
Special delEVery: Extracellular Vesicles as Promising Delivery Platform to the Brain



Abstract
:1. Introduction
2. Barriers in Brain Drug Delivery
2.1. The Blood–Brain Barrier
2.2. The Blood–Cerebrospinal Fluid Barrier
2.3. Circumventing the Barriers
3. Extracellular Vesicles as Drug Delivery Vehicle
3.1. Extracellular Vesicles
3.2. Extracellular Vesicles as Brain Drug Delivery System

| Cell Type | EV Source | Isolation Technique | EV Characterization | Experimental Set Ups | In Vivo EV Dose | Administration Route | MoA | Label or Loaded Cargo (Method) | Proof Brain /CNS Localization | Ref | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Morphology | Protein Markers | EV Size/Charge | ||||||||||
| Brain cells | bEnd.3 endothelial cells (mouse) | Total Exosome RNA and Protein Isolation Kit | SEM | +CD9 +CD63 +CD81 | <150 nm (SEM, Nano C nanosizing system–Beckman Coulter) | In vitro: bEnd.3 cells In vivo: Brain cancer zebrafish embryo’s | 4 nl of 200 μg/mL EVs | IV (common cardial vein injection) | Active receptor-mediated endocytosis, not further specified | Rhodamine 123 label (incubation) Doxorubicin, Paclitaxel (incubation) | Yes, brain detection rhodamine labelled EVs (fluorescent confocal imaging) + effect tumor cells | Yang et al. [67] |
| Total Exosome RNA and Protein Isolation Kit | ND (cfr. Yang et al. [67]) | ND (cfr. Yang et al. [67]) | ND (cfr. Yang et al. [67]) | In vitro: bEnd.3 cells and astrocytes In vivo: Brain cancer zebrafish embryo’s | 4 nl of 200 μg/mL EVs | IV (common cardial vein injection) | Possible involvement high expression CD63 | Rhodamine 123 label (incubation) Anti-VEGF siRNA (EV transfection) | Yes, brain detection siRNA (fluorescent confocal imaging) + effect tumor cells | Yang et al. [86] | ||
| ExoQuick-TC Exosome Precipitation Solution | ND | ND | ND | In vivo: Photothrombic stroke in type 2 diabetes mellitus mouse | 3 × 1010 particles/mouse (qNano, iZon) | IV | ND | PKH26 label (incubation) | Yes, brain sections PKH26 labelled EVs (laser scanning confocal imaging) + functional effects endogenous EV miR-126 | Venkat et al. [97] | ||
| BV2 microglia (mouse) | Total Exosome Isolation Reagent and UC | TEM | +CD9 +CD63 +CD81 | 30–100 nm (TEM) 96 nm (NTA) | In vitro: Hippocampal neuron cells In vivo: Repeated mild traumatic brain injury mouse model | 3 × 1010 particles in 200 μL/mouse, 35 days post-injury | IV | ND | PKH26 label (incubation) miR-124-3p (transfection source cells) | Yes, brain sections PKH26 labelled EVs (confocal imaging) | Ge et al. [98] | |
| BV2 microglia, M2 polarized (mouse) | UC | TEM | +CD9 +CD63 +TSG101 | 30–120 nm (TEM, NTA) | In vitro: Primary neural cells (OGD) In vivo: Transient MCAO mouse model | 100 μg/dose/day/mouse, right after model induction, 3 consequent days | IV | ND | PKH26 label (incubation) | Yes, brain sections PKH26 labelled EVs (confocal imaging) + functional effects endogenous miR-124 | Song et al. [99] | |
| Primary astrocytes (mouse) | UC | TEM | +CD9 +CD63 +ALIX | 40–160 nm (DLS, Nanosizer) | In vitro: HT-22 neurons (OGD) In vivo: MCAO rat model | 80 μg/2 mL, 1 h after ligation operation | IV | ND | Dil (only in vitro) (incubation) | Only functional effects | Pei et al. [100] | |
| Primary astrocytes, ischemic preconditioned (mouse) | UC | TEM | +CD9 +CD63 +ALIX +TSG101 | 50–150 nm (DLS, Zetasizer) | In vitro: Primary neural cells (OGD) In vivo: MCAO mouse model | 100 μg EVs/day, 3 injections per day for total of 3 days, immediately after MCAO. | IV | ND | Dil (only in vitro) (incubation) | Dil labelled EV detection in brain mentioned + functional effects | Chen et al. [101] | |
| Primary pericytes (mouse) | UC | TEM | +CD9 +CD81 | 30–200 nm (NTA) | In vitro: Primary spinal cord endothelial cells. In vivo: SCI mouse model | 20 μg EVs, 1 h after SCI | IV | ND | ND | Only functional effects (spinal cord) | Yuan et al. [102] | |
| Cancer cells | MDA-MB-231 breast cancer cell line (brain seeking variant only) (human) | Ultracentrifugation and OptiPrep gradient UC | TEM | +CD9 +CD63 +ALIX -GM130 | ~158 nm (NTA) | In vitro: Static and microfluidic human brain endothelial cell models In vivo: Nu/Nu mice MDA-MB-231 cell injection model and zebrafish BBB model | 3 μg EVs (3–4 × 109 particles/100 μl) EVs, injected retro-orbitally, every 2 days for a total of 10 injections (mice) 5 nl of 400 μg/mL EV stock (zebrafish) | IV (retro-orbital) (Distribution studies and BBB integrity) Intracardiac (Transcytosis and BBB integrity) | Involvement of clathrin-dependent but not caveolin-dependent uptake for transcytosis | Gaussia luciferase/Palm TdTomato (Transduced source cells) | Yes, Mouse brain sections TdTomato labelled EV uptake Zebrafish EV transcytosis live imaging | Morad et al. [66] |
| MDA-MB-231 breast cancer cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1e6 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after LPS stimulus possibly indicates involvement of selectins, cytokines, enhanced adsorptive transcytosis, insulin transport or disrupted barrier transport | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| SK-Mel-28 melanoma cell line (human) | ExoQuick-TC Exosome Precipitation Solution or MagCapture Exosome Isolation Kit PS | ND | +ALIX -GRP78 (SRM Analysis) | ~105 nm (NTA) | In vitro: hCMEC/D3 cell model | NA | NA | Identification CD46 major receptor for uptake in human blood–brain barrier endothelial cells | PKH67 label (only in vitro) (incubation) | NA | Kuroda et al. [64] | |
| EL-4 lymphoblast cell line (mouse) | 10,000 g centrifugation pellet + sucrose gradient centrifugation (post-loading) | ND | ND | ND | In vivo: 3 therapeutic models: -LPS brain inflammation -MOG-peptide induced EAE MS mouse model and -GL26-Luciferase brain tumor-bearing model mouse | 10 μg EV protein/mouse | IN | ND | IRDye800 label DiR label PKH26 label (incubation) Curcumin or JSI-124 (incubation) | DiR labelled EV detection in brain (Odyssey laser scanning imager, Carestream Molecular Imaging system) Brain sections PKH26 labelled EVs (confocal imaging) Curcumin load detection in brain + functional effects | Zhuang et al. [103] | |
| SCCVII oral squamous cancer cells, (mouse) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after WGA stimulus suggests binding to brain endothelial cell glycoproteins containing sialic acid or N-acetyl-d-glucosamine + decreased uptake after LPS stimulus | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| MEL526 melanoma cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | No specific mechanism identified | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| PCI-30 Human HPV (-) HNSCC cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after LPS stimulus possibly indicates involvement of selectins, cytokines, enhanced adsorptive transcytosis, insulin transport or disrupted barrier transport | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| SSC-90 Human HPV (+) HNSCC cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | No specific mechanism identified | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| Kasumi Leukemic cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after WGA stimulus suggests binding to brain endothelial cell glycoproteins containing sialic acid or N-acetyl-d-glucosamine | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| Stem cells | NSC (human) | Not described | TEM | +CD63 +CD81 (routinely detected–data not shown) | NSC < 200 nm (NTA) | In vitro: Differentiated neural cells In vivo: MCAO mouse model | 3 doses (not specified) at 2, 14 and 48 h post TE_MCAO in young mice Or 6, 24 and 48 h post stroke (In aged mice) | IV | ND | Indium-111 Dil label (only in vitro (incubation) | Radioactively labelled EV detection 1 h post -TE-MCAO (SPECT) + functional effects | Webb et al. [104] |
| UF | (cfr. Webb et al. [104]) | +CD81 +NSC EV marker profile (MACSPlex exosome kit) | NSC < 200 nm (NTA) | In vitro: Human umbilical MSC. In vivo: MCAO porcine model | 2.7 × 1010 particles/kg EVs in 50 mL, administered at 2, 14 and 24 h post-MCAO | IV (peripheral ear vein) | ND | Dil (only in vitro) (incubation) | Only functional effects | Webb et al. [105] | ||
| UC | TEM | ND | ~147 nm (NTA) | In vivo: 5xFAD AD mouse model | 2.25 × 107 particles, 1 or 2 injections | IV (retro-orbital) | ND | NA | Only functional effects | Apodaca et al. [106] | ||
| UC | ND | ND | ND | In vivo: Wild type mice | 6.70 × 106 particles | IV, IN and hippocampal injection | ND | PKH26 (incubation) | PKH26 labelled EV detection in brain sections (confocal imaging) | Ioannides et al. [107] | ||
| NSC (mouse) | PEG complexing and centrifugation | ND | ND | 100–200 nm (NTA) | In vitro: Primary cortical astrocyte or neuronal cultures (OGD) In vivo: MCAO mouse model | 10 μg of total EV protein, 2 h after transient MCAO | IV | ND | NA | Only functional effects | Sun X et al. [108] | |
| Urine stem cells (human) | DC/UC | TEM | +CD9 +ALIX +TSG101 -GM130 | ~74 nm (Flow nanoanalyzer) | In vitro: Neural stem (OGD) study In vivo: Rat MCAO stroke model | 1 × 1011 particles, 1 injection, 4 h post-MCAO | IV | ND | DiR (only in vitro) (incubation) | DiR labelled EV biodistribution (IVIS Spectrum) | Ling et al. [109] | |
| Blood cells | Raw 264.7 macrophage cell line (mouse) | UC and SEC (post labeling) | TEM | +ALIX +TSG101 +LAMP2B | ~90 nm (NTA) ~130 nm (DLS) −18 mV (DLS) | In vitro: hCMEC/D3 model In vivo: Wild type mice | 4 × 105 cpm of Iodine-125-labelled EVs (65 μg or 3 × 1011 EVs per batch) | IV | LFA-1 (EV) with ICAM-1 and C-type lectin receptor on brain endothelial cells | Iodine-125-label (Chloramine-T method) CM-Dil label (only in vitro) BDNF (incubation) | Radioactive labelled EV delivery Radioactive labelled BDNF EV cargo brain delivery | Yuan et al. [110] |
| UC | TEM and AFM | +ALIX +CD63 -CANX | ~110 nm, after loading 117 nm (NTA) −4.5 mV, after loading −4.9 mV (DLS) | In vitro: hCMEC/D3 model In vivo: -SD rats for tissue distribution and bioavailability study -C57BL/6 mice model -okadiak injection AD mouse model | Curcumin-EVs at 0.4 mg/kg (rat) Curcumin-EVs at 20 μg curcumin load/dose. 1 injection/day for 7 days (mouse) | Rat: IV Mouse: IP | LFA-1 (EV) with ICAM-1 on brain endothelial cells | Fluorescent curcumin (source cell incubation) | Fluorescent EV cargo detected in brain sections (confocal imaging) and brain tissue (IVIS spectrum imaging) | Wang et al. [111] | ||
| Gradient centrifugation + SEC (Sepharose 6 BCL) | TEM and AFM | +TSG101 | 100 nm, after loading 100–200 nm (NTA) 100 nm, after loading 100-200 nm (DLS) | In vitro: PC12 neuronal cells In vivo: 6-OHDA injection PD mouse model | 2.4 × 1010 particles/mouse for biodistribution. 1.2 × 109 particles, 10 times every other day for PD mice treatment. | IN | ND | Dil label (incubation) Catalase (incubation, freeze/thaw, extrusion + in vivo read-out: saponin and sonication) | DiR labelled EV brain sections (confocal imaging) + functional effects catalase loading | Haney et al. [91] | ||
| J774A.1 macrophage cell line (mouse) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Involvement of 6-mannose-receptor + Increased uptake after WGA stimulus suggests binding to brain endothelial cell glycoproteins containing sialic acid or N-acetyl-d-glucosamine | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| Primary T cells (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after LPS stimulus possibly indicates involvement of selectins, cytokines, enhanced adsorptive transcytosis, insulin transport or disrupted barrier transport | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| Autologous DC (mouse) | Differential UC and sucrose gradient (post-labelling) | TEM | +ALIX +TSG101 | ~100 nm (NTA) | In vivo: wild type NMRI or C57BL/6 mice | 1 × 1010 particles/g mouse | IV | ND | DiR label (incubation) | DiR labelled EV biodistribution (IVIS spectrum) | Wiklander et al. [85] | |
| Bloodserum derived (predominantly produced by reticulocytes) | UC | TEM | +CD9 +CD63 +CD81 | 40–200 nm (TRPS, Izon science) | In vitro: bEnd.3 and SH-SY5Y cell line In vivo: 6-OHDA injection PD mouse model | 18 mg/kg (with 1 mg EVs = about 4.16 × 1011 blood particles) | IV | Transferrin–transferrin receptor interaction (on both EVs and brain endothelial cells) | PKH67/PKH26 (in vitro only) (incubation) DiD label (incubation) Dopamine (incubation) | DiD labelled EVs in brain sections (confocal imaging) | Qu et al. [63] | |
| Reticulocytes naieve or PD patients (human) | SEC | ND | +ALIX | ~200 nm (NTA) | In vitro: Primary mouse brain endothelial cells, N9 microglia In vivo: CD-1 wild type mice + LPS stimulus | 300,000 cpm of labelled EVs | IV (jugular vein) | Adsorptive transcytosis | -Iodine-125 label (chloramine-T method) -Dil label (incubation) | Radioactively labelled EV measurement of whole brain Dil labelled EVs were detected on brain slices–only in LPS condition (confocal microscopy) | Matsumoto et al. [112] | |
| Other cell types | HEKT293 cells (human) | UC | ND | +CD9 +CD63 +CD81 | ~ 96 nm, with luciferase construct addition 80 nm (NTA) | In vitro: Brain microvascular endothelial cells (BMEC cell line) | NA | NA | Internalization via clathrin-dependent and caveolae-dependent routes | PKH67 and PKH26 label (in vitro only) (incubation) Lactadherin-Gaussia luciferase (Transduction source cells) | NA | Chen et al. [62] |
| NIH-3T3 fibroblast cell line (mouse) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Inidcations involvement of mannose-6-phosphate receptor + Increased uptake after WGA stimulus suggests binding to brain endothelial cell glycoproteins containing sialic acid or N-acetyl-d-glucosamine | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| HaCaT keratinocyte cell line (human) | Mini SEC | TEM | +CD9 +CD63 +ALIX +TSG101 -CANX -GRP94 | 76–130 nm (TRPS, Izon) | In vivo: CD-1 wild type mouse injections + LPS/WGA/M6P IP injection | 1 × 106 cpm of Iodine-125 radioactively labelled EVs | IV (left jugular vein) | Increased uptake after WGA stimulus suggests binding to brain endothelial cell glycoproteins containing sialic acid or N-acetyl-d-glucosamine | Iodine-125 label (chloramine-T method) | Radioactively labelled EV measurement of whole brain and different brain regions | Banks et al. [65] | |
| Targeting Ligand | EV Source | Isolation Technique | EV Characterization | Experimental Set Ups | In Vivo EV Dose | Administration Route | MoA | Label or Loaded Cargo (Method) | Proof Brain/CNS Localization | Ref | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Morphology | Protein Markers | EV Size/Charge | ||||||||||
| RVG | Immature DCs (mouse) | UC | TEM | +LAMP2B | 80 nm (NTA, TEM) | In vitro: Neuro2a cells and C2C12 cells In vivo: Wild type mice | 150 μg EVs with 150 μg siRNA cargo | IV | nAchRs targeting | -(Cy5 /cy3 labeled) GAPDH siRNA -BACE1 siRNA (electroporation) -RVG-lamp2b (transfection) | Cy3 GAPDH siRNA RVG EV cargo detection in coronal brain sections (confocal imaging) + Functional siRNA cargo delivery | Alvarez-Erviti et al. [90] |
| UC | ND | ND | 100 nm (NTA) | In vitro: Human SH-SY5Y cells expressing mouse α-syn-HA In vivo: Wild type and Tg13 PD mouse model | 150 μg EVs with 150 μg siRNA cargo | IV | nAchRs targeting | Anti-α-syn siRNA (electroporation) -RVG-lamp2b (transfection) | Functional siRNA cargo delivery | Cooper et al. [113] | ||
| Primary DCs (mouse) | UC | ND | ND | ~90 nm (NTA) | In vitro: SH-SY5Y cells expressing GFP or S129D α-syn In vivo: C57BL6/C3H F1 mice α-syn PFF PD mouse model. | 150 μg EV/dose loaded with 150 μg shRNA Set up 1: 1 injection, read-out after 45 days Set up 2: Second injection day 45, read-out day 90 | IV | nAchRs targeting | -Anti-GFP shRNA -Anti- α-syn shRNA (electroporation) -RVG-lamp2b (transfection) | Functional effects cargo delivery | Izco et al. [114] | |
| HEK 293T cells (human) | Gradient centrifugation and UC (post-loading/post-labelling) | TEM | +ALIX | 100 nm (TEM) | In vitro: Neuro2a cells, C2C12 control cells, primary neurons In vivo: α-syn PFF PD mouse model. | 120 μg/mouse, weekly, during 4 weeks | IP | nAchRs targeting | -(Fluorescently labelled) F5R2 aptamer (PEI method) -CellVue Claret EV label (incubation) -RVG-lamp2b (transfection) | Labelled EV detection in cortex and midbrain on brain sections (confocal imaging) + Functional effects F5R2 | Ren et al. [115] | |
| Exosome isolation kit (Invitrogen) | TEM | ND | 90 nm (TEM) 85 nm (NTA) | In vitro: Neuro2 cells In vivo: Morphine injection addiction mouse model | 200 μg EVs/ mouse optimal dose. Loaded with 0.14 pmol/μg siRNA 4 injections, every 2 days | IV | nAchRs targeting | -Opioid receptor Mu siRNA (transfection) -RVG-lamp2b (transfection) | Functional effects cargo delivery | Liu et al. [116] | ||
| Differential UC and sucrose gradient (post-labelling) | TEM | +ALIX +TSG101 | ~100 nm (NTA) | In vivo: Wild type NMRI or C57BL/6 mice | 1 × 1010 particles/g mouse | IV | nAchRs targeting | -DiR label (incubation) -RVG-lamp2b (transfection) | DiR labelled EV biodistribution (IVIS spectrum) | Wiklander et al. [85] | ||
| UC | TEM | +CD63 +ALIX +TSG101 +LAMP2B -GM130 | ~100 nm (NTA) | In vitro: HEK293 cells In vivo: Photothrombosis stroke mouse model | 200 μg EVs, 24 h post ischemia | IV | nAchRs targeting | Dil label (incubation) -Nerve growth factor protein and mRNA (transfection) -RVG-Lamp2b (transfection) | Dil labelled EV detection in brain and different cell types (fluorescence imaging) | Yang et al. [117] | ||
| UC | TEM | +CD9 +CD63 +TGS101 +LAMP2B | ~117 nm (NTA/DLS) | In vivo: Rodent and non-human primate ischemic stroke models | 12 mg EVs/kg | IV | nAchRs targeting | -Dil label (incubation) -RVG-Lamp2b (transfection) -circSCMH1 (transfection) | -Dil labelled EV detection in brain and different cell types (fluorescence imaging) -qPCR detection circSCMH1 RNA in brain tissue -Functional effects cargo delivery | Yang et al. [118] | ||
| HEK293T cells (human) (+partially validated in MSCs (human)) | -For In vitro: CM without purification (debris spins only) -For NTA: SEC (Exo-Spin kit) -For in vivo: Donor cells implantation | ND | +CD9 +TSG101 +HSP90B (unpurified CM) (ELISA) | ~100 nm (NTA) | In vitro: -nAchRs expressing HEK 293T cells -Neuro2A cell cultures + 6-OHDA In vivo: Striatal 6-OHDA injection PD mouse model. | NA | Subcutaneous donor cell implantation | nAchRs targeting | “EXOtic” delivery system: -EV booster genes at C-terminus CD63 -L7Ae at C-terminus CD63 -C/D box in 3′UTR NanoLuc/catalase mRNA -Cx43 mutant -lamp2b-RVG construct (all transfection) -RVG-lamp2b (transfection) | Functional effects cargo delivery | Kojima et al. [88] | |
| Optimized RVG (higher degradation resistance) | HEK293FT cells (human) | UC | TEM | CD63 | ~130 nm (NTA) | In vitro: neuroblastoma cells | NA | NA | nAchRs targeting | PKH67 label (incubation) -RVG-lamp2b (transfection) | NA | Hung et al. [119] |
| T7-peptide or RVG | HEK293T cells (human) | ExoEasy Maxi Kit (Qiagen) + UC (post-loading) | SEM | ND | ND | In vitro: C6 neural cells In vivo: Intracranial tumor rat model | 20 μg EVs loaded with 20 μg miRNA-21 anti-sense oligonucleotides | IV | T7 peptide: Transferrin receptor (on both BBB and glioblastoma tumor cells) RVG: nAchRs targeting | -Dil label (incubation) -(Fluorescently labelled) miRNA 21 anti-sense oligonucleotide (electroporation) -T7-peptide-Lamp2b (transfection) OR -RVG-Lamp2b (transfection) | Detection DiR labelled EVs on brain section (confocal imaging) and brain tissue (IVIS spectrum) + functional effects cargo delivery | Kim et al. [120] |
| 4F-LDL peptide | Fibroblast cells L929 (mouse) | UC (max 14,000 g) | TEM | ND | 200–300 nm (TEM), 300–325 nm (DLS), −10 mV zeta potential | In vitro: U87 cells and U87 glioma 3D spheroids In vivo: U87 glioma injection BALB/c nude mice | 5 mg/kg mouse MTX, EV dose not specified | IV | LDLR overexpresssion on the BBB and GBM cell lines | -PKH26 label/DiR label (incubation) -4F-LDL peptide (EV membrane inserted via ApoA-I mimetic peptide 4F) -Surface KLA (pro-apoptotic) therapeutic glioblastoma peptide (4F EV insertion) -MTX (source cell loading) | DiR labelled EV detection in brain (IVIS Spectrum) + Functional effects MTX cargo | Ye et al. [121] |
| RGD-4C peptide | ReNcell VM, neural progenitor cell line (human) | UC | TEM | +ALIX +TSG101 -CANX | <200 nm (NTA) | In vitro: BV2 microglia In vivo: MCAO mouse model | 100 μg total protein = 2.5–3.7 × 1010 particles per mouse. After 1 h of MCAO and 12 h of reperfusion Study therapeutic potential: 300 μg EVs, 12 h after reperfusion (in 200 μL). | IV | Targeting the ischemic lesion region (integrins activated endothelial cells) | -CSFE label -Cy5.5 label (click chemistry) -RGD-4C peptide (phosphatidylserine binding domains of lactadherin) (incubation) -TdTomato-labeled or Gluc-display (transduction) | Detection Cy5.5 labeled EVs in brain tissue (IVIS spectrum) Detection TdTomato labelled EVs in brain sections (confocal fluorescence imaging) | Tian et al. [122] |
| c(RGDyK) peptide | H9 ESC (human) | UC | TEM | +CD63 +ALIX +TSG101 -GM130 | 70 nm (unmodified and PTX loaded EVs (with peptide) 107 nm (no peptide) 125 nm (with peptide) (Flow nanoanalyzer) | In vitro: different cancer cell lines for EV uptake, glioblastoma cell lines (U87 U251 for anti-proliferative characteristics In vivo: Glioma mouse model | 1 × 1011 particles/mL, 125 μL) every other day, during 2 weeks | IV (caudal vein) | Targeting αVβ3 integrin receptors, overexpressed on the surface of proliferating glioblastoma tumor endothelium | Dil label (incubation) -Paclitaxel (incubation) -c(RGD) peptide (chemical crosslining) | Detection Dil labeled EVs in brain tissue (IVIS spectrum) | Zhu et al. [123] |
| CDX peptide CREKA peptide | Embryonic fibroblasts (mouse) (MEF) Mouse bone marrow derived DCs | UC and OptiPrep density gradient UC | Cryo-EM | MEF: +CD9 +CD63 +TSG101 | MEF: ~20–50 nm unmodified, ~70–110 nm (after CNP) (DLS) | In vitro: U87 and GL261 cells In vivo: U87 tumor model and GL261 tumor model | 1 × 1012 particles, every 3 days, 10 days after tumor cell implantation | IV | CDX peptide: U87 tumor cell targeting CREKA peptide: GL261 tumor cell targeting | PKH26 label (incubation) Tumor targeting peptides, both in fusion with N-terminus CD47 transmembrane protein (CNP) mRNA (CNP) | Detection PKH26 labelled EV brain tumor uptake (IVIS Spectrum, Two-photon imaging) + functional effects | Yang et al. [124] |
| RGERPPR peptide (tumor targeting) | Raw 264.7 macrophage cell line (mouse) | UF, UC, and filtration sequentially | TEM | +CD63 +CD81 | ~120 nm (NTA) zetapotential -25 mV | In vitro: U251 and Bel-7404 cell target receptor cells In vivo: Glioma bearing BALB/c nude mice | 200 μg (with 800 μg of both curcumin and SPION) | IV | Glioma NRP-1 receptor | -Dil label (incubation) -RGERPPR peptide (click chemistry) -Superparamagnetic iron oxide nanoparticles (SPION) and curcumin (electroporation) | Detection DiR labelled EVs in brain tissue +SPION detection (MRI) + functional effects | Jia et al. [125] |
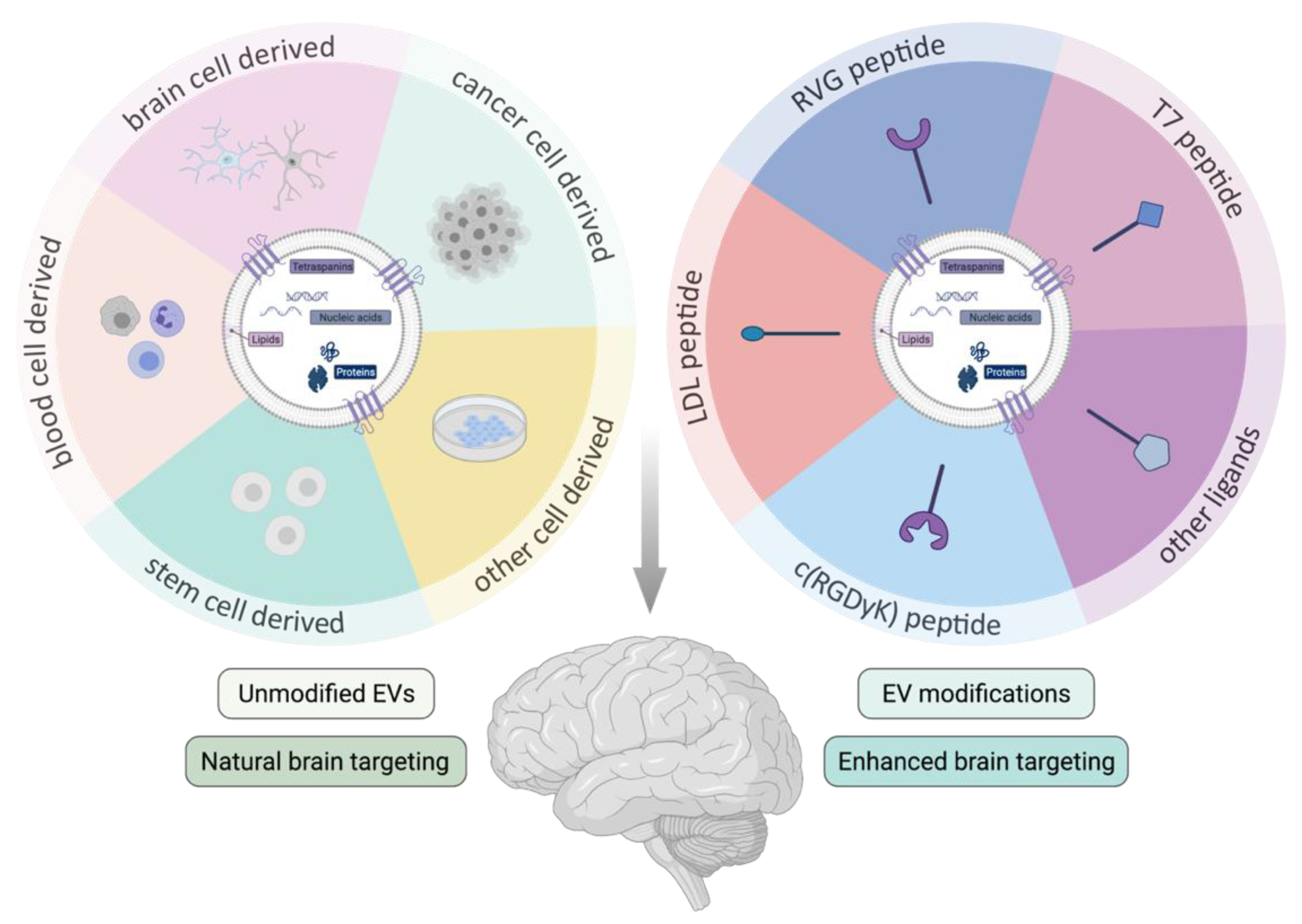
3.3. Extracellular Vesicle Brain Targeting
3.3.1. Unmodified Extracellular Vesicles
- Brain (cancer) cell-derived EVs
- b.
- Cancer cell derived EVs
- c.
- Stem cell derived EVs
- d.
- Blood cell derived EVs
- e.
- Other cellular sources
3.3.2. Modified Extracellular Vesicles
- Rabies virus glycoprotein modified EVs
- b.
- Other targeting strategies
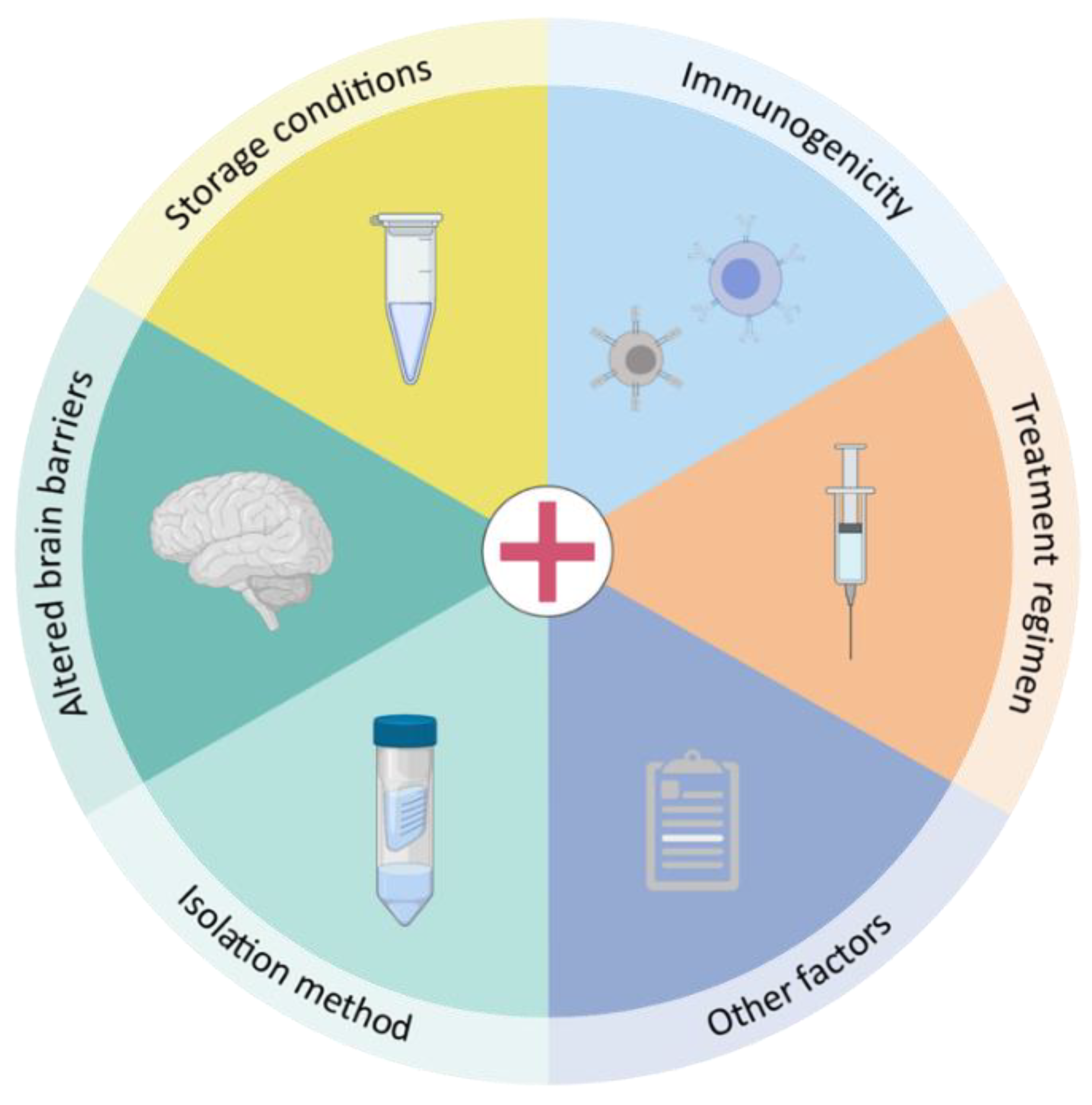
4. Practical Considerations towards EVs as a Therapeutic Delivery Platform to the Brain
4.1. Storage Conditions
4.2. EV Isolation Techniques
4.3. Cellular Source and Immunogenic Potential
4.4. Alteration of Brain Barriers in Neurological Disorders
4.5. Other Important Factors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow. Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier endogenous transporters as therapeutic targets: A new model for small molecule CNS drug discovery. Expert. Opin. Ther. Targets 2015, 19, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, M.; Vandenbroucke, R.E.; Decrock, E.; Culot, M.; Cecchelli, R.; Leybaert, L. A new angle on blood-CNS interfaces: A role for connexins? FEBS Lett. 2014, 588, 1259–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Engelhardt, B.; Sorokin, L. The blood–brain and the blood–cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Saraiva, C.; Praca, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, M.; Abramovitz, L.; Peer, D. Precision nanomedicine in neurodegenerative diseases. ACS Nano 2014, 8, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Zhou, J.; Piepmeier, J.M.; Saltzman, W.M. Polymeric nanoparticles for drug delivery to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 701–705. [Google Scholar] [CrossRef] [Green Version]
- van der Meel, R.; Fens, M.H.; Vader, P.; van Solinge, W.W.; Eniola-Adefeso, O.; Schiffelers, R.M. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J. Control. Release 2014, 195, 72–85. [Google Scholar] [CrossRef]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.P.; Fenart, L. Modelling of the blood-brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef]
- Fischer, H.; Gottschlich, R.; Seelig, A. Blood-brain barrier permeation: Molecular parameters governing passive diffusion. J. Membr. Biol. 1998, 165, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef]
- Banks, W.A.; Moinuddin, A.; Morley, J.E. Regional transport of TNF-alpha across the blood-brain barrier in young ICR and young and aged SAMP8 mice. Neurobiol. Aging 2001, 22, 671–676. [Google Scholar] [CrossRef]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Inoue, T.; Ohtsuki, S.; Terasaki, T. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J. Pharm. Sci. 2013, 102, 3343–3355. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Tachikawa, M.; Obuchi, W.; Hoshi, Y.; Tomioka, Y.; Ohtsuki, S.; Terasaki, T. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: Application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood-brain barrier in ddY, FVB, and C57BL/6J mice. Fluids Barriers CNS 2013, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Devel. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.I.; Sei, Y.J.; Park, H.J.; Kim, J.; Ryu, Y.; Choi, J.J.; Sung, H.J.; MacDonald, T.J.; Levey, A.I.; Kim, Y. Microengineered human blood-brain barrier platform for understanding nanoparticle transport mechanisms. Nat. Commun. 2020, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Strazielle, N.; Ghersi-Egea, J.F. Potential Pathways for CNS Drug Delivery Across the Blood-Cerebrospinal Fluid Barrier. Curr. Pharm. Des. 2016, 22, 5463–5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryniarski, M.A.; Ren, T.; Rizvi, A.R.; Snyder, A.M.; Morris, M.E. Targeting the Choroid Plexuses for Protein Drug Delivery. Pharmaceutics 2020, 12, 963. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Choroid plexus in the central nervous system: Biology and physiopathology. J. Neuropathol. Exp. Neurol. 2000, 59, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Keep, R.F.; Jones, H.C. A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Brain Res. Dev. Brain Res. 1990, 56, 47–53. [Google Scholar] [CrossRef]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert. Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Nagaraja, T.N.; Patel, P.; Gorski, M.; Gorevic, P.D.; Patlak, C.S.; Fenstermacher, J.D. In normal rat, intraventricularly administered insulin-like growth factor-1 is rapidly cleared from CSF with limited distribution into brain. Cereb. Fluid Res. 2005, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Matheson, C.; Sun, J.; Radeke, M.J.; Feinstein, S.C.; Miller, J.A. Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. Exp. Neurol. 1994, 127, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Slavc, I.; Cohen-Pfeffer, J.L.; Gururangan, S.; Krauser, J.; Lim, D.A.; Maldaun, M.; Schwering, C.; Shaywitz, A.J.; Westphal, M. Best practices for the use of intracerebroventricular drug delivery devices. Mol. Genet. Metab. 2018, 124, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Ghersi-Egea, J.F.; Monkkonen, K.S.; Schmitt, C.; Honnorat, J.; Fevre-Montange, M.; Strazielle, N. Blood-brain interfaces and cerebral drug bioavailability. Rev. Neurol. 2009, 165, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Ghersi-Egea, J.F.; Finnegan, W.; Chen, J.L.; Fenstermacher, J.D. Rapid distribution of intraventricularly administered sucrose into cerebrospinal fluid cisterns via subarachnoid velae in rat. Neuroscience 1996, 75, 1271–1288. [Google Scholar] [CrossRef]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef]
- Grapp, M.; Wrede, A.; Schweizer, M.; Huwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Buckers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef] [Green Version]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Balusu, S.; Van Cauwenberghe, C.; Brkic, M.; Pauwels, M.; Plehiers, N.; Bruggeman, A.; Dujardin, P.; Van Imschoot, G.; Van Wonterghem, E.; et al. Importance of extracellular vesicle secretion at the blood-cerebrospinal fluid interface in the pathogenesis of Alzheimer’s disease. Acta. Neuropathol. Commun. 2021, 9, 143. [Google Scholar] [CrossRef]
- Lepko, T.; Pusch, M.; Muller, T.; Schulte, D.; Ehses, J.; Kiebler, M.; Hasler, J.; Huttner, H.B.; Vandenbroucke, R.E.; Vandendriessche, C.; et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO J. 2019, 38, e100481. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid Transport within Cerebral Perivascular Spaces Underlies Widespread Tracer Distribution in the Brain after Intranasal Administration. J. Cereb. Blood Flow Metabolism. 2014, 35, 371–381. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliver. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Davis, T.P. Perivascular and Perineural Pathways Involved in Brain Delivery and Distribution of Drugs after Intranasal Administration. Pharmaceutics 2019, 11, 598. [Google Scholar] [CrossRef] [Green Version]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, K.; Jiang, X. From nose to brain: Understanding transport capacity and transport rate of drugs. Expert. Opin. Drug Del. 2008, 5, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Bentivoglio, M.; Kristensson, K.; Rottenberg, M.E. Circumventricular Organs and Parasite Neurotropism: Neglected Gates to the Brain? Front. Immunol. 2018, 9, 2877. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Banks, W.A. Burger’s Medicinal Chemistry and Drug Discovery; John Wiley & Sons: New York, NY, USA, 2021; pp. 1–22. [Google Scholar] [CrossRef]
- de Jong, O.G.; Kooijmans, S.A.A.; Murphy, D.E.; Jiang, L.; Evers, M.J.W.; Sluijter, J.P.G.; Vader, P.; Schiffelers, R.M. Drug Delivery with Extracellular Vesicles: From Imagination to Innovation. Acc. Chem. Res. 2019, 52, 1761–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niel, G.; D′Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; de Jong, O.G.; Schiffelers, R.M. Exploring interactions between extracellular vesicles and cells for innovative drug delivery system design. Adv. Drug Deliv. Rev. 2021, 173, 252–278. [Google Scholar] [CrossRef] [PubMed]
- Samir, E.L.A.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.A.; Lotvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, 492. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef] [Green Version]
- Rene, C.A.; Parks, R.J. Delivery of Therapeutic Agents to the Central Nervous System and the Promise of Extracellular Vesicles. Pharmaceutics 2021, 13, 492. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Segaliny, A.; et al. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef]
- Kuroda, H.; Tachikawa, M.; Yagi, Y.; Umetsu, M.; Nurdin, A.; Miyauchi, E.; Watanabe, M.; Uchida, Y.; Terasaki, T. Cluster of Differentiation 46 Is the Major Receptor in Human Blood-Brain Barrier Endothelial Cells for Uptake of Exosomes Derived from Brain-Metastatic Melanoma Cells (SK-Mel-28). Mol. Pharm. 2019, 16, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; Toth, E.A.; Sodar, B.W.; Szabo-Taylor, K.E. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.W.; Nguyen, J. Exosomes as therapeutics: The implications of molecular composition and exosomal heterogeneity. J. Control. Release 2016, 228, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mager, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtio, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Jang, S.C.; Konecna, B.; Hoog, J.L.; Sihlbom, C.; Lasser, C.; Lotvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles-Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 6, 36338. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Fernandes, L.; Rocha, V.B.; Carregari, V.C.; Urbani, A.; Palmisano, G. A Perspective on Extracellular Vesicles Proteomics. Front. Chem. 2017, 5, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Zlokovic, B.V. Remote control of BBB: A tale of exosomes and microRNA. Cell Res. 2017, 27, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhang, Y.; Du, X.F.; Li, J.; Zi, H.X.; Bu, J.W.; Yan, Y.; Han, H.; Du, J.L. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017, 27, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, L.; Li, L.; Cao, Y. Exosomes Derived from Brain Metastatic Breast Cancer Cells Destroy the Blood-Brain Barrier by Carrying lncRNA GS1-600G8.5. Biomed. Res. Int. 2020, 2020, 7461727. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.U.; Allen, T.; Shen, D.; et al. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474–3487. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Villa, A.; Crescenti, D.; Marzagalli, M.; Kuryk, L.; Limonta, P.; Mazzaferro, V.; Ciana, P. Heterologous and cross-species tropism of cancer-derived extracellular vesicles. Theranostics 2019, 9, 5681–5693. [Google Scholar] [CrossRef]
- Emam, S.E.; Abu Lila, A.S.; Elsadek, N.E.; Ando, H.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Ishida, T. Cancer cell-type tropism is one of crucial determinants for the efficient systemic delivery of cancer cell-derived exosomes to tumor tissues. Eur. J. Pharm. Biopharm. 2019, 145, 27–34. [Google Scholar] [CrossRef]
- Villa, A.; Garofalo, M.; Crescenti, D.; Rizzi, N.; Brunialti, E.; Vingiani, A.; Belotti, P.; Sposito, C.; Franzè, S.; Cilurzo, F.; et al. Transplantation of autologous extracellular vesicles for cancer-specific targeting. Theranostics 2021, 11, 2034–2047. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; Schiffelers, R.M.; Zarovni, N.; Vago, R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacol. Res. 2016, 111, 487–500. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Fogarty, B.; LaForge, B.; Aziz, S.; Pham, T.; Lai, L.; Bai, S. Delivery of Small Interfering RNA to Inhibit Vascular Endothelial Growth Factor in Zebrafish Using Natural Brain Endothelia Cell-Secreted Exosome Nanovesicles for the Treatment of Brain Cancer. AAPS J. 2017, 19, 475–486. [Google Scholar] [CrossRef]
- Silva, A.M.; Lazaro-Ibanez, E.; Gunnarsson, A.; Dhande, A.; Daaboul, G.; Peacock, B.; Osteikoetxea, X.; Salmond, N.; Friis, K.P.; Shatnyeva, O.; et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J. Extracell. Vesicles 2021, 10, e12130. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.; El-Baba, M.D.; Saxena, P.; Auslander, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [Green Version]
- Didiot, M.C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release. 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Jones, T.W.; Dutta, S.; Zhu, Y.; Wang, X.; Narayanan, S.P.; Fagan, S.C.; Zhang, D. Overview and Update on Methods for Cargo Loading into Extracellular Vesicles. Processes 2021, 9, 356. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release. 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Rankin-Turner, S.; Vader, P.; O’Driscoll, L.; Giebel, B.; Heaney, L.M.; Davies, O.G. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Adv. Drug Deliv. Rev. 2021, 173, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.P.; Holme, M.N.; Stevens, M.M. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano 2017, 11, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Vader, P.; Fuhrmann, G. Approaches to surface engineering of extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 173, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Cui, C.; Chopp, M.; Zacharek, A.; Wang, F.; Landschoot-Ward, J.; Shen, Y.; Chen, J. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke 2019, 50, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Guo, M.; Hu, T.; Li, W.; Huang, S.; Yin, Z.; Li, Y.; Chen, F.; Zhu, L.; Kang, C.; et al. Increased Microglial Exosomal miR-124-3p Alleviates Neurodegeneration and Improves Cognitive Outcome after rmTBI. Mol. Ther. 2020, 28, 503–522. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Li, Z.; He, T.; Qu, M.; Jiang, L.; Li, W.; Shi, X.; Pan, J.; Zhang, L.; Wang, Y.; et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910–2923. [Google Scholar] [CrossRef]
- Pei, X.; Li, Y.; Zhu, L.; Zhou, Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp. Cell Res. 2019, 382, 111474. [Google Scholar] [CrossRef]
- Chen, W.; Wang, H.; Zhu, Z.; Feng, J.; Chen, L. Exosome-Shuttled circSHOC2 from IPASs Regulates Neuronal Autophagy and Ameliorates Ischemic Brain Injury via the miR-7670-3p/SIRT1 Axis. Mol. Ther. Nucleic Acids 2020, 22, 657–672. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, Q.; Wang, P.; Jing, Y.; Yao, H.; Tang, Y.; Li, Z.; Zhang, H.; Xiu, R. Exosomes Derived From Pericytes Improve Microcirculation and Protect Blood-Spinal Cord Barrier After Spinal Cord Injury in Mice. Front. Neurosci. 2019, 13, 319. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.L.; Kaiser, E.E.; Scoville, S.L.; Thompson, T.A.; Fatima, S.; Pandya, C.; Sriram, K.; Swetenburg, R.L.; Vaibhav, K.; Arbab, A.S.; et al. Human Neural Stem Cell Extracellular Vesicles Improve Tissue and Functional Recovery in the Murine Thromboembolic Stroke Model. Transl. Stroke Res. 2018, 9, 530–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, R.L.; Kaiser, E.E.; Jurgielewicz, B.J.; Spellicy, S.; Scoville, S.L.; Thompson, T.A.; Swetenburg, R.L.; Hess, D.C.; West, F.D.; Stice, S.L. Human Neural Stem Cell Extracellular Vesicles Improve Recovery in a Porcine Model of Ischemic Stroke. Stroke 2018, 49, 1248–1256. [Google Scholar] [CrossRef]
- Apodaca, L.A.; Baddour, A.A.D.; Garcia, C., Jr.; Alikhani, L.; Giedzinski, E.; Ru, N.; Agrawal, A.; Acharya, M.M.; Baulch, J.E. Human neural stem cell-derived extracellular vesicles mitigate hallmarks of Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 57. [Google Scholar] [CrossRef]
- Ioannides, P.; Giedzinski, E.; Limoli, C.L. Evaluating different routes of extracellular vesicle administration for cranial therapies. J. Cancer Metastasis Treat. 2020, 6. [Google Scholar] [CrossRef]
- Sun, X.; Jung, J.H.; Arvola, O.; Santoso, M.R.; Giffard, R.G.; Yang, P.C.; Stary, C.M. Stem Cell-Derived Exosomes Protect Astrocyte Cultures From in vitro Ischemia and Decrease Injury as Post-stroke Intravenous Therapy. Front. Cell Neurosci. 2019, 13, 394. [Google Scholar] [CrossRef]
- Ling, X.; Zhang, G.; Xia, Y.; Zhu, Q.; Zhang, J.; Li, Q.; Niu, X.; Hu, G.; Yang, Y.; Wang, Y.; et al. Exosomes from human urine-derived stem cells enhanced neurogenesis via miR-26a/HDAC6 axis after ischaemic stroke. J. Cell Mol. Med. 2020, 24, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Wang, H.; Sui, H.; Zheng, Y.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3beta pathway. Nanoscale 2019, 11, 7481–7496. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of alpha-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson’s disease? Acta. Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.M.; Wiklander, P.B.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izco, M.; Blesa, J.; Schleef, M.; Schmeer, M.; Porcari, R.; Al-Shawi, R.; Ellmerich, S.; de Toro, M.; Gardiner, C.; Seow, Y.; et al. Systemic Exosomal Delivery of shRNA Minicircles Prevents Parkinsonian Pathology. Mol. Ther. 2019, 27, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhao, Y.; Xue, F.; Zheng, Y.; Huang, H.; Wang, W.; Chang, Y.; Yang, H.; Zhang, J. Exosomal DNA Aptamer Targeting alpha-Synuclein Aggregates Reduced Neuropathological Deficits in a Mouse Parkinson′s Disease Model. Mol. Ther. Nucleic Acids 2019, 17, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, D.; Liu, Z.; Zhou, Y.; Chu, D.; Li, X.; Jiang, X.; Hou, D.; Chen, X.; Chen, Y.; et al. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci. Rep. 2015, 5, 17543. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, S.; Hou, L.; Zhu, D.; Yin, S.; Yang, G.; Wang, Y. Therapeutic Effects of Simultaneous Delivery of Nerve Growth Factor mRNA and Protein via Exosomes on Cerebral Ischemia. Mol. Ther. Nucleic Acids 2020, 21, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, B.; Zhang, Z.; Wang, S.; Bai, Y.; Zhang, Y.; Tang, Y.; Du, L.; Xu, L.; Wu, F.; et al. Extracellular Vesicle-Mediated Delivery of Circular RNA SCMH1 Promotes Functional Recovery in Rodent and Nonhuman Primate Ischemic Stroke Models. Circulation 2020, 142, 556–574. [Google Scholar] [CrossRef]
- Hung, M.E.; Leonard, J.N. Stabilization of exosome-targeting peptides via engineered glycosylation. J. Biol. Chem. 2015, 290, 8166–8172. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J. Control. Release. 2020, 317, 273–281. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, T.; He, W.; Jin, H.; Liu, C.; Yang, Z.; Ren, J. Methotrexate-Loaded Extracellular Vesicles Functionalized with Therapeutic and Targeted Peptides for the Treatment of Glioblastoma Multiforme. ACS Appl. Mater. Interfaces 2018, 10, 12341–12350. [Google Scholar] [CrossRef]
- Tian, T.; Cao, L.; He, C.; Ye, Q.; Liang, R.; You, W.; Zhang, H.; Wu, J.; Ye, J.; Tannous, B.A.; et al. Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics 2021, 11, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ling, X.; Yang, Y.; Zhang, J.; Li, Q.; Niu, X.; Hu, G.; Chen, B.; Li, H.; Wang, Y.; et al. Embryonic Stem Cells-Derived Exosomes Endowed with Targeting Properties as Ch.emotherapeutics Delivery Vehicles for Glioblastoma Therapy. Adv. Sci. 2019, 6, 1801899. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Escude Martinez de Castilla, P.; Tong, L.; Huang, C.; Sofias, A.M.; Pastorin, G.; Chen, X.; Storm, G.; Schiffelers, R.M.; Wang, J.W. Extracellular vesicles as a drug delivery system: A systematic review of preclinical studies. Adv. Drug Deliv. Rev. 2021, 175, 113801. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Jordan, V.; Blenkiron, C.; Chamley, L.W. Biodistribution of extracellular vesicles following administration into animals: A systematic review. J. Extracell. Vesicles 2021, 10, e12085. [Google Scholar] [CrossRef]
- Lazaro-Ibanez, E.; Faruqu, F.N.; Saleh, A.F.; Silva, A.M.; Tzu-Wen Wang, J.; Rak, J.; Al-Jamal, K.T.; Dekker, N. Selection of Fluorescent, Bioluminescent, and Radioactive Tracers to Accurately Reflect Extracellular Vesicle Biodistribution in Vivo. ACS Nano 2021, 15, 3212–3227. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Garcia-Romero, N.; Carrion-Navarro, J.; Esteban-Rubio, S.; Lazaro-Ibanez, E.; Peris-Celda, M.; Alonso, M.M.; Guzman-De-Villoria, J.; Fernandez-Carballal, C.; de Mendivil, A.O.; Garcia-Duque, S.; et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget 2017, 8, 1416–1428. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem. Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Campanella, C.; Caruso Bavisotto, C.; Logozzi, M.; Marino Gammazza, A.; Mizzoni, D.; Cappello, F.; Fais, S. On the Choice of the Extracellular Vesicles for Therapeutic Purposes. Int. J. Mol. Sci. 2019, 20, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazifar, M.; Mohammadi, M.R.; Pone, E.J.; Yeri, A.; Lasser, C.; Segaliny, A.I.; McIntyre, L.L.; Shelke, G.V.; Hutchins, E.; Hamamoto, A.; et al. Stem Cell-Derived Exosomes as Nanotherapeutics for Autoimmune and Neurodegenerative Disorders. ACS Nano 2019, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Barreto, G.E.; Read, M.I.; Tafti, H.A.; Sahebkar, A. The Therapeutic Potential of Mesenchymal Stem Cell-Derived Exosomes in Treatment of Neurodegenerative Diseases. Mol. Neurobiol. 2019, 56, 8157–8167. [Google Scholar] [CrossRef]
- Guy, R.; Offen, D. Promising Opportunities for Treating Neurodegenerative Diseases with Mesenchymal Stem Cell-Derived Exosomes. Biomolecules 2020, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Fayazi, N.; Sheykhhasan, M.; Soleimani Asl, S.; Najafi, R. Stem Cell-Derived Exosomes: A New Strategy of Neurodegenerative Disease Treatment. Mol. Neurobiol. 2021, 58, 3494–3514. [Google Scholar] [CrossRef]
- Araldi, R.P.; D’Amelio, F.; Vigerelli, H.; de Melo, T.C.; Kerkis, I. Stem Cell-Derived Exosomes as Therapeutic Approach for Neurodegenerative Disorders: From Biology to Biotechnology. Cells 2020, 9, 2663. [Google Scholar] [CrossRef]
- Betzer, O.; Perets, N.; Angel, A.; Motiei, M.; Sadan, T.; Yadid, G.; Offen, D.; Popovtzer, R. In Vivo Neuroimaging of Exosomes Using Gold Nanoparticles. ACS Nano 2017, 11, 10883–10893. [Google Scholar] [CrossRef] [PubMed]
- Perets, N.; Betzer, O.; Shapira, R.; Brenstein, S.; Angel, A.; Sadan, T.; Ashery, U.; Popovtzer, R.; Offen, D. Golden Exosomes Selectively Target Brain Pathologies in Neurodegenerative and Neurodevelopmental Disorders. Nano Lett. 2019, 19, 3422–3431. [Google Scholar] [CrossRef]
- Vogel, A.; Upadhya, R.; Shetty, A.K. Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders. EBioMedicine 2018, 38, 273–282. [Google Scholar] [CrossRef]
- Zhang, G.; Zhu, Z.; Wang, H.; Yu, Y.; Chen, W.; Waqas, A.; Wang, Y.; Chen, L. Exosomes derived from human neural stem cells stimulated by interferon gamma improve therapeutic ability in ischemic stroke model. J. Adv. Res. 2020, 24, 435–445. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Harrison, E.B.; Mahajan, V.; Ahmed, S.; He, Z.; Suresh, P.; Hingtgen, S.D.; Klyachko, N.L.; Mosley, R.L.; et al. Specific transfection of inflamed brain by macrophages: A new therapeutic strategy for neurodegenerative diseases. PLoS ONE 2013, 8, e61852. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Haney, M.J.; Gupta, R.; Bohnsack, J.P.; He, Z.; Kabanov, A.V.; Batrakova, E.V. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS ONE 2014, 9, e106867. [Google Scholar] [CrossRef]
- He, R.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-laden exosomes target ischemic brain tissue and alleviate cerebral ischemia-reperfusion injury by inhibiting ROS-mediated mitochondrial apoptosis. Mater. Sci. Eng. C. Mater. Biol. Appl. 2020, 117, 111314. [Google Scholar] [CrossRef]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Publisher Correction: Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusic, A.D.; Pusic, K.M.; Clayton, B.L.; Kraig, R.P. IFNgamma-stimulated dendritic cell exosomes as a potential therapeutic for remyelination. J. Neuroimmunol. 2014, 266, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Pusic, A.D.; Kraig, R.P. Youth and environmental enrichment generate serum exosomes containing miR-219 that promote CNS myelination. Glia 2014, 62, 284–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsugi-Kobukai, S.; Fujimaki, H.; Hotta, C.; Nakazawa, M.; Minami, M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol. Lett. 2003, 89, 125–131. [Google Scholar] [CrossRef]
- Han, C.; Xiong, N.; Guo, X.; Huang, J.; Ma, K.; Liu, L.; Xia, Y.; Shen, Y.; Li, J.; Jiang, H.; et al. Exosomes from patients with Parkinson’s disease are pathological in mice. J. Mol. Med. 2019, 97, 1329–1344. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.; Jia, L.; Lyu, J.; Sun, Y.; Yu, H.; Li, H.; Liu, W.; Weng, Y.; Yu, W. Exosomes Mediate Hippocampal and Cortical Neuronal Injury Induced by Hepatic Ischemia-Reperfusion Injury through Activating Pyroptosis in Rats. Oxid Med. Cell Longev. 2019, 2019, 3753485. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, G.; Hwang, D.W.; Lee, M. Delivery of High Mobility Group Box-1 siRNA Using Brain-Targeting Exosomes for Ischemic Stroke Therapy. J. Biomed. Nanotechnol. 2019, 15, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol. Ther. Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Zhang, H.X.; He, C.P.; Fan, S.; Zhu, Y.L.; Qi, C.; Huang, N.P.; Xiao, Z.D.; Lu, Z.H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Deville, S.; Berckmans, P.; Hoof, R.V.; Lambrichts, I.; Salvati, A.; Nelissen, I. Comparison of extracellular vesicle isolation and storage methods using high-sensitivity flow cytometry. PLoS ONE 2021, 16, e0245835. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Li, Y.-J.; Hu, X.-B.; Huang, S.; Xiang, D.-X. Preservation of small extracellular vesicles for functional analysis and therapeutic applications: A comparative evaluation of storage conditions. Drug Delivery. 2021, 28, 162–170. [Google Scholar] [CrossRef]
- Lőrincz, Á.M.; Timár, C.I.; Marosvári, K.A.; Veres, D.S.; Otrokocsi, L.; Kittel, Á.; Ligeti, E. Effect of storage on physical and functional properties of extracellular vesicles derived from neutrophilic granulocytes. J. Extracell. Vesicles 2014, 3, 25465. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. Jci. Insight. 2018, 3, e99263. [Google Scholar] [CrossRef]
- Park, S.J.; Jeon, H.; Yoo, S.-M.; Lee, M.-S. The effect of storage temperature on the biological activity of extracellular vesicles for the complement system. Vitro Cell Dev. Biol. Anim. 2018, 54, 423–429. [Google Scholar] [CrossRef]
- Sokolova, V.; Ludwig, A.-K.; Hornung, S.; Rotan, O.; Horn, P.A.; Epple, M.; Giebel, B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces 2011, 87, 146–150. [Google Scholar] [CrossRef]
- Cheng, Y.; Zeng, Q.; Han, Q.; Xia, W. Effect of pH, temperature and freezing-thawing on quantity changes and cellular uptake of exosomes. Protein Cell 2019, 10, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroto, R.; Zhao, Y.; Jamaluddin, M.; Popov, V.L.; Wang, H.; Kalubowilage, M.; Zhang, Y.; Luisi, J.; Sun, H.; Culbertson, C.T.; et al. Effects of storage temperature on airway exosome integrity for diagnostic and functional analyses. J. Extracell. Vesicles 2017, 6, 1359478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Ban, J.-J.; Im, W.; Kim, M. Influence of storage condition on exosome recovery. Biotechnol. Bioproc. E. 2016, 21, 299–304. [Google Scholar] [CrossRef]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Hoen, E.N.N.-t.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Bosch, S.; Beaurepaire, L.d.; Allard, M.; Mosser, M.; Heichette, C.; Chrétien, D.; Jegou, D.; Bach, J.-M. Trehalose prevents aggregation of exosomes and cryodamage. Sci. Rep. 2016, 6, 36162. [Google Scholar] [CrossRef] [Green Version]
- Neupane, Y.R.; Huang, C.; Wang, X.; Chng, W.H.; Venkatesan, G.; Zharkova, O.; Wacker, M.G.; Czarny, B.; Storm, G.; Wang, J.-W.; et al. Lyophilization Preserves the Intrinsic Cardioprotective Activity of Bioinspired Cell-Derived Nanovesicles. Pharmaceutics 2021, 13, 1052. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, G.D.; Barabadi, M.; Tan, J.L.; Morton, D.A.V.; Frith, J.E.; Lim, R. To Protect and to Preserve: Novel Preservation Strategies for Extracellular Vesicles. Front. Pharmacol. 2018, 9, 1199. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Li, Y.-M.; Wang, Z. Preserving extracellular vesicles for biomedical applications: Consideration of storage stability before and after isolation. Drug Deliv. 2021, 28, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Bahr, M.M.; Amer, M.S.; Abo-El-Sooud, K.; Abdallah, A.N.; El-Tookhy, O.S. Preservation techniques of stem cells extracellular vesicles: A gate for manufacturing of clinical grade therapeutic extracellular vesicles and long-term clinical trials. Int. J. Vet. Sci. Medicine 2020, 8, 1–8. [Google Scholar] [CrossRef]
- Jeyaram, A.; Jay, S.M. Preservation and Storage Stability of Extracellular Vesicles for Therapeutic Applications. AAPS J. 2017, 20, 1. [Google Scholar] [CrossRef]
- Clayton, A.; Boilard, E.; Buzas, E.I.; Cheng, L.; Falcón-Perez, J.M.; Gardiner, C.; Gustafson, D.; Gualerzi, A.; Hendrix, A.; Hoffman, A.; et al. Considerations towards a roadmap for collection, handling and storage of blood extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1647027. [Google Scholar] [CrossRef] [Green Version]
- Erdbrügger, U.; Blijdorp, C.J.; Bijnsdorp, I.V.; Borràs, F.E.; Burger, D.; Bussolati, B.; Byrd, J.B.; Clayton, A.; Dear, J.W.; Falcón-Pérez, J.M.; et al. Urinary extracellular vesicles: A position paper by the Urine Task Force of the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12093. [Google Scholar] [CrossRef] [PubMed]
- Roux, Q.; Deun, J.; Dedeyne, S.; Hendrix, A. The EV-TRACK summary add-on: Integration of experimental information in databases to ensure comprehensive interpretation of biological knowledge on extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1699367. [Google Scholar] [CrossRef] [PubMed]
- Veerman, R.E.; Teeuwen, L.; Czarnewski, P.; Akpinar, G.G.; Sandberg, A.; Cao, X.; Pernemalm, M.; Orre, L.M.; Gabrielsson, S.; Eldh, M. Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. J. Extracell. Vesicles 2021, 10, e12128. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Song, Y.; Park, C.H.; Choi, C. Platform technologies and human cell lines for the production of therapeutic exosomes. Extracell. Vesicles Circ. Nucleic Acids 2021, 2, 3–17. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K.L.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Saleh, A.F.; Lázaro-Ibáñez, E.; Forsgard, M.A.M.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Yan, I.K.; Parasramka, M.; Mohankumar, S.; Matsuda, A.; Patel, T. In vitro toxicology studies of extracellular vesicles. J. Appl. Toxicol. 2017, 37, 310–318. [Google Scholar] [CrossRef]
- Tan, T.T.; Lai, R.C.; Padmanabhan, J.; Sim, W.K.; Choo, A.B.H.; Lim, S.K. Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV). Pharmaceuticals 2021, 14, 345. [Google Scholar] [CrossRef]
- Driedonks, T.; Jiang, L.; Carlson, B.; Han, Z.; Liu, G.; Queen, S.E.; Shirk, E.N.; Gololobova, O.; Nyberg, L.; Lima, G.; et al. Pharmacokinetics and biodistribution of extracellular vesicles administered intravenously and intranasally to Macaca nemestrina. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gorlé, N.; Cauwenberghe, C.V.; Libert, C.; Vandenbroucke, R.E. The effect of aging on brain barriers and the consequences for Alzheimer’s disease development. Mamm. Genome. 2016, 27, 407–420. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, I.; Ek, J.; Stolp, H. The molecular anatomy and functions of the choroid plexus in healthy and diseased brain. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183430. [Google Scholar] [CrossRef] [PubMed]
- Ghersi-Egea, J.-F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta. Neuropathologica. 2018, 135, 337–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; Andaloussi, S.E.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef]
- Gupta, D.; Zickler, A.M.; Andaloussi, S.E.L. Dosing Extracellular Vesicles. Adv. Drug Deliver. Rev. 2021, 178, 113961. [Google Scholar] [CrossRef]
- Belhadj, Z.; He, B.; Deng, H.; Song, S.; Zhang, H.; Wang, X.; Dai, W.; Zhang, Q. A combined “eat me/don’t eat me” strategy based on extracellular vesicles for anticancer nanomedicine. J. Extracell. Vesicles 2020, 9, 1806444. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; Meel, R.v.d.; Fens, M.H.A.M.; Heijnen, H.F.G.; Henegouwen, P.M.P.v.B.e.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Parada, N.; Romero-Trujillo, A.; Georges, N.; Alcayaga-Miranda, F. Camouflage strategies for therapeutic exosomes evasion from phagocytosis. J. Adv. Res. 2021, 31, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Portillo, H.A.d.; et al. Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauwels, M.J.; Vandendriessche, C.; Vandenbroucke, R.E. Special delEVery: Extracellular Vesicles as Promising Delivery Platform to the Brain. Biomedicines 2021, 9, 1734. https://doi.org/10.3390/biomedicines9111734
Pauwels MJ, Vandendriessche C, Vandenbroucke RE. Special delEVery: Extracellular Vesicles as Promising Delivery Platform to the Brain. Biomedicines. 2021; 9(11):1734. https://doi.org/10.3390/biomedicines9111734
Chicago/Turabian StylePauwels, Marie J., Charysse Vandendriessche, and Roosmarijn E. Vandenbroucke. 2021. "Special delEVery: Extracellular Vesicles as Promising Delivery Platform to the Brain" Biomedicines 9, no. 11: 1734. https://doi.org/10.3390/biomedicines9111734
APA StylePauwels, M. J., Vandendriessche, C., & Vandenbroucke, R. E. (2021). Special delEVery: Extracellular Vesicles as Promising Delivery Platform to the Brain. Biomedicines, 9(11), 1734. https://doi.org/10.3390/biomedicines9111734