
Pattern of Repetitive Element Transcription Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus


Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. RE Annotation and Classification
2.3. Bulk RNA-Seq Analysis
2.4. Defining the Transcriptionally Active RE Instances
2.5. Analysis of scRNA-Seq Data
2.6. Code Availability
3. Results
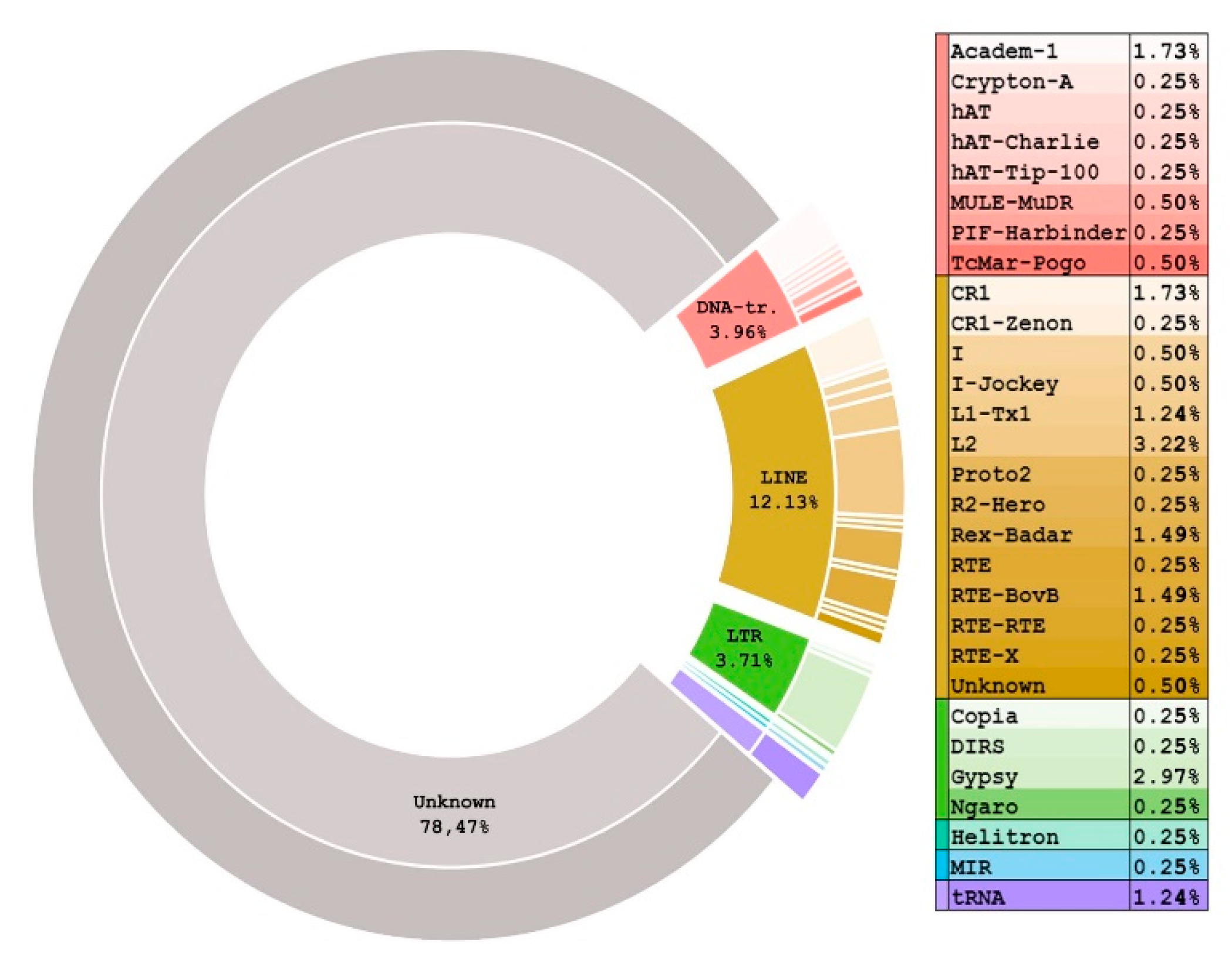
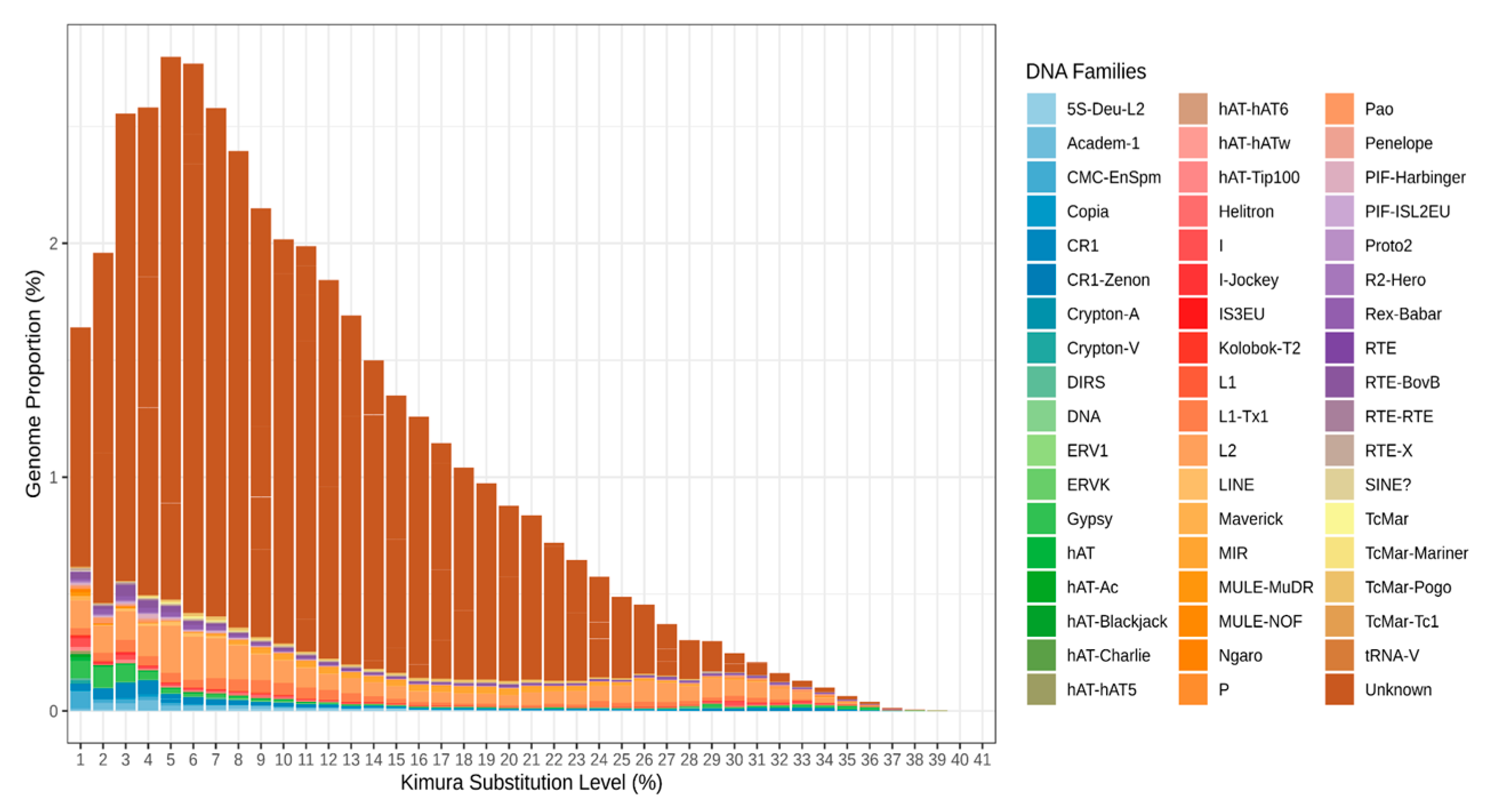
3.1. Unknown REs Contribute the Largest Portion to the S. purpuratus Genome REs
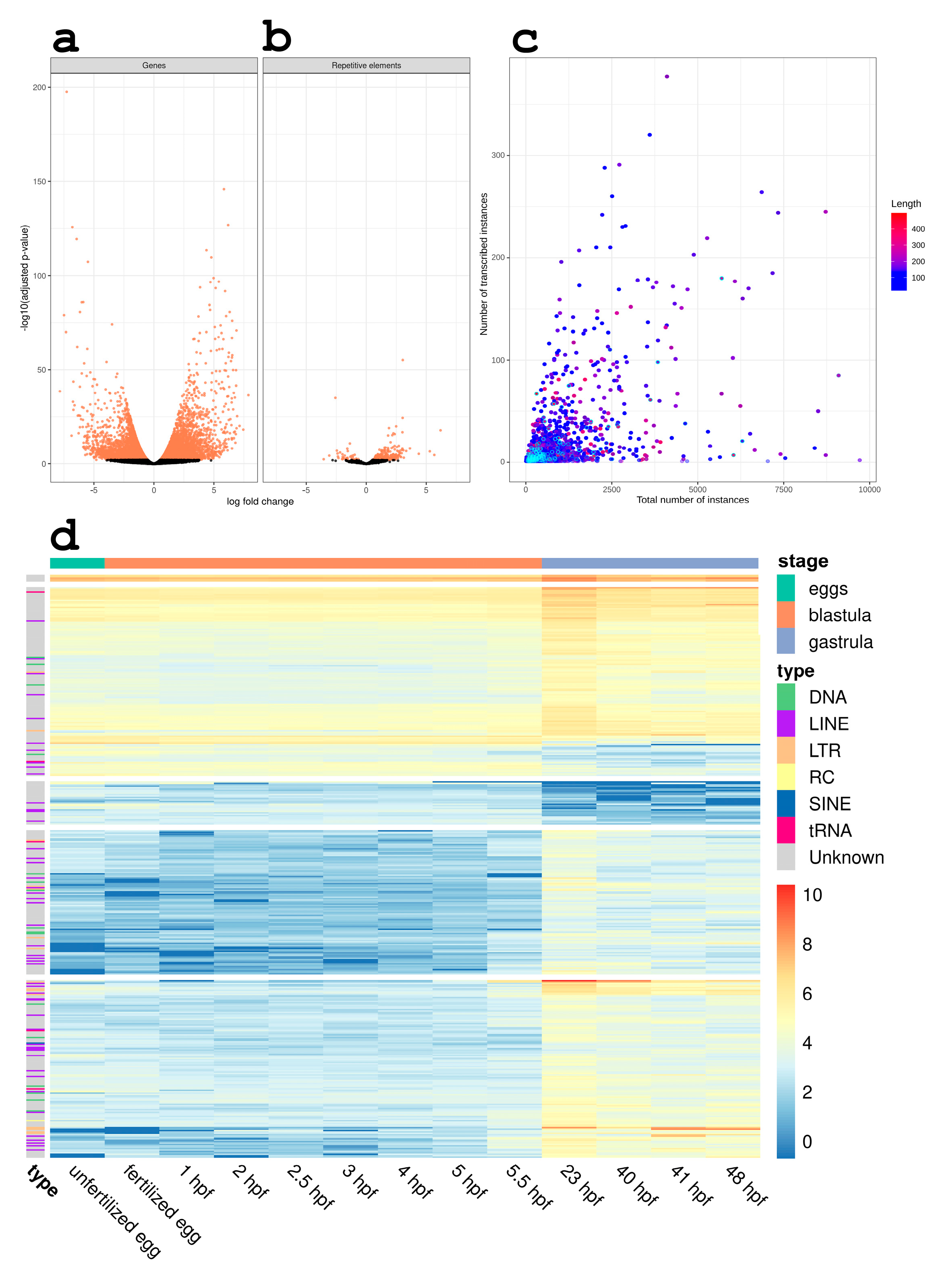
3.2. RE Expression Significantly Changes during the Gastrulation Process
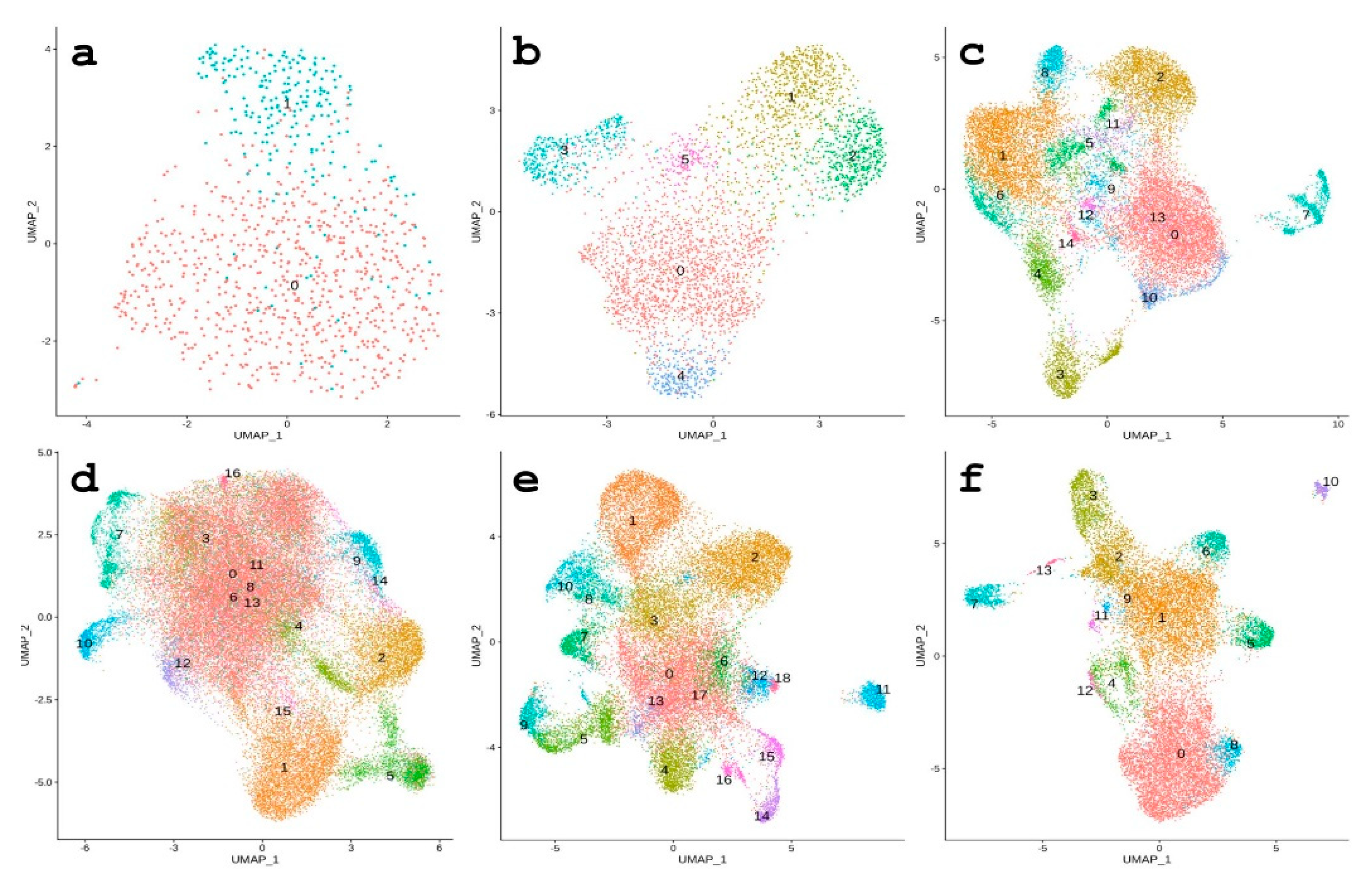
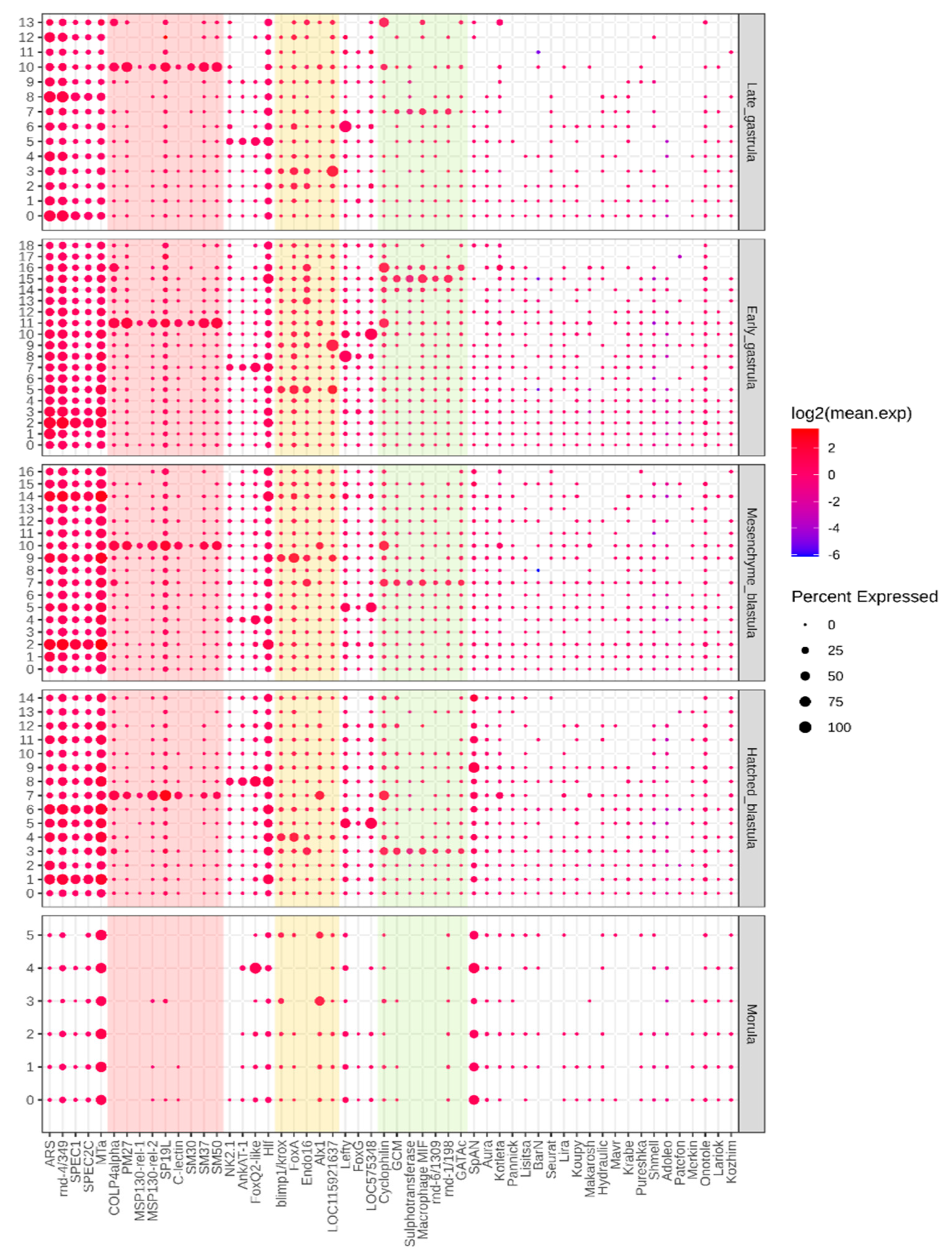
3.3. REs Serve as Markers of Cell Populations According to scRNA-seq Data
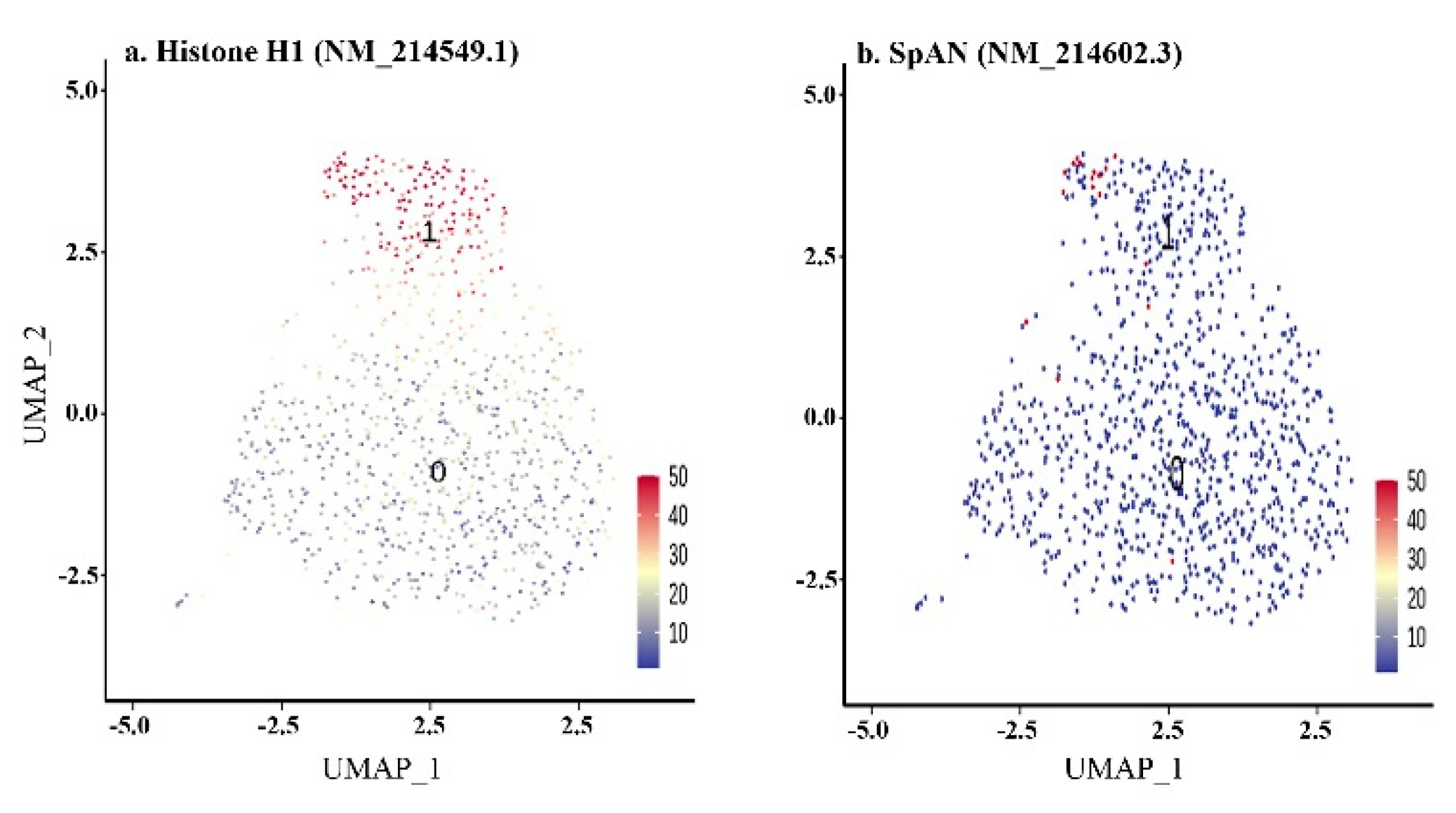
3.3.1. 64-Cell Stage
3.3.2. Morula Stage
3.3.3. From Hatched Blastula to Late Gastrula Stages
3.3.4. Aboral Ectoderm Domain
3.3.5. Apical Plate
3.3.6. Endoderm Lineage
3.3.7. Primary Mesenchyme Cells
3.3.8. Secondary Mesenchyme Cells
3.3.9. Oral Ectoderm
3.3.10. SFE1-Positive Population
4. Discussion
4.1. Gene Regulatory Networks (GRNs), Primary Mesenchyme Cells (PMC)
4.2. Repetitive Elements as the Precursor of Functional ncRNA
4.3. scRNA-Seq Results
4.4. Repeats as the Putative Evolutionary Force
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katsura, Y.; Ikemura, T.; Kajitani, R.; Toyoda, A.; Itoh, T.; Ogata, M. Comparative genomics of Glandirana rugosa using unsu-pervised AI reveals a high CG frequency. Life Sci Alliance 2021, 1, 4. [Google Scholar]
- Meyer, A.; Schloissnig, S.; Franchini, P.; Du, K.; Woltering, J.M.; Irisarri, I. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 2021, 590, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Timoshevskaya, N.; Timoshevskiy, V.A.; Keinath, M.C.; Hardy, D.; Voss, S.R. A chromosome-scale assembly of the axolotl genome. Genome Res. 2019, 29, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 3765. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.D.P.; Mattick, J. Noncoding RNA in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar] [CrossRef]
- Waring, M.; Britten, R.J.; Etzler, M.E.; Moog, F. Nucleotide Sequence Repetition: A Rapidly Reassociating Fraction of Mouse DNA. Science 1966, 154, 791–794. [Google Scholar] [CrossRef]
- Britten, R.J.; Kohne, D.E. Repeated Sequences in DNA. Science 1968, 161, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B. A unified classification system for eukaryotic trans-posable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Makałowski, W.; Gotea, V.; Pande, A.; Makałowska, I. Transposable Elements: Classification, Identification, and Their Use As a Tool For Comparative Genomics. Methods Mol. Biol. 2019, 1910, 177–207. [Google Scholar] [CrossRef]
- Mita, P.; Boeke, J.D. How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Franke, V.; Ganesh, S.; Karlic, R.; Malik, R.; Pasulka, J.; Horvat, F. Long terminal repeats power evolution of genes and gene ex-pression programs in mammalian oocytes and zygotes. Genome Res. 2017, 27, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rishishwar, L.; Mariño-Ramírez, L.; Jordan, I.K. Human population-specific gene expression and transcriptional net-work modification with polymorphic transposable elements. Nucleic Acids Res. 2017, 45, 2318–2328. [Google Scholar]
- Venuto, D.; Bourque, G. Identifying co-opted transposable elements using comparative epigenomics. Dev. Growth Differ. 2018, 60, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Bouckenheimer, J.; Assou, S.; Riquier, S.; Hou, C.; Philippe, N.; Sansac, C. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum. Reprod. Update 2017, 23, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, Z.; Krause, H.M. Long Noncoding RNAs and Repetitive Elements: Junk or Intimate Evolutionary Partners? Trends Genet. 2019, 35, 892–902. [Google Scholar] [CrossRef]
- Hadjiargyrou, M.; Delihas, N. The Intertwining of Transposable Elements and Non-Coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef]
- Hirose, T.; Mishima, Y.; Tomari, Y. Elements and machinery of non-coding RNAs: Toward their taxonomy. EMBO Rep. 2014, 15, 489–507. [Google Scholar] [CrossRef]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 2016, 37, 3–9. [Google Scholar] [CrossRef]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expres-sion analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Fablet, M.; Modolo, L.; Lopez-Maestre, H.; Vieira, C. TEtools facilitates big data expression analysis of transposable ele-ments and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2017, 45, e17. [Google Scholar] [PubMed]
- Jeong, H.-H.; Yalamanchili, H.K.; Guo, C.; Shulman, J.M.; Liu, Z. An ultra-fast and scalable quantification pipeline for transposable elements from next generation sequencing data. Biocomputing 2001 2017, 23, 168–179. [Google Scholar] [CrossRef]
- Treiber, C.D.; Waddell, S. Transposon expression in the Drosophila brain is driven by neighboring genes and diversifies the neural transcriptome. Genome Res. 2020, 30, 1559–1569. [Google Scholar] [CrossRef]
- Ansaloni, F.; Scarpato, M.; Di Schiavi, E.; Gustincich, S.; Sanges, R. Exploratory analysis of transposable elements expression in the C. elegans early embryo. BMC Bioinform. 2019, 20, 484. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Babarinde, I.A.; Sun, L.; Xu, S.; Chen, R.; Shi, J. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun. 2021, 12, 1456. [Google Scholar] [CrossRef]
- Shao, W.; Wang, T. Transcript assembly improves expression quantification of transposable elements in single-cell RNA-seq data. Genome Res. 2021, 31, 88–100. [Google Scholar] [CrossRef]
- Lyons, D.C.; Kaltenbach, S.L.; Mcclay, D.R. Morphogenesis in sea urchin embryos: Linking cellular events to gene regulatory net-work states. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 231–252. [Google Scholar] [CrossRef]
- Adonin, L.; Drozdov, A.; Barlev, N.A. Sea Urchin as a Universal Model for Studies of Gene Networks. Front. Genet. 2021, 11. [Google Scholar] [CrossRef]
- Ransick, A.; Rast, J.P.; Minokawa, T.; Calestani, C.; Davidson, E.H. New Early Zygotic Regulators Expressed in Endomesoderm of Sea Urchin Embryos Discovered by Differential Array Hybridization. Dev. Biol. 2002, 246, 132–147. [Google Scholar] [CrossRef]
- Revilla-I-Domingo, R.; Oliveri, P.; Davidson, E.H. A missing link in the sea urchin embryo gene regulatory network: HesC and the double-negative specification of micromeres. Proc. Natl. Acad. Sci. USA 2007, 104, 12383–12388. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-H.; Li, E.; Geiss, G.K.; Longabaugh, W.J.; Krämer, A.; Davidson, E.H. A perturbation model of the gene regulatory network for oral and aboral ectoderm specification in the sea urchin embryo. Dev. Biol. 2009, 329, 410–421. [Google Scholar] [CrossRef]
- Peter, I.S.; Davidson, E.H. Evolution of Gene Regulatory Networks Controlling Body Plan Development. Cell 2011, 144, 970–985. [Google Scholar] [CrossRef]
- Foster, S.; Oulhen, N.; Wessel, G. A single cell RNA sequencing resource for early sea urchin development. Development 2020, 147. [Google Scholar] [CrossRef]
- Melsted, P.; Booeshaghi, A.S.; Liu, L.; Gao, F.; Lu, L.; Min, K.H.J. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. 2021, 39, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Bisgrove, B.W.; Burke, R.D. Development of Serotonergic Neurons in Embryos of the Sea Urchin, Strongylocentrotus purpuratus. (serotonergic/neural development/embryo/echinoid). Dev. Growth Differ. 1986, 28, 569–574. [Google Scholar] [CrossRef]
- Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C. The Genome of the Sea Urchin Stron-gylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef]
- Yaguchi, S.; Yaguchi, J.; Wei, Z.; Shiba, K.; Angerer, L.M.; Inaba, K. ankAT-1 is a novel gene mediating the apical tuft formation in the sea urchin embryo. Dev. Biol. 2010, 348, 67–75. [Google Scholar] [CrossRef]
- Tu, Q.; Brown, C.T.; Davidson, E.H.; Oliveri, P. Sea urchin Forkhead gene family: Phylogeny and embryonic expression. Dev. Biol. 2006, 300, 49–62. [Google Scholar] [CrossRef]
- Barresi, M.J.; Gilbert, S.F. Developmental Biology, 12th ed.; Oxford University Press: Oxford, UK, 2013; 888p. [Google Scholar]
- Amore, G.; Davidson, E.H. cis-Regulatory control of cyclophilin, a member of the ETS-DRI skeletogenic gene battery in the sea urchin embryo. Dev. Biol. 2006, 293, 555–564. [Google Scholar] [CrossRef]
- Wessel, G.M.; Conner, S.; Laidlaw, M.; Harrison, J.; LaFleur, G.J. SFE1, A Constituent of the Fertilization Envelope in the Sea Urchin Is Made by Oocytes and Contains Low-Density Lipoprotein-Receptor-Like Repeats1. Biol. Reprod. 2000, 63, 1706–1712. [Google Scholar] [CrossRef]
- Levine, M.; Davidson, E.H. Gene regulatory networks for development. Proc. Natl. Acad. Sci. USA 2005, 102, 4936–4942. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.S.; Davidson, E.H. The endoderm gene regulatory network in sea urchin embryos up to mid-blastula stage. Dev. Biol. 2010, 340, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Tadros, W.; Lipshitz, H.D. The maternal-to-zygotic transition: A play in two acts. Development 2009, 136, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Kipryushina, Y.O.; Yakovlev, K.V. Maternal control of early patterning in sea urchin embryos. Differentiation 2020, 113, 28–37. [Google Scholar] [CrossRef]
- Martik, M.L.; Lyons, D.C.; McClay, D.R. Developmental gene regulatory networks in sea urchins and what we can learn from them. F1000Research 2016, 5, 203. [Google Scholar] [CrossRef]
- Massri, A.J.; Greenstreet, L.; Afanassiev, A.; Escobar, A.B.; Wray, G.M.; Schiebinger, G. Developmental Single-cell transcriptomics in the Lytechinus variegatus Sea Urchin Embryo. Development 2021, 148, dev198614. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Cameron, R.A.; Worley, K.C.; Gibbs, R.A.; Davidson, E.H. Gene structure in the sea urchin Strongylocentrotus purpuratus based on transcriptome analysis. Genome Res. 2012, 22, 2079–2087. [Google Scholar] [CrossRef]
- Wei, Z.; Angerer, R.C.; Angerer, L.M. Identification of a New Sea Urchin Ets Protein, SpEts4, by Yeast One-Hybrid Screening with the Hatching Enzyme Promoter. Mol. Cell. Biol. 1999, 19, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Romancino, D.P.; Anello, L.; Lavanco, A.; Buffa, V.; Di Bernardo, M.; Bongiovanni, A. A sea urchin in vivo model to evaluate Epithe-lial-Mesenchymal Transition. Dev. Growth Differ. 2017, 59, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Katow, H. Mechanisms of the epithelial-to-mesenchymal transition in sea urchin embryos. Tissue Barriers 2015, 3, e1059004. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okazaki, K. Spicule Formation by Isolated Micromeres of the Sea Urchin Embryo. Am. Zoöl. 1975, 15, 567–581. [Google Scholar] [CrossRef]
- Oliveri, P.; Davidson, E.H.; McClay, D.R. Activation of pmar1 controls specification of micromeres in the sea urchin embryo. Dev. Biol. 2003, 258, 32–43. [Google Scholar] [CrossRef]
- Zhu, X.; Mahairas, G.; Illies, M.; Cameron, R.A.; Davidson, E.H.; Ettensohn, C.A. A large-scale analysis of mRNAs expressed by pri-mary mesenchyme cells of the sea urchin embryo. Development 2001, 128, 2615–2627. [Google Scholar] [CrossRef]
- Takacs, C.M.; Amore, G.; Oliveri, P.; Poustka, A.J.; Wang, D.; Burke, R.D. Expression of an NK2 homeodomain gene in the apical ectoderm defines a new territory in the early sea urchin embryo. Dev. Biol. 2004, 269, 152–164. [Google Scholar] [CrossRef]
- Fort, V.; Khelifi, G.; Hussein, S.M. Long non-coding RNAs and transposable elements: A functional relationship. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2021, 1868, 118837. [Google Scholar] [CrossRef]
- Hinman, V.F.; Davidson, E.H. Evolutionary plasticity of developmental gene regulatory network architecture. Proc. Natl. Acad. Sci. USA 2007, 104, 19404–19409. [Google Scholar] [CrossRef]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef]
- Campo-Paysaa, F.; Sémon, M.; Cameron, R.A.; Peterson, K.J.; Schubert, M. microRNA complements in deuterostomes: Origin and evolution of microRNAs. Evol. Dev. 2011, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Song, J.L.; Stoeckius, M.; Maaskola, J.; Friedländer, M.; Stepicheva, N.; Juliano, C. Select microRNAs are essential for early de-velopment in the sea urchin. Dev. Biol. 2012, 362, 104–113. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Jin, P.; Zhou, X.; Chen, L.; Ma, F. The Role of Transposable Elements in the Origin and Evolution of MicroRNAs in Human. PLoS ONE 2015, 10, e0131365. [Google Scholar] [CrossRef]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.-H.; Thomas, K.; Park, I.H. Large intergenic non-coding RNA-RoR modulates re-programming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef]
- Bhat, S.A.; Ahmad, S.M.; Mumtaz, P.T.; Malik, A.A.; Dar, M.A.; Urwat, U. Long non-coding RNAs: Mechanism of action and functional utility. Non-Coding RNA Res. 2016, 1, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Elisaphenko, E.A.; Kolesnikov, N.N.; Shevchenko, A.I.; Rogozin, I.B.; Nesterova, T.B.; Brockdorff, N. A Dual Origin of the Xist Gene from a Protein-Coding Gene and a Set of Transposable Elements. PLoS ONE 2008, 3, e2521. [Google Scholar] [CrossRef]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J. Kcnq1ot1 Antisense Noncoding RNA Mediates Lineage-Specific Transcriptional Silencing through Chromatin-Level Regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R. The Air Noncoding RNA Epigenetically Silences Transcription by Targeting G9a to Chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef]
- Delás, M.J.; Hannon, G.J. lncRNAs in development and disease: From functions to mechanisms. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of Long Noncoding RNA Evolution De-rived from Direct Comparison of Transcriptomes in 17 Species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef]
- Kutter, C.; Watt, S.; Stefflova, K.; Wilson, M.D.; Goncalves, A.; Ponting, C.P. Rapid Turnover of Long Noncoding RNAs and the Evolution of Gene Expression. PLoS Genet. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mam-mals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Guigó, R. The RIDL hypothesis: Transposable elements as functional domains of long noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G. Transposable Elements Are Major Contributors to the Origin, Diversification, and Regulation of Vertebrate Long Noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Carlevaro-Fita, J.; Polidori, T.; Das, M.; Navarro, C.; Zoller, T.I.; Johnson, R. Ancient exapted transposable elements promote nuclear enrichment of human long noncoding RNAs. Genome Res. 2019, 29, 208–222. [Google Scholar] [CrossRef]
- Schartl, M.; Kneitz, S.; Volkoff, H.; Adolfi, M.; Schmidt, C.; Fischer, P. The Piranha Genome Provides Molecular Insight Associated to Its Unique Feeding Behavior. Genome Biol. Evol. 2019, 11, 2099–2106. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | Lineage | Stage | Clusters | Marker Genes | Number of Marker RE | |
|---|---|---|---|---|---|---|
| Upregulated | Downregulated | |||||
| Ectoderm | Aboral ectoderm | HB | 1 | Spec1A, Spec2C, MTa | 1 | 0 |
| MB | 1, 2 | Spec1A, Spec2C, MTa, ARS | 1 | 3 | ||
| EG | 1, 2 | Spec1A, Spec2C, ARS | 2 | 1 | ||
| LG | 0, 8 | Spec1A, Spec2C, ARS | 10 | 0 | ||
| Apical plate | HB | 8 | Hlf, AnkAT-1, FoxQ2-like | 1 | 6 | |
| MB | 4 | FoxQ2-like | 0 | 0 | ||
| EG | 7 | FoxQ2-like, AnkAT-1 | 2 | 3 | ||
| LG | 5 | Hlf, AnkAT-1, NK2.1, FoxQ2-like | 5 | 1 | ||
| Oral ectoderm | HB | 5 | Lefty | 0 | 0 | |
| MB | 5 | Lefty | 0 | 0 | ||
| EG | 8 | Lefty | 0 | 0 | ||
| LG | 6 | Lefty, FoxA | 6 | 3 | ||
| Endoderm | Endoderm | HB | 4 | FoxA, blimp1/krox | 0 | 0 |
| MB | 9 | FoxA | 0 | 7 | ||
| EG | 5 | FoxA | 0 | 10 | ||
| LG | 2, 3 | FoxA | 6 | 3 | ||
| Mesoderm | PMC | HB | 7 | MSP130-rel-2, P19, Col4alpha, SM37, Alx1, Cyclophilin, PM27, C-lectin-like, SM37 | 7 | 9 |
| MB | 10 | MSP130-rel-2, P19, Col4alpha, SM37, Alx1, Cyclophilin, PM27, C-lectin-like, SM37 | 4 | 14 | ||
| EG | 11 | MSP130-rel-2, P19, Col4alpha, SM37, Alx1, Cyclophilin, PM27, C-lectin-like, SM37, SM29, SM50 | 24 | 13 | ||
| LG | 10 | MSP130-rel-2, P19, Col4alpha, SM37, Alx1, Cyclophilin, PM27, C-lectin-like, SM37, SM29, SM50 | 6 | 1 | ||
| SMC | HB | 3 | gcm, GATAc, cyclophilin | 6 | 1 | |
| MB | 7 | Sulphotransferase | 3 | 15 | ||
| EG | 15, 16 | Sulphotransferase, gcm, endo16, GATAc | 25 | 47 | ||
| LG | 7 | Sulphotransferase, gcm, endo16, GATAc, Macrophage MIF | 6 | 2 | ||
| SFE1-positive | HB | - | - | - | - | |
| MB | 12 | SFE1 | 0 | 0 | ||
| EG | 4 | SFE1 | 0 | 0 | ||
| LG | 4 | SFE1 | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panyushev, N.; Okorokova, L.; Danilov, L.; Adonin, L. Pattern of Repetitive Element Transcription Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus. Biomedicines 2021, 9, 1736. https://doi.org/10.3390/biomedicines9111736
Panyushev N, Okorokova L, Danilov L, Adonin L. Pattern of Repetitive Element Transcription Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus. Biomedicines. 2021; 9(11):1736. https://doi.org/10.3390/biomedicines9111736
Chicago/Turabian StylePanyushev, Nick, Larisa Okorokova, Lavrentii Danilov, and Leonid Adonin. 2021. "Pattern of Repetitive Element Transcription Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus" Biomedicines 9, no. 11: 1736. https://doi.org/10.3390/biomedicines9111736
APA StylePanyushev, N., Okorokova, L., Danilov, L., & Adonin, L. (2021). Pattern of Repetitive Element Transcription Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus. Biomedicines, 9(11), 1736. https://doi.org/10.3390/biomedicines9111736