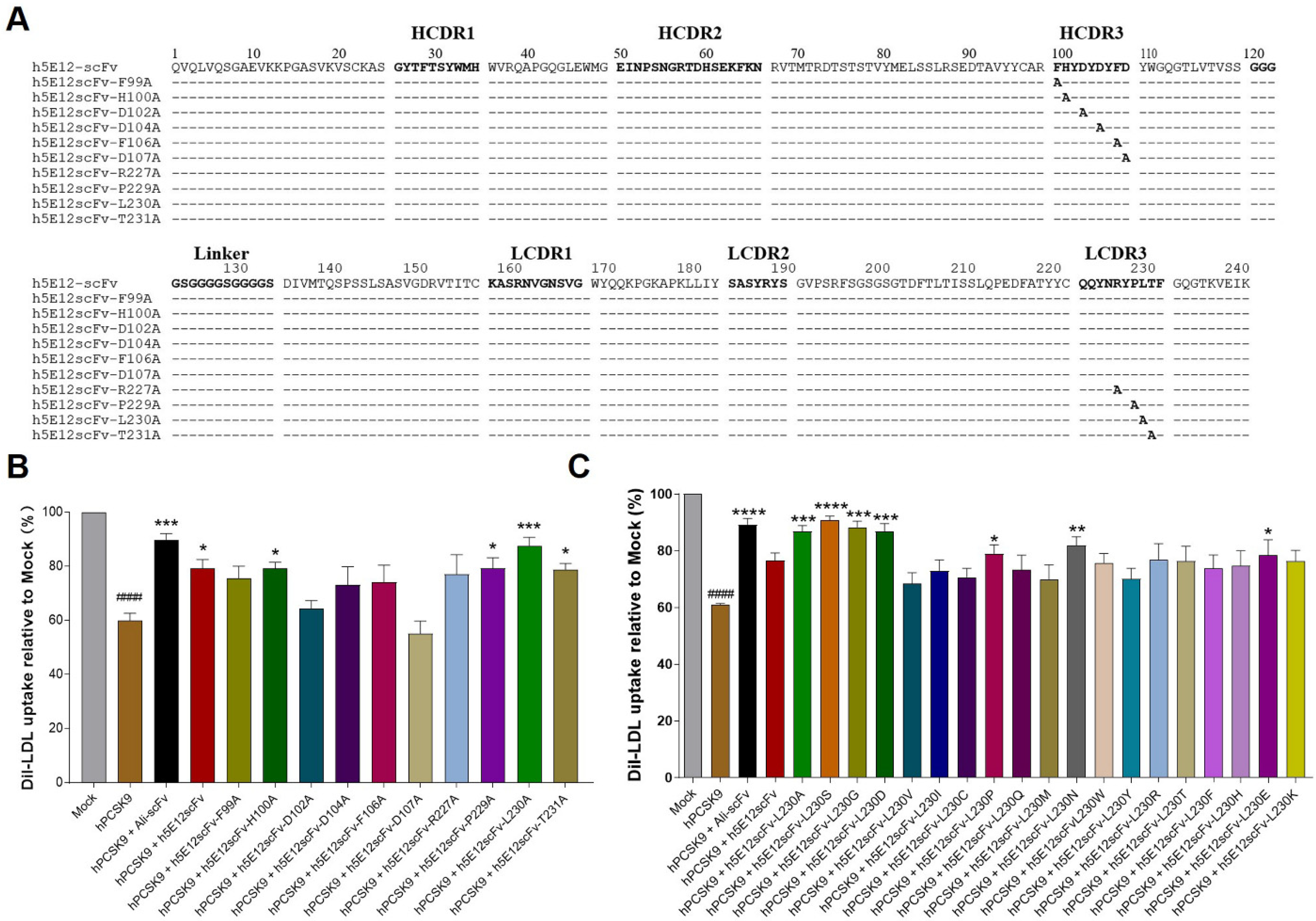
3.5. Affinity Maturation of h5E12scFv In Vitro
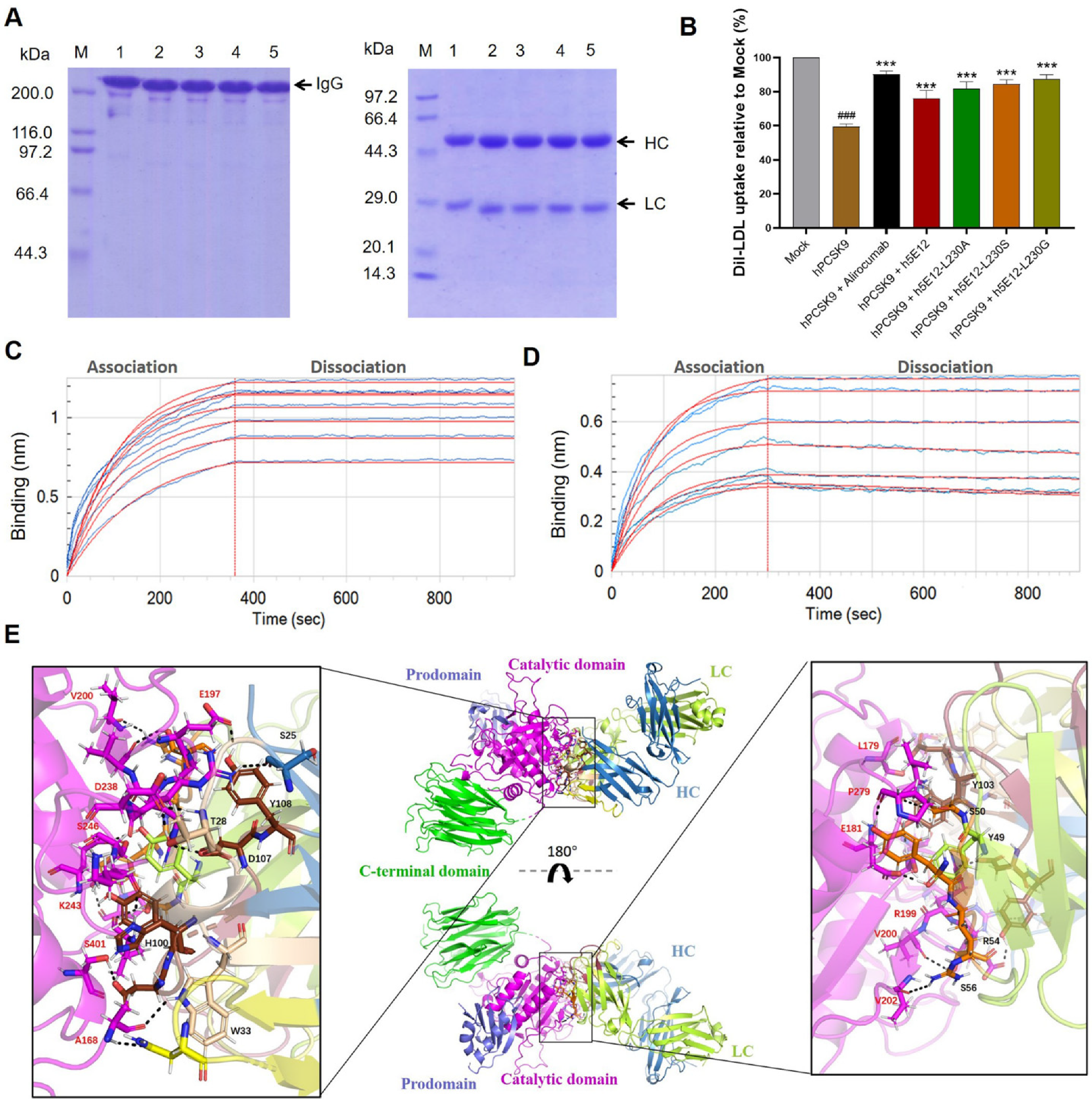
3.6. Generation and Characterization of Full-Length Anti-PCSK9 Antibodies
3.7. Hypolipidemic Effect of h5E12-L230G in Mice Over-Expressing hPCSK9
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tietge, U.J. Hyperlipidemia and cardiovascular disease: Inflammation, dyslipidemia, and atherosclerosis. Curr. Opin. Lipidol. 2014, 25, 94–95. [Google Scholar] [CrossRef]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [PubMed]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egom, E.E.; Pharithi, R.B.; Hesse, S.; Starr, N.; Armstrong, R.; Sulaiman, H.M.; Gazdikova, K.; Mozos, I.; Caprnda, M.; Kubatka, P.; et al. Latest Updates on Lipid Management. High Blood Press Cardiovasc. Prev. 2019, 26, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A Receptor-Mediated Pathway for Cholesterol Homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassoury, N.; Blasiole, D.A.; Tebon Oler, A.; Benjannet, S.; Hamelin, J.; Poupon, V.; McPherson, P.S.; Attie, A.D.; Prat, A.; Seidah, N.G. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 2007, 8, 718–732. [Google Scholar] [CrossRef]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 2007, 14, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Mayer, G.; Poupon, V.; McPherson, P.S.; Desjardins, R.; Ly, K.; Asselin, M.C.; Day, R.; Duclos, F.J.; Witmer, M.; et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: Evidence for an intracellular route. J. Biol. Chem. 2009, 284, 28856–28864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.; Chao, G.; Sitkoff, D.; Lo, F.; Monshizadegan, H.; Meyers, D.; Low, S.; Russo, K.; DiBella, R.; Denhez, F.; et al. Pharmacologic Profile of the Adnectin BMS-962476, a Small Protein Biologic Alternative to PCSK9 Antibodies for Low-Density Lipoprotein Lowering. J. Pharmacol. Exp. Ther. 2014, 350, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, M.W.; Elmen, J.; Fisker, N.; Hansen, H.F.; Persson, R.; Moller, M.R.; Rosenbohm, C.; Orum, H.; Straarup, E.M.; Koch, T. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol. Ther. 2012, 20, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: Rationale and design of the ODYSSEY Outcomes trial. Am. Heart J. 2014, 168, 682–689.e1. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [Green Version]
- Yokote, K.; Kanada, S.; Matsuoka, O.; Sekino, H.; Imai, K.; Tabira, J.; Matsuoka, N.; Chaudhuri, S.; Teramoto, T. Efficacy and Safety of Bococizumab (RN316/PF-04950615), a Monoclonal Antibody Against Proprotein Convertase Subtilisin/Kexin Type 9, in Hypercholesterolemic Japanese Subjects Receiving a Stable Dose of Atorvastatin or Treatment-Naive- Results From a Randomized, Placebo-Controlled, Dose-Ranging Study. Circ. J. 2017, 81, 1496–1505. [Google Scholar] [PubMed] [Green Version]
- Bergeron, N.; Phan, B.A.; Ding, Y.; Fong, A.; Krauss, R.M. Proprotein convertase subtilisin/kexin type 9 inhibition: A new therapeutic mechanism for reducing cardiovascular disease risk. Circulation 2015, 132, 1648–1666. [Google Scholar] [CrossRef] [PubMed]
- KÖHler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Ren, K.; Wang, B.; Qi, Q. Development of a new EGFR antibody antagonist which exhibits potential biological effects against laryngeal cancer. Ann. Transl. Med. 2021, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Cohan, S.L.; Lucassen, E.B.; Romba, M.C.; Linch, S.N. Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines 2019, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Cheng, S.C.; Xie, H.; Xie, Y. A humanized monoclonal antibody constructed from intronless expression vectors targets human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 284, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—a review and update. Biotechnology Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidar, J.N.; Yuan, Q.A.; Zeng, L.; Snavely, M.; Luna, X.; Zhang, H.; Zhu, W.; Ludwig, D.L.; Zhu, Z. A universal combinatorial design of antibody framework to graft distinct CDR sequences: A bioinformatics approach. Proteins 2012, 80, 896–912. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Glover, D.R. Engineering therapeutic proteins. Curr. Opin. Struct. Biol. 2000, 10, 417–420. [Google Scholar] [CrossRef]
- Ersching, J.; Efeyan, A.; Mesin, L.; Jacobsen, J.T.; Pasqual, G.; Grabiner, B.C.; Dominguez-Sola, D.; Sabatini, D.M.; Victora, G.D. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity 2017, 46, 1045–1058.e6. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, C.; MacKenzie, C.R.; Hall, J.C. Isolation and affinity maturation of hapten-specific antibodies. Biotechnol. Adv. 2007, 25, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.M.; Weiss, G.A. Optimizing the affinity and specificity of proteins with molecular display. Mol Biosyst 2006, 2, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.; Lee, S.; Park, J.P.; Choo, J.; Lee, E.K. Modification of phage display technique for improved screening of high-affinity binding peptides. J. Biotechnol. 2019, 289, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lei, G.; Chen, M.; Wang, K.; Lv, W.; Zhang, P.; Hu, T.; Gao, J.; Lu, C.; Mei, Y.; et al. Development of a novel, fully human, anti-PCSK9 antibody with potent hypolipidemic activity by utilizing phage display-based strategy. EBioMedicine 2021, 65, 103250. [Google Scholar] [CrossRef] [PubMed]
- Stuible, M.; Burlacu, A.; Perret, S.; Brochu, D.; Paul-Roc, B.; Baardsnes, J.; Loignon, M.; Grazzini, E.; Durocher, Y. Optimization of a high-cell-density polyethylenimine transfection method for rapid protein production in CHO-EBNA1 cells. J. Biotechnol. 2018, 281, 39–47. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Stojadinovic, A.; Izadjoo, M.J. Immunization, Hybridoma Generation, and Selection for Monoclonal Antibody Production. Methods Mol. Biol. 2014, 1131, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.D.; Beatty, B.G.; Vlahos, W.G. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. J. Immunol. Methods 1987, 100, 173–179. [Google Scholar] [CrossRef]
- Gu, L.; Ye, P.; Li, H.; Wang, Y.; Xu, Y.; Tian, Q.; Lei, G.; Zhao, C.; Gao, Z.; Zhao, W.; et al. Lunasin attenuates oxidant-induced endothelial injury and inhibits atherosclerotic plaque progression in ApoE(-/-) mice by up-regulating heme oxygenase-1 via PI3K/Akt/Nrf2/ARE pathway. FASEB J. 2019, 33, 4836–4850. [Google Scholar] [CrossRef]
- Ly, K.; Saavedra, Y.G.; Canuel, M.; Routhier, S.; Desjardins, R.; Hamelin, J.; Mayne, J.; Lazure, C.; Seidah, N.G.; Day, R. Annexin A2 reduces PCSK9 protein levels via a translational mechanism and interacts with the M1 and M2 domains of PCSK9. J. Biol. Chem. 2014, 289, 17732–17746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Raifu, M.; Howard, M.; Smith, L.; Hansen, D.; Goldsby, R.; Ratner, D. Universal PCR amplification of mouse immunoglobulin gene variable regions: The design of degenerate primers and an assessment of the effect of DNA polymerase 3′ to 5′ exonuclease activity. J. Immunol. Methods 2000, 233, 167–177. [Google Scholar] [CrossRef]
- Verhoeyen, M.; Milstein, C.; Winter, G. Reshaping human antibodies: Grafting an antilysozyme activity. Science 1988, 239, 1534–1536. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Henry, A.H.; Searle, S.J.; Guild, B.C.; Roguska, M.; Rees, A.R. Comparison of Surface Accessible Residues in Human and Murine Immunoglobulin Fv Domains. J. Mol. Biol. 1994, 235, 959–973. [Google Scholar] [CrossRef]
- Foote, J.; Winter, G. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 1992, 224, 487–499. [Google Scholar] [CrossRef]
- De Haard, H.; Kazemier, B.; Arie, V.D.B.; Oudshoorn, P.; Boender, P.; Arends, J.W.; Van Gemen, B.J.I. Vernier zone residue 4 of mouse subgroup II kappa light chains is a critical determinant for antigen recognition. Immunotechnology 1999, 4, 203–215. [Google Scholar] [CrossRef]
- Sundberg, E.J.; Mariuzza, R.A. Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 2002, 61, 119–160. [Google Scholar]
- Zheng, L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiba, H.; Tsumoto, K. Thermodynamics of antibody-antigen interaction revealed by mutation analysis of antibody variable regions. J. Biochem 2015, 158, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Xiao, S.; Yuan, M.; Gao, Y.; Sun, J.; Xue, C. Using overlap-extension PCR technique to fusing genes for constructing recombinant plasmids. J. Basic Microbiol. 2018, 58, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Hessell, A.J.; Hangartner, L.; Hunter, M.; Havenith, C.E.; Beurskens, F.J.; Bakker, J.M.; Lanigan, C.M.; Landucci, G.; Forthal, D.N.; Parren, P.W.; et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 2007, 449, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef]
- Jacobsen, F.W.; Stevenson, R.; Li, C.; Salimi-Moosavi, H.; Liu, L.; Wen, J.; Luo, Q.; Daris, K.; Buck, L.; Miller, S.; et al. Engineering an IgG Scaffold Lacking Effector Function with Optimized Developability. J. Biol. Chem. 2017, 292, 1865–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, L.W.; Jr Qiu, D.; Mahon, D.; Adamo, M.; Cheng, K.C. C-terminal lysine variants in fully human monoclonal antibodies: Investigation of test methods and possible causes. Biotechnol. Bioeng. 2008, 100, 1132–1143. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.H.; Thompson, A.R.; Loeb, K.; Ye, X. Long-term and therapeutic-level hepatic gene expression of human factor IX after naked plasmid transfer in vivo. Mol. Ther. 2001, 3, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Vauquelin, G.; Charlton, S.J. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 2010, 161, 488–508. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A. The dynamics of drug-target interactions: Drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 2010, 5, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Heinonen, T.; Orloff, D.; Libby, P. Vascular biomarkers and surrogates in cardiovascular disease. Circulation 2006, 113, 2936–2942. [Google Scholar] [CrossRef] [Green Version]
- Miller, M. ACC/AHA lipids & ASCVD guidelines: 2018 update. Metabolism 2019, 99, 116–118. [Google Scholar] [PubMed]
- Mampuya, W.M.; Frid, D.; Rocco, M.; Huang, J.; Brennan, D.M.; Hazen, S.L.; Cho, L. Treatment strategies in patients with statin intolerance: The Cleveland Clinic experience. Am. Heart J. 2013, 166, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, G.J.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.; Pearson, G.; et al. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Canadian Consensus Working Group Update. Can. J. Cardiol. 2016, 32, S35–S65. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. PCSK9: A convertase that coordinates LDL catabolism. J. Lipid Res. 2009, 50, S172–S177. [Google Scholar] [CrossRef] [Green Version]
- He, N.Y.; Li, Q.; Wu, C.Y.; Ren, Z.; Gao, Y.; Pan, L.H.; Wang, M.M.; Wen, H.Y.; Jiang, Z.S.; Tang, Z.H.; et al. Lowering serum lipids via PCSK9-targeting drugs: Current advances and future perspectives. Acta Pharmacol. Sin. 2017, 38, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.; Meredith, I.T.; Nasis, A. PCSK9 Monoclonal Antibodies in 2016: Current Status and Future Challenges. Heart Lung Circ. 2017, 26, 786–798. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Nissen, S.E.; Rader, D.J.; Hovingh, G.K.; Wang, M.-D.; Shen, T.; Krueger, K.A. Safety and efficacy of LY3015014, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 (PCSK9): A randomized, placebo-controlled Phase 2 study. Eur. Heart J. 2016, 37, 1360–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gao, X.; Gong, R. Engineering of Fc Fragments with Optimized Physicochemical Properties Implying Improvement of Clinical Potentials for Fc-Based Therapeutics. Front. Immunol. 2017, 8, 1860. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.Y.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.G.; Condra, J.H.; Orsatti, L.; Shen, X.; Di Marco, S.; Pandit, S.; Bottomley, M.J.; Ruggeri, L.; Cummings, R.T.; Cubbon, R.M.; et al. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 2010, 285, 12882–12891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosztyu, P.; Kuchar, M.; Cerny, J.; Barkocziova, L.; Maly, M.; Petrokova, H.; Czernekova, L.; Liskova, V.; Raskova Kafkova, L.; Knotigova, P.; et al. Proteins mimicking epitope of HIV-1 virus neutralizing antibody induce virus-neutralizing sera in mice. EBioMedicine 2019, 47, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, S.M.; Supolikova, L.; Molcanova, L.; Smejkal, K.; Bednar, D.; Slaninova, I. Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines 2021, 9, 357. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Dose (mg/kg) | LDL-C (mmol/L) | TC (mmol/L) | HDLC (mmol/L) | TG (mmol/L) | |
|---|---|---|---|---|---|---|
| Normal group | / | 0.624 ± 0.021 | 3.642 ± 0.161 | 1417 ± 0.042 | 0.0921 ± 0.028 | |
| Model group 1 | / | 1560 ± 0.037 ### | 5.250 ± 0.212 ### | 1340 ± 0.036 | 1351 ± 0.049 ### | |
| Alirocumab group | Low dose | 1 | 1506 ± 0.164 | 4.903 ± 0.071 | 1275 ± 0.066 | 1320 ± 0.052 |
| Medium dose | 3 | 1122 ± 0.122 ** | 4650 ± 0.066 ** | 1417 ± 0.088 | 1211 ± 0.048 | |
| High dose | 10 | 0.964 ± 0.024 *** | 4276 ± 0.062 *** | 1287 ± 0.054 | 1162 ± 0.037 * | |
| h5E12-L230G group | Low dose | 1 | 1480 ± 0.0262 | 4.770 ± 0.045 * | 1462 ± 0.084 | 1312 ± 0.026 |
| Medium dose | 3 | 1093 ± 0.0395 ** | 4658 ± 0.022 ** | 1333 ± 0.071 | 1196 ± 0.048 | |
| High dose | 10 | 0.996 ± 0.0644 *** | 4469 ± 0.063 *** | 1257 ± 0.044 | 1155 ± 0.037 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Z.; Xu, M.; Mei, Y.; Hu, T.; Zhang, P.; Chen, M.; Lv, W.; Lu, C.; Tan, S. Generation of a Novel High-Affinity Antibody Binding to PCSK9 Catalytic Domain with Slow Dissociation Rate by CDR-Grafting, Alanine Scanning and Saturated Site-Directed Mutagenesis for Favorably Treating Hypercholesterolemia. Biomedicines 2021, 9, 1783. https://doi.org/10.3390/biomedicines9121783
Bai Z, Xu M, Mei Y, Hu T, Zhang P, Chen M, Lv W, Lu C, Tan S. Generation of a Novel High-Affinity Antibody Binding to PCSK9 Catalytic Domain with Slow Dissociation Rate by CDR-Grafting, Alanine Scanning and Saturated Site-Directed Mutagenesis for Favorably Treating Hypercholesterolemia. Biomedicines. 2021; 9(12):1783. https://doi.org/10.3390/biomedicines9121783
Chicago/Turabian StyleBai, Zhengli, Menglong Xu, Ying Mei, Tuo Hu, Panpan Zhang, Manman Chen, Wenxiu Lv, Chenchen Lu, and Shuhua Tan. 2021. "Generation of a Novel High-Affinity Antibody Binding to PCSK9 Catalytic Domain with Slow Dissociation Rate by CDR-Grafting, Alanine Scanning and Saturated Site-Directed Mutagenesis for Favorably Treating Hypercholesterolemia" Biomedicines 9, no. 12: 1783. https://doi.org/10.3390/biomedicines9121783
APA StyleBai, Z., Xu, M., Mei, Y., Hu, T., Zhang, P., Chen, M., Lv, W., Lu, C., & Tan, S. (2021). Generation of a Novel High-Affinity Antibody Binding to PCSK9 Catalytic Domain with Slow Dissociation Rate by CDR-Grafting, Alanine Scanning and Saturated Site-Directed Mutagenesis for Favorably Treating Hypercholesterolemia. Biomedicines, 9(12), 1783. https://doi.org/10.3390/biomedicines9121783