
Human iPSC-Derived Neurons as A Platform for Deciphering the Mechanisms behind Brain Aging

 ,
,
Abstract
1. Introduction
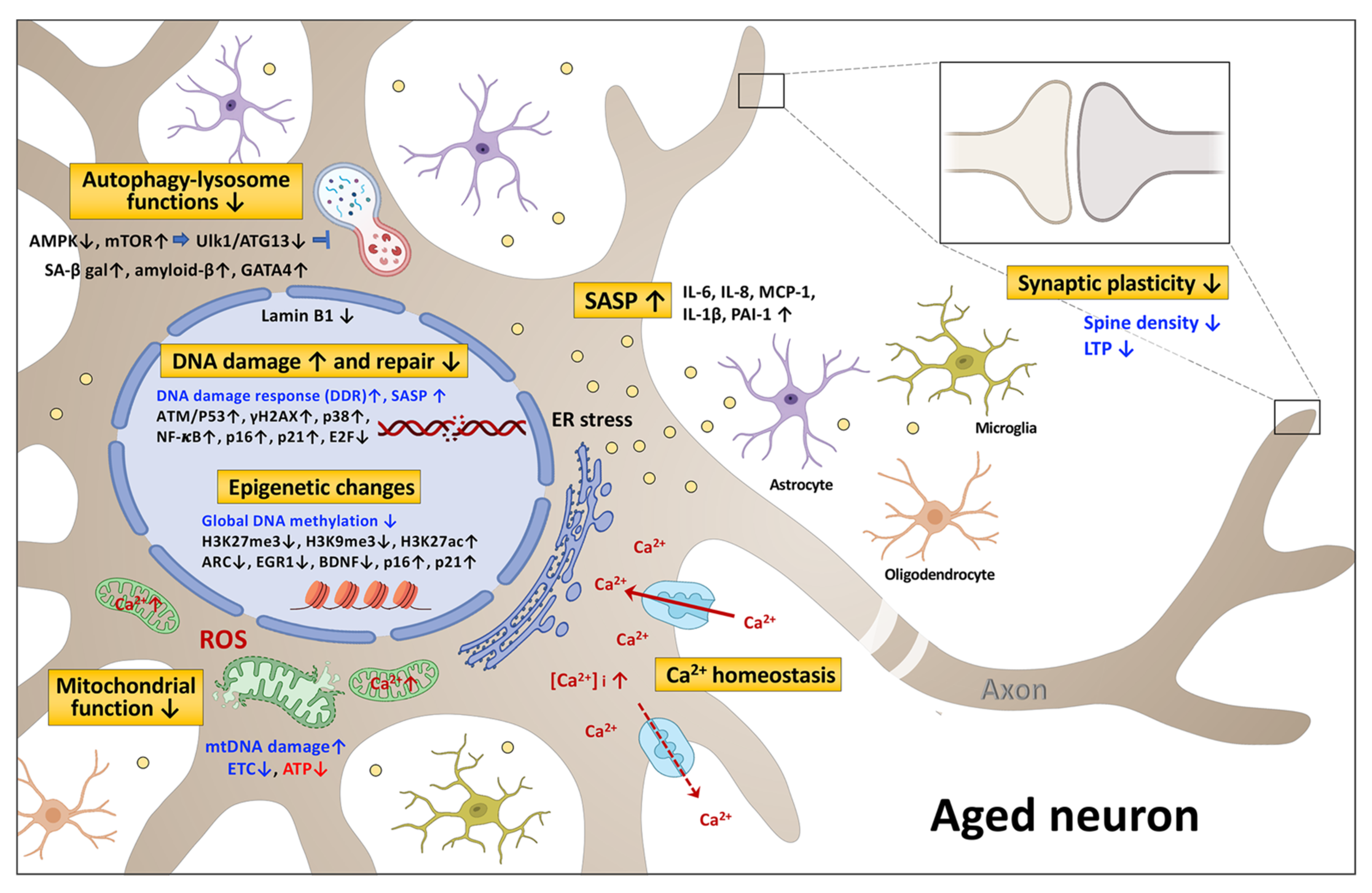
2. The Signaling Pathways Associated with Neuronal Senescence and Brain Aging
2.1. DNA Damage and Repair
2.2. Epigenetic Changes
2.2.1. DNA Methylation
2.2.2. Histone Modification
2.3. Mitochondrial Dysfunction
2.4. Autophagy-Lysosome Dysfunction
2.5. Senescence-Associated Secretory Phenotype (SASP)
2.6. Calcium Homeostasis and Synaptic Plasticity
3. Overview of In Vitro Cell Models for Neuronal Senescence
3.1. Immortalized Cell Lines
3.2. Primary Neuronal Cultures
3.3. Human Neurons Directly Converted from Fibroblast (Fib-iNs)
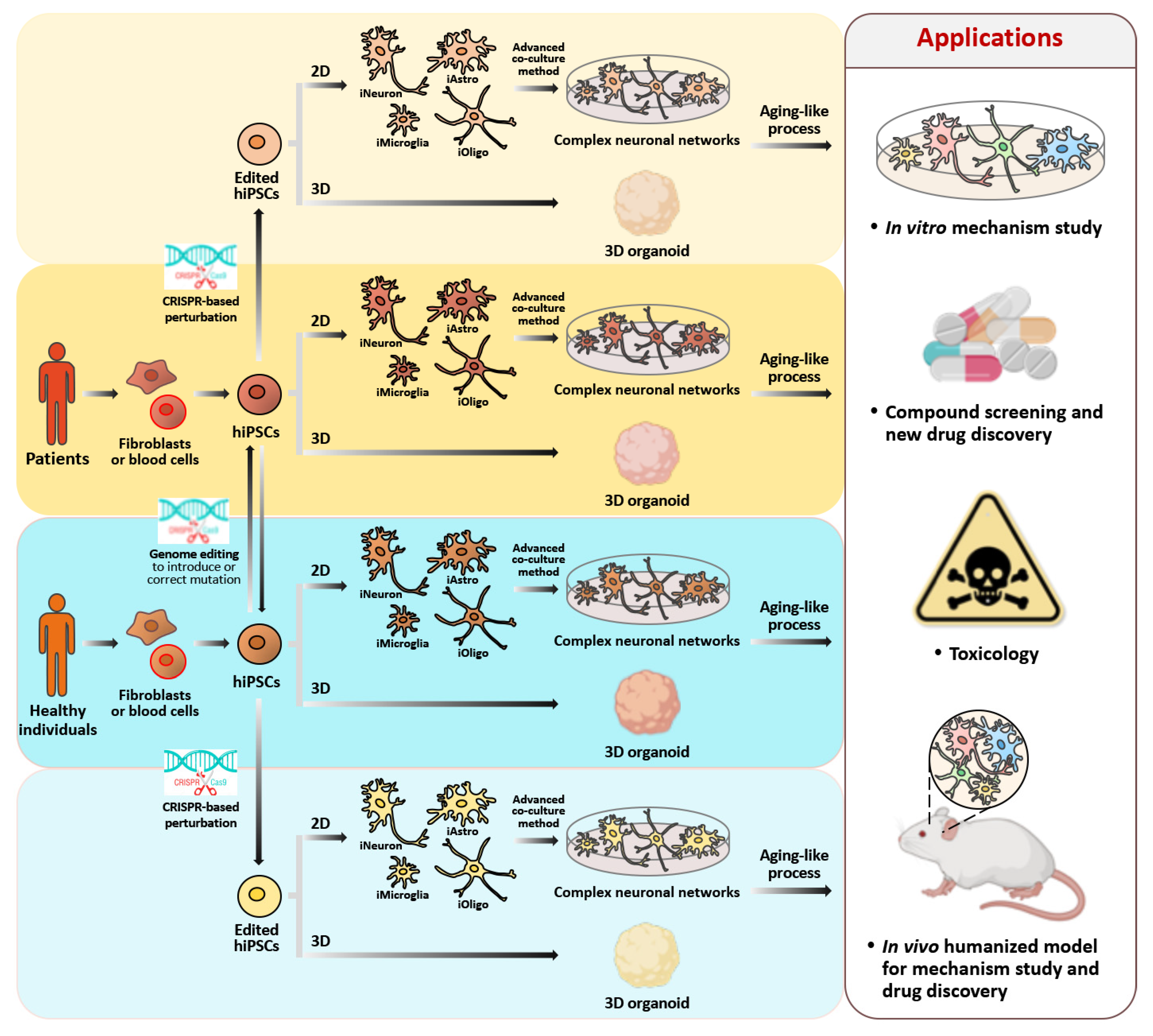
3.4. Human iPSC-Derived Neurons (hiPSC-iNs)
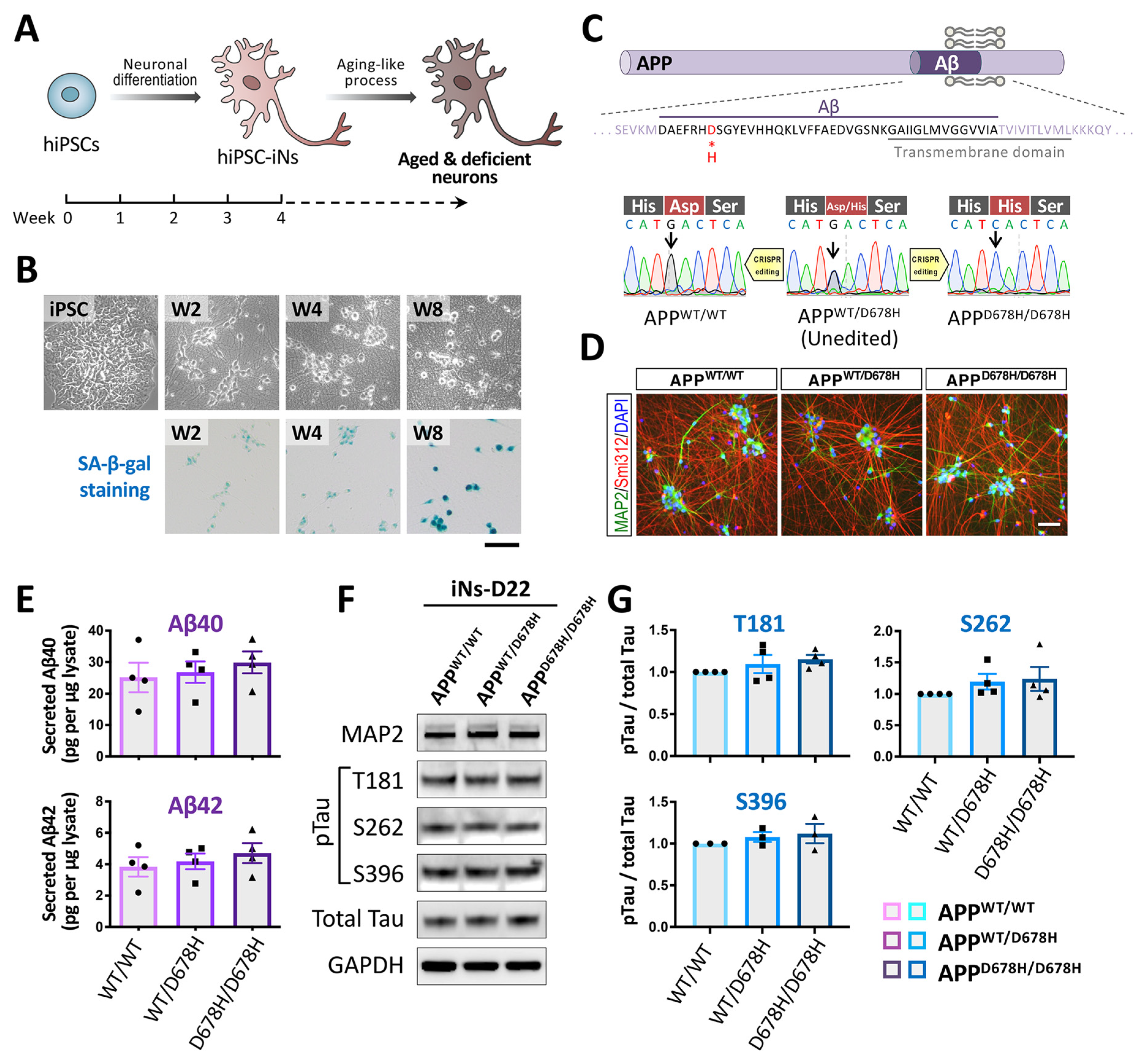
3.4.1. Neuronal Differentiation of hiPSCs
3.4.2. Approaches for Carrying out an In Vitro Aging-like Process Using hiPSC-Derived Neurons
4. Application of Genome Editing Technology to hiPSC-Derived Neurons in Order to Study Cellular Senescence and Carry out Drug Discovery
4.1. The CRISPR/Cas9-Mediated Genome Editing and Gene Expression in hiPSCs
4.2. The Inducible CRISPR/Cas9 Systems in hiPSC-iNs
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P. Investigational drugs and nutrients for human longevity. Recent clinical trials registered in ClinicalTrials.gov and clinicaltrialsregister.eu. Expert Opin. Investig. Drugs 2021, 30, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zuo, X.; Ouyang, Z.; Qiao, P.; Wang, F. A systematic review of antiaging effects of 23 traditional chinese medicines. Evid.-Based Complement. Altern. Med. 2021, 2021, 1–13. [Google Scholar] [CrossRef]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front. Endocrinol. 2021, 12, 718942. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Leung, C.; Jia, Z. Mouse genetic models of human brain disorders. Front. Genet. 2016, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Buffalo, E.A.; Movshon, J.A.; Wurtz, R.H. From basic brain research to treating human brain disorders. Proc. Natl. Acad. Sci. USA 2019, 116, 26167–26172. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. 2021, 7, e12179. [Google Scholar]
- Si, Z.; Sun, L.; Wang, X. Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomed. Pharmacother. 2021, 137, 111327. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Rueda, N. Cellular senescence in neurodegenerative diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Wan, T.; Miwa, S. Senescence in post-mitotic cells: A driver of aging? Antioxid. Redox Signal. 2021, 34, 308–323. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular senescence in brain aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef]
- Sharma, S. Age-related nonhomologous end joining activity in rat neurons. Brain Res. Bull. 2007, 73, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. Somatic mutations and aging: A re-evaluation. Mutat. Res. Mol. Mech. Mutagen. 2000, 447, 117–135. [Google Scholar] [CrossRef]
- Lombard, D.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Shiloh, Y.; Kastan, M.B. ATM: Genome stability, neuronal development, and cancer cross paths. Adv. Cancer Res. 2001, 83, 209–254. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; Van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nat. Cell Biol. 2006, 444, 1038–1043. [Google Scholar] [CrossRef]
- Jaarsma, D.; Van Der Pluijm, I.; De Waard, M.C.; Haasdijk, E.D.; Brandt, R.; Vermeij, M.; Rijksen, Y.; Maas, A.; Van Steeg, H.; Hoeijmakers, J.H.J.; et al. Age-related neuronal degeneration: Complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology. PLoS Genet. 2011, 7, e1002405. [Google Scholar] [CrossRef]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhan, M.; Duan, W.; Prabhu, V.; Brenneman, R.; Wood, W.; Firman, J.; Li, H.; Zhang, P.; Ibe, C.; et al. Gene expression atlas of the mouse central nervous system: Impact and interactions of age, energy intake and gender. Genome Biol. 2007, 8, R234. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, L.; De Filippis, L.; Lecis, D.; Vescovi, A.; Delia, D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ. 2009, 16, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Liu, Z.; Peng, L.; Tang, X.; Meng, F.; Ao, Y.; Zhou, M.; Wang, M.; Cao, X.; Qin, B.; et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 2018, 7, e34836. [Google Scholar] [CrossRef]
- Aguado, J.; Chaggar, H.K.; Gómez-Inclán, C.; Shaker, M.R.; Leeson, H.C.; Mackay-Sim, A.; Wolvetang, E.J. Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell 2021, 20, e13468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Furukawa, K.; Opresko, P.L.; Xu, X.; Bohr, V.A.; Mattson, M.P. TRF2 dysfunction elicits DNA damage responses associated with senescence in proliferating neural cells and differentiation of neurons. J. Neurochem. 2006, 97, 567–581. [Google Scholar] [CrossRef]
- Cheng, A.; Shin-Ya, K.; Wan, R.; Tang, S.C.; Miura, T.; Tang, H.; Khatri, R.; Gleichman, M.; Ouyang, X.; Liu, N.; et al. Telomere protection mechanisms change during neurogenesis and neuronal maturation: Newly generated neurons are hypersensitive to telomere and DNA damage. J. Neurosci. 2007, 27, 3722–3733. [Google Scholar] [CrossRef]
- Wagner, K.D.; Ying, Y.; Leong, W.; Jiang, J.; Hu, X.; Chen, Y.; Michiels, J.F.; Lu, Y.; Gilson, E.; Wagner, N.; et al. The differential spatiotemporal expression pattern of shelterin genes throughout lifespan. Aging 2017, 9, 1219–1232. [Google Scholar] [CrossRef]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Elkahloun, A.G.; Saavedra, J.M. Candesartan neuroprotection in rat primary neurons negatively correlates with aging and senescence: A transcriptomic analysis. Mol. Neurobiol. 2020, 57, 1656–1673. [Google Scholar] [CrossRef]
- Guo, Z.; Tian, Y.; Guo, Y.; Li, B.; Liu, X.; Xie, K.; Song, Y.; Wang, D. RAD6B plays a critical role in neuronal DNA damage response to resist neurodegeneration. Front. Cell. Neurosci. 2019, 13, 392. [Google Scholar] [CrossRef]
- Vanyushin, B.F.; E Nemirovsky, L.; Klimenko, V.V.; Vasiliev, V.K.; Belozersky, A.N. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontology 1973, 19, 138–152. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Prasad, R.; Jho, E.H. A concise review of human brain methylome during aging and neurodegenerative diseases. BMB Rep. 2019, 52, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W. The link between epigenetic clocks for aging and senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A. Cortical DNA methylation maintains remote memory. Nat. Neurosci. 2010, 13, 664–666. [Google Scholar] [CrossRef]
- Ianov, L.; Riva, A.; Kumar, A.; Foster, T.C. DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age-related cognitive impairment. Front. Aging Neurosci. 2017, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Khare, T.; Pai, S.; Koncevicius, K.; Pal, M.; Kriukiene, E.; Liutkeviciute, Z.; Irimia, M.; Jia, P.; Ptak, C.; Xia, M.; et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 2012, 19, 1037–1043. [Google Scholar] [CrossRef]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (Cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; van Groen, T.; Kadish, I.; Li, Y.; Wang, D.; James, S.R.; Karpf, A.R.; Tollefsbol, T.O. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenet. 2011, 2, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. DNA methylation and cognitive aging. Oncotarget 2015, 6, 13922–13932. [Google Scholar] [CrossRef]
- Haberman, R.; Quigley, C.; Gallagher, M. Characterization of CpG island DNA methylation of impairment-related genes in a rat model of cognitive aging. Epigenetics 2012, 7, 1008–1019. [Google Scholar] [CrossRef]
- Penner, M.; Roth, T.; Chawla, M.; Hoang, L.; Roth, E.; Lubin, F.; Sweatt, J.; Worley, P.; Barnes, C. Age-related changes in Arc transcription and DNA methylation within the hippocampus. Neurobiol. Aging 2011, 32, 2198–2210. [Google Scholar] [CrossRef]
- Penner, M.; Parrish, R.R.; Hoang, L.; Roth, T.; Lubin, F.; Barnes, C. Age-related changes inEgr1 transcription and DNA methylation within the hippocampus. Hippocampus 2016, 26, 1008–1020. [Google Scholar] [CrossRef]
- Hernandez, D.G.; Nalls, M.A.; Gibbs, J.R.; Arepalli, S.; van der Brug, M.; Chong, S.; Moore, M.; Longo, D.L.; Cookson, M.R.; Traynor, B.J.; et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011, 20, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Masser, D.R.; Logan, S.; Wronowski, B.; Mangold, C.A.; Clark, N.; Otalora, L.; Unnikrishnan, A.; Ford, M.M.; Giles, C.B.; et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenet. Chromatin 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, Z.; Ren, Y.; Wu, X.; Liu, Y.; Wang, T.; Li, Y.; Cong, Y.; Guo, Y. Neuroprotective effects of salidroside on ageing hippocampal neurons and naturally ageing mice via the PI3K /Akt/ TERT pathway. Phytother. Res. 2021, 35, 5767–5780. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- So, A.Y.; Jung, J.W.; Lee, S.; Kim, H.S.; Kang, K.S. DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through MicroRNAs. PLoS ONE 2011, 6, e19503. [Google Scholar] [CrossRef]
- Zheng, Q.H.; Ma, L.W.; Zhu, W.G.; Zhang, Z.Y.; Tong, T.J. p21Waf1/Cip1 plays a critical role in modulating senescence through changes of DNA methylation. J. Cell. Biochem. 2006, 98, 1230–1248. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, D.; Zhao, H.; Yang, L. NRSF: An angel or a devil in neurogenesis and neurological diseases. J. Mol. Neurosci. 2014, 56, 131–144. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef]
- Song, Z.; Shah, S.Z.; Yang, W.; Dong, H.; Yang, L.; Zhou, X.; Zhao, D. Downregulation of the repressor element 1-silencing transcription factor (rest) is associated with Akt-mTOR and wnt-beta-catenin signaling in prion diseases models. Front. Mol. Neurosci. 2017, 10, 128. [Google Scholar] [CrossRef]
- Xiao, N.M.; Zhang, Y.M.; Zheng, Q.; Gu, J. Klotho is a serum factor related to human aging. Chin. Med. J. 2004, 117, 742–747. [Google Scholar] [PubMed]
- King, G.D.; Rosene, D.L.; Abraham, C.R. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. AGE 2012, 34, 1405–1419. [Google Scholar] [CrossRef]
- Shaker, M.R.; Aguado, J.; Chaggar, H.K.; Wolvetang, E.J. Klotho inhibits neuronal senescence in human brain organoids. Npj Aging Mech. Dis. 2021, 7, 1–12. [Google Scholar] [CrossRef]
- Chen, K.; Sun, Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell 2018, 17, e12762. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Heyn, H.; Vidal, E.; Moran, S.; Sayols, S.; Delgado-Morales, R.; Schultz, M.D.; Ansoleaga, B.; Esparcia, P.G.; Espinal, M.P.; et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl. Psychiatry 2016, 6, e718. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Jin, Y.; Allen, E.G.; Jin, P. Diverse and dynamic DNA modifications in brain and diseases. Hum. Mol. Genet. 2019, 28, R241–R253. [Google Scholar] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Singh, P.P.; Mahmoudi, S.; Harel, I.; Casey, K.M.; Dulken, B.W.; Kundaje, A.; Brunet, A. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019, 29, 697–709. [Google Scholar] [CrossRef]
- Barter, J.D.; Foster, T.C. Aging in the brain: New roles of epigenetics in cognitive decline. Neuroscientist 2018, 24, 516–525. [Google Scholar] [CrossRef]
- Benayoun, B.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Qian, H.; Ertl, R.; Astle, C.M.; Wang, G.G.; Harrison, D.E.; Xu, X. Histone modifications change with age, dietary restriction and rapamycin treatment in mouse brain. Oncotarget 2015, 6, 15882–15890. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Feller, C.; Ladurner, A.; Imhof, A. The metabolic impact on histone acetylation and transcription in ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- Gadecka, A.; Bielak-Zmijewska, A. Slowing down ageing: The role of nutrients and microbiota in modulation of the epigenome. Nutrients 2019, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic mechanisms of longevity and aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- McCauley, B.S.; Dang, W. Histone methylation and aging: Lessons learned from model systems. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014, 1839, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Sen, P. The senescent cell epigenome. Aging 2018, 10, 3590–3609. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Prieto, G.A.; Petrosyan, A.; Loertscher, B.M.; Dieskau, A.P.; Overman, L.E.; Cotman, C.W. H3K9me3 inhibition improves memory, promotes spine formation, and increases BDNF levels in the aged hippocampus. J. Neurosci. 2016, 36, 3611–3622. [Google Scholar] [CrossRef]
- Wang, C.M.; Na Tsai, S.; Yew, T.W.; Kwan, Y.W.; Ngai, S.M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 2009, 11, 87–102. [Google Scholar] [CrossRef]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic regulation of cellular senescence and aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef]
- Jęśko, H.; Wencel, P.; Strosznajder, R.; Strosznajder, J. Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- Chouliaras, L.; Hove, D.V.D.; Kenis, G.; Draanen, M.; Hof, P.; van Os, J.; Steinbusch, H.; Schmitz, C.; Rutten, B. Histone deacetylase 2 in the mouse hippocampus: Attenuation of age-related increase by caloric restriction. Curr. Alzheimer Res. 2013, 10, 868–876. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, P.; Thakur, M.K. Histone deacetylase 2 inhibition attenuates downregulation of hippocampal plasticity gene expression during aging. Mol. Neurobiol. 2017, 55, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Martín-Segura, A.; Baliyan, S.; Ahmed, T.; Balschun, D.; Venero, C.; Martin, M.G.; Dotti, C.G. Aging triggers a repressive chromatin state at BDNF promoters in hippocampal neurons. Cell Rep. 2016, 16, 2889–2900. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than Just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 1–30. [Google Scholar] [CrossRef]
- Houtkooper, R.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.W.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nat. Cell Biol. 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Hoppel, C.L. Oxidative phosphorylation and aging. Ageing Res. Rev. 2006, 5, 402–433. [Google Scholar] [CrossRef]
- Takeda, K.; Ichijo, H. Neuronal p38 MAPK signalling: An emerging regulator of cell fate and function in the nervous system. Genes Cells 2002, 7, 1099–1111. [Google Scholar] [CrossRef]
- Jezek, P.; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Madamanchi, N.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, M.; Larsson, N.-G. Mitochondrial dysfunction in mammalian ageing. Biol. IGF-1 Its Interact. Insul. Health Malig. States 2007, 287, 197–213. [Google Scholar] [CrossRef]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.; Sefiani, A.; Horvat, D.; Huntington, T.; Lei, Y.; West, A.; Geoffroy, C. Age-dependent decline in neuron growth potential and mitochondria functions in cortical neurons. Cells 2021, 10, 1625. [Google Scholar] [CrossRef]
- Granzotto, A.; Bomba, M.; Castelli, V.; Navarra, R.; Massetti, N.; D’Aurora, M.; Onofrj, M.; Cicalini, I.; DEL Boccio, P.; Gatta, V.; et al. Inhibition of de novo ceramide biosynthesis affects aging phenotype in an in vitro model of neuronal senescence. Aging 2019, 11, 6336–6357. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.D.; He, L.; Tu, C.; Guo, X.A.; Yu, S.; Liu, K.; Gong, S. Mitochondrial DNA 3,860-bp deletion increases with aging in the auditory nervous system of C57BL/6J mice. ORL 2019, 81, 92–100. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD+ metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.H.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in aging: Molecular mechanisms and translational implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Alcain, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Chen, C.; Si, E.; Cai, Y.; Wang, J.; Huang, W.; Li, D.; Wang, Y.; Chen, X. Mitochondrial effects of PGC-1alpha silencing in MPP+ treated human SH-SY5Y neuroblastoma cells. Front. Mol. Neurosci. 2017, 10, 164. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Xia, D.Y.; Huang, X.; Bi, C.F.; Mao, L.L.; Peng, L.J.; Qian, H.R. PGC-1alpha or FNDC5 is involved in modulating the effects of Abeta1-42 oligomers on suppressing the expression of BDNF, a beneficial factor for inhibiting neuronal apoptosis, abeta deposition and cognitive decline of APP/PS1 Tg mice. Front. Aging Neurosci. 2017, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Masciopinto, F.; Silvestri, E.; Del Viscovo, A.; Lattanzio, R.; La Sorda, R.; Ciavardelli, D.; Goglia, F.; Piantelli, M.; Canzoniero, L.M.T.; et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010, 1, e91. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Grady, J.P.; Turnbull, D.M. Respiratory chain deficiency in aged spinal motor neurons. Neurobiol. Aging 2014, 35, 2230–2238. [Google Scholar] [CrossRef]
- Course, M.M.; Wang, X. Transporting mitochondria in neurons. F1000Research 2016, 5, 1735. [Google Scholar] [CrossRef]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, X.; Zheng, Y.; Chen, Z. Regulation of mitophagy in ischemic brain injury. Neurosci. Bull. 2015, 31, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Wang, B.; Huang, M.; Shang, D.; Yan, X.; Zhao, B.; Zhang, X. Mitochondrial behavior in axon degeneration and regeneration. Front. Aging Neurosci. 2021, 13, 650038. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Feng, Z.; Hanson, R.W.; Berger, N.A.; Trubitsyn, A. Reprogramming of energy metabolism as a driver of aging. Oncotarget 2016, 7, 15410–15420. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.J.; Pamplona, R.; Buffenstein, R.; Buttemer, W. Life and death: Metabolic rate, membrane composition, and life span of animals. Physiol. Rev. 2007, 87, 1175–1213. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Arias, E.; Kwon, H.J.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; E Pessin, J.; Singh, R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012, 13, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113 Pt 20, 3613–3622. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, H.; Rubinsztein, D.C.; Lee, J.E. Autophagy, cellular aging and age-related human diseases. Exp. Neurobiol. 2019, 28, 643–657. [Google Scholar] [CrossRef]
- Anderson, R.M.; Weindruch, R. The caloric restriction paradigm: Implications for healthy human aging. Am. J. Hum. Biol. 2012, 24, 101–106. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Yu, B.P.; Chung, H.Y. Stress resistance by caloric restriction for longevity. Ann. N. Y. Acad. Sci. 2006, 928, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Rusli, F.; Lute, C.; Boekschoten, M.V.; Van Dijk, M.; Van Norren, K.; Menke, A.L.; Muller, M.; Steegenga, W.T. Intermittent calorie restriction largely counteracts the adverse health effects of a moderate-fat diet in aging C57BL/6J mice. Mol. Nutr. Food Res. 2017, 61, 1600677. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.M. Effect of restricted feeding upon aging and chronic diseases in rats and dogs. Am. J. Public Health Nations Health 1947, 37, 521–528. [Google Scholar] [CrossRef]
- Ross, M.H. Length of life and nutrition in the rat. J. Nutr. 1961, 75, 197–210. [Google Scholar] [CrossRef]
- Ross, M.H. Length of life and caloric intake. Am. J. Clin. Nutr. 1972, 25, 834–838. [Google Scholar] [CrossRef]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Anderson, R.M.; Weindruch, R. Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol. Metab. 2010, 21, 134–141. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 2009, 20, 325–331. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef]
- Pan, D.A.; Hardie, D.G. A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. Biochem. J. 2002, 367 Pt 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Ruiz, Y.R.; Otten, E.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Shi, L.; Zhong, D.; Li, Y.; Li, J.; Jin, R. AMPK: Potential therapeutic target for Alzheimer’s disease. Curr. Protein Pept. Sci. 2020, 21, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Mugume, Y.; Kazibwe, Z.; Bassham, D. Target of rapamycin in control of autophagy: Puppet master and signal integrator. Int. J. Mol. Sci. 2020, 21, 8259. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Arriola Apelo, S.I.; Lamming, D.W. Rapamycin: An InhibiTOR of aging emerges from the soil of easter island. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2016, 71, 841–849. [Google Scholar] [CrossRef]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage alzheimer-type tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Malaquin, N.; Martinez, A.; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef] [PubMed]
- Pitler, T.A.; Landfield, P.W. Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Res. 1990, 508, 1–6. [Google Scholar] [CrossRef]
- Thibault, O.; Landfield, P.W. Increase in single L-type calcium channels in hippocampal neurons during aging. Science 1996, 272, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; O’Connor, J.; Moynagh, P. Dissection of tumor-necrosis factor-α inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early—but not late—phase LTP. Neuroscience 2004, 124, 319–326. [Google Scholar] [CrossRef]
- Maglione, M.; Kochlamazashvili, G.; Eisenberg, T.; Rácz, B.; Michael, E.; Toppe, D.; Stumpf, A.; Wirth, A.; Zeug, A.; Müller, F.E.; et al. Spermidine protects from age-related synaptic alterations at hippocampal mossy fiber-CA3 synapses. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- De Brabander, J.M.; Kramers, R.J.; Uylings, H.B. Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. Eur. J. Neurosci. 1998, 10, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Tsuzuki, M.; Keino, H.; Satoh, M.; Chiba, Y.; Saitoh, Y.; Hosokawa, M. Apical vulnerability to dendritic retraction in prefrontal neurones of ageing SAMP10 mouse: A model of cerebral degeneration. Neuropathol. Appl. Neurobiol. 2006, 32, 1–14. [Google Scholar] [CrossRef]
- Dickstein, D.; Weaver, C.; Luebke, J.; Hof, P. Dendritic spine changes associated with normal aging. Neuroscience 2013, 251, 21–32. [Google Scholar] [CrossRef]
- Adams, M.; Donohue, H.; Linville, M.; Iversen, E.; Newton, I.; Brunso-Bechtold, J. Age-related synapse loss in hippocampal CA3 is not reversed by caloric restriction. Neuroscience 2010, 171, 373–382. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, Y. L-histidine and L-carnosine exert anti-brain aging effects in D-galactose-induced aged neuronal cells. Nutr. Res. Pract. 2020, 14, 188–202. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.S.; Kim, E.; Kim, Y.; Kim, Y. Curcumin and hesperetin attenuate D-galactose-induced brain senescence in vitro and in vivo. Nutr. Res. Pract. 2020, 14, 438–452. [Google Scholar] [CrossRef]
- Ho, D.H.; Nam, D.; Seo, M.K.; Park, S.W.; Seol, W.; Son, I. LRRK2 kinase inhibitor rejuvenates oxidative stress-induced cellular senescence in neuronal cells. Oxidative Med. Cell. Longev. 2021, 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; He, Y.; Wang, L.; Mo, C.; Zhang, J.; Zhang, W.; Li, J.; Liao, Z.; Tang, X.; Xiao, H. D-galactose induces necroptotic cell death in neuroblastoma cell lines. J. Cell. Biochem. 2011, 112, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.L.; Lin, S.-B.; Yu, Y.L.; Chien, M.-H.; Su, K.J.; Lin, C.-J.; Way, T.D.; Yiang, G.T.; Lin, C.C.; Chan, D.C.; et al. Isochaihulactone protects PC12 cell against H2O2 induced oxidative stress and exerts the potent anti-aging effects in D-galactose aging mouse model. Acta Pharmacol. Sin. 2010, 31, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Jembrek, M.J.; Vuković, L.; Puhović, J.; Erhardt, J.; Oršolić, N. Neuroprotective effect of quercetin against hydrogen peroxide-induced oxidative injury in P19 neurons. J. Mol. Neurosci. 2012, 47, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Bhanu, M.U.; Mandraju, R.K.; Bhaskar, C.; Kondapi, A.K. Cultured cerebellar granule neurons as an in vitro aging model: Topoisomerase IIbeta as an additional biomarker in DNA repair and aging. Toxicol. Vitr. 2010, 24, 1935–1945. [Google Scholar] [CrossRef]
- Bigagli, E.; Luceri, C.; Scartabelli, T.; Dolara, P.; Casamenti, F.; Pellegrini-Giampietro, D.E.; Giovannelli, L. Long-term neuroglial cocultures as a brain aging model: Hallmarks of senescence, MicroRNA expression profiles, and comparison with in vivo models. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2015, 71, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Cheng, S.; Huang, F.; Fan, W.; Chen, Y.; Shi, H.; He, H. Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med. Sci. Monit. 2011, 17, BR91–BR96. [Google Scholar] [CrossRef]
- Geng, Y.-Q.; Guan, J.-T.; Xu, X.-H.; Fu, Y.-C. Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem. Biophys. Res. Commun. 2010, 396, 866–869. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ishikawa, F. Proteostasis failure and cellular senescence in long-term cultured postmitotic rat neurons. Aging Cell 2019, 19, e13071. [Google Scholar] [CrossRef]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, A.; Stella, A.M.G.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999, 13, 2225–2234. [Google Scholar] [CrossRef]
- Copani, A.; Melchiorri, D.; Caricasole, A.; Martini, F.; Sale, P.; Carnevale, R.; Gradini, R.; Sortino, M.A.; Lenti, L.; De Maria, R.; et al. β-amyloid-induced synthesis of the ganglioside GD3 is a requisite for cell cycle reactivation and apoptosis in neurons. J. Neurosci. 2002, 22, 3963–3968. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Wersto, R.P.; Cardozo-Pelaez, F.; Smilenov, L.; Chan, S.L.; Chrest, F.J.; Emokpae, R., Jr.; Gorospe, M.; Mattson, M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 2004, 41, 549–561. [Google Scholar] [CrossRef]
- Copani, A.; Hoozemans, J.J.; Caraci, F.; Calafiore, M.; Van Haastert, E.S.; Veerhuis, R.; Rozemuller, A.J.; Aronica, E.; Sortino, M.A.; Nicoletti, F. DNA polymerase-beta is expressed early in neurons of Alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with beta-amyloid. J. Neurosci. 2006, 26, 10949–10957. [Google Scholar] [CrossRef]
- Majd, S.; Zarifkar, A.; Rastegar, K.; Takhshid, M.A. Different fibrillar Abeta 1-42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res. 2008, 1218, 224–229. [Google Scholar] [CrossRef]
- Caraci, F.; Battaglia, G.; Busceti, C.; Biagioni, F.; Mastroiacovo, F.; Bosco, P.; Drago, F.; Nicoletti, F.; Sortino, M.A.; Copani, A. TGF-beta 1 protects against Abeta-neurotoxicity via the phosphatidylinositol-3-kinase pathway. Neurobiol. Dis. 2008, 30, 234–242. [Google Scholar] [CrossRef]
- Modi, P.K.; Komaravelli, N.; Singh, N.; Sharma, P. Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol. Biol. Cell 2012, 23, 3722–3730. [Google Scholar] [CrossRef]
- Merlo, S.; Basile, L.; Giuffrida, M.L.; Sortino, M.A.; Guccione, S.; Copani, A. Identification of 5-methoxyflavone as a novel DNA polymerase-beta inhibitor and neuroprotective agent against beta-amyloid toxicity. J. Nat. Prod. 2015, 78, 2704–2711. [Google Scholar] [CrossRef]
- Zhao, X.; Fang, J.; Li, S.; Gaur, U.; Xing, X.; Wang, H.; Zheng, W. Artemisinin attenuated hydrogen peroxide (H2O2)-induced oxidative injury in SH-SY5Y and hippocampal neurons via the activation of AMPK pathway. Int. J. Mol. Sci. 2019, 20, 2680. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 2016, 5, e18648. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.; Blithikioti, C.; Ku, M.; et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, M.L.; Zang, T.; Zhang, C.L. Direct reprogramming rather than iPSC-based reprogramming maintains aging hallmarks in human motor neurons. Front. Mol. Neurosci. 2017, 10, 359. [Google Scholar] [CrossRef]
- Dong, C.M.; Wang, X.L.; Wang, G.M.; Zhang, W.J.; Zhu, L.; Gao, S.; Yang, D.J.; Qin, Y.; Liang, Q.J.; Chen, Y.L.; et al. Erratum: A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis. 2017, 8, e3041. [Google Scholar] [CrossRef]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Vera, E.; Bosco, N.; Studer, L. Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep. 2016, 17, 1184–1192. [Google Scholar] [CrossRef]
- McBurney, M.W.; Rogers, B.J. Isolation of male embryonal carcinoma cells and their chromosome replication patterns. Dev. Biol. 1982, 89, 503–508. [Google Scholar] [CrossRef]
- Augusti-Tocco, G.; Sato, G. Establishment of functional clonal lines of neurons from mouse neuroblastoma. Proc. Natl. Acad. Sci. USA 1969, 64, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978, 38 Pt 1, 3751–3757. [Google Scholar] [PubMed]
- Jones-Villeneuve, E.M.V.; McBurney, M.W.; Rogers, K.A.; Kalnins, V.I. Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J. Cell Biol. 1982, 94, 253–262. [Google Scholar] [CrossRef]
- Breen, K.C.; Anderton, B.H. Cyclic AMP-dependent expression of the heavy neurofilament (NF-H) polypeptide in differentiating neuroblastoma cells. Mol. Brain Res. 1990, 7, 161–165. [Google Scholar] [CrossRef]
- Breen, K.C.; Anderton, B.H. Temporal expression of neurofilament polypeptides in differentiating neuroblastoma cells. NeuroReport 1991, 2, 21–24. [Google Scholar] [CrossRef]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef]
- Morton, A.J.; Buss, T.N. Accelerated differentiation in response to retinoic acid after retrovirally mediated gene transfer of GAP-43 into mouse neuroblastoma cells. Eur. J. Neurosci. 1992, 4, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Blanco, V.; Camelo, J.L.; Carri, N.G. Growth inhibition, morphological differentiation and stimulation of survival in neuronal cell type (Neuro-2a) TREATED with trophic molecules. Cell Biol. Int. 2001, 25, 909–917. [Google Scholar] [CrossRef]
- van Esbroeck, A.; Kantae, V.; Di, X.; van der Wel, T.; den Dulk, H.; Stevens, A.F.; Singh, S.; Bakker, A.T.; Florea, B.I.; Stella, N.; et al. Identification of alpha, beta-hydrolase domain containing protein 6 as a diacylglycerol lipase in neuro-2a Cells. Front. Mol. Neurosci. 2019, 12, 286. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.Y.; Lin, Y.C.; Chang, C.L.; Lu, H.T.; Chin, C.H.; Hsu, T.T.; Chu, D.; Sun, S.H. Functional decreases in P2X7 receptors are associated with retinoic acid-induced neuronal differentiation of Neuro-2a neuroblastoma cells. Cell. Signal. 2009, 21, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Loomis, P.; Yoon, M.; Goldman, R.D. Rapid transport of neural intermediate filament protein. J. Cell Sci. 2003, 116 Pt 11, 2345–2359. [Google Scholar] [CrossRef]
- Amino, S.; Itakura, M.; Ohnishi, H.; Tsujimura, J.; Koizumi, S.; Takei, N.; Takahashi, M. Nerve growth factor enhances neurotransmitter release from PC12 cells by increasing Ca2+-responsible secretory vesicles through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J. Biochem. 2002, 131, 887–894. [Google Scholar] [CrossRef]
- Chu, E.; Chu, J.; Chu, T.-C.; Socci, R.R. 7-OH-DPAT-induced inhibition of norepinephrine release in PC12 cells. Pharmacology 2004, 70, 130–139. [Google Scholar] [CrossRef] [PubMed]
- A Greene, L.; Rukenstein, A. Regulation of acetylcholinesterase activity by nerve growth factor. Role of transcription and dissociation from effects on proliferation and neurite outgrowth. J. Biol. Chem. 1981, 256, 6363–6367. [Google Scholar] [CrossRef]
- Liu, H.; Nowak, R.; Chao, W.; Bloch, K.D. Nerve growth factor induces anti-apoptotic heme oxygenase-1 in rat pheochromocytoma PC12 cells. J. Neurochem. 2003, 86, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, S.P.; Borwege, S.; Millan, M.J.; Joyce, J.N. Involvement of dopamine D2/D3 receptors and BDNF in the neuroprotective effects of S32504 and pramipexole against 1-methyl-4-phenylpyridinium in terminally differentiated SH-SY5Y cells. Exp. Neurol. 2004, 190, 157–170. [Google Scholar] [CrossRef]
- Bellucci, A.; Collo, G.; Sarnico, I.; Battistin, L.; Missale, C.; Spano, P. Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2D3receptor activation. J. Neurochem. 2008, 106, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Kawato, Y.; Osakada, F.; Izumi, Y.; Katsuki, H.; Nakagawa, T.; Kaneko, S.; Niidome, T.; Takada-Takatori, Y.; Akaike, A. Dibutyryl cyclic AMP induces differentiation of human neuroblastoma SH-SY5Y cells into a noradrenergic phenotype. Neurosci. Lett. 2008, 443, 199–203. [Google Scholar] [CrossRef]
- Cernaianu, G.; Brandmaier, P.; Scholz, G.; Ackermann, O.P.; Alt, R.; Rothe, K.; Cross, M.; Witzigmann, H.; Tröbs, R.-B. All-trans retinoic acid arrests neuroblastoma cells in a dormant state. Subsequent nerve growth factor/brain-derived neurotrophic factor treatment adds modest benefit. J. Pediatr. Surg. 2008, 43, 1284–1294. [Google Scholar] [CrossRef]
- Giménez-Cassina, A.; Lim, F.; Diaz-Nido, J. Differentiation of a human neuroblastoma into neuron-like cells increases their susceptibility to transduction by herpesviral vectors. J. Neurosci. Res. 2006, 84, 755–767. [Google Scholar] [CrossRef]
- Pahlman, S.; Ruusala, A.I.; Abrahamsson, L.; Mattsson, M.E.; Esscher, T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell Differ. 1984, 14, 135–144. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Lau, W.K.W.; Yu, M.S.; Lai, C.S.W.; Yeung, S.C.; So, K.F.; Chang, R.C.-C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. NeuroToxicology 2009, 30, 127–135. [Google Scholar] [CrossRef]
- Paik, S.; Somvanshi, R.; Kumar, U. Somatostatin-mediated changes in microtubule-associated proteins and retinoic acid–induced neurite outgrowth in SH-SY5Y cells. J. Mol. Neurosci. 2019, 68, 120–134. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Mao, Y.; Li, B.; Zhu, Y.; Zhang, S.; Wang, S.; Jiang, Y.; Xu, N.; Xie, Y.; et al. NEAT1 regulates microtubule stabilization via FZD3/GSK3beta/P-tau pathway in SH-SY5Y cells and APP/PS1 mice. Aging 2020, 12, 23233–23250. [Google Scholar]
- Shen, K.; Liu, X.; Chen, D.; Chang, J.; Zhang, Y.; Kou, X. Voluntary wheel-running exercise attenuates brain aging of rats through activating miR-130a-mediated autophagy. Brain Res. Bull. 2021, 172, 203–211. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W. Cell death of dopamine neurons in aging and Parkinson’s disease. Mech. Ageing Dev. 1999, 111, 175–188. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Dragicevic, N.B.; Deng, J.H.; Bai, Y.; Dimayuga, E.; Ding, Q.; Chen, Q.; Bruce-Keller, A.J.; Keller, J. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J. Biol. Chem. 2004, 279, 20699–20707. [Google Scholar] [CrossRef]
- Araki, W.; Minegishi, S.; Motoki, K.; Kume, H.; Hohjoh, H.; Araki, Y.M.; Tamaoka, A. Disease-associated mutations of TDP-43 promote turnover of the protein through the proteasomal pathway. Mol. Neurobiol. 2014, 50, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; McGeer, P.L. Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol. Aging 2002, 23, 787–794. [Google Scholar] [CrossRef]
- Gao, K.; Liu, M.; Ding, Y.; Yao, M.; Zhu, Y.; Zhao, J.; Cheng, L.; Bai, J.; Wang, F.; Cao, J.; et al. A phenolic amide (LyA) isolated from the fruits of Lycium barbarum protects against cerebral ischemia-reperfusion injury via PKCepsilon/Nrf2/HO-1 pathway. Aging 2019, 11, 12361–12374. [Google Scholar] [CrossRef]
- Sedighi, M.; Baluchnejadmojarad, T.; Afshin-Majd, S.; Amiri, M.; Aminzade, M.; Roghani, M. Anti-aging klotho protects SH-SY5Y cells against amyloid β1–42 neurotoxicity: Involvement of Wnt1/pCREB/Nrf2/HO-1 signaling. J. Mol. Neurosci. 2021, 71, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Jeong, K.; Kim, K.; Lee, Y.-S.; Jeong, S.; Kim, S.S.; Yoon, K.-S.; Ha, J.; Kang, I.; Choe, W. Cyclophilin B protects SH-SY5Y human neuroblastoma cells against MPP+-induced neurotoxicity via JNK pathway. Biochem. Biophys. Res. Commun. 2016, 478, 1396–1402. [Google Scholar] [CrossRef]
- Buttiglione, M.; Vitiello, F.; Sardella, E.; Petrone, L.; Nardulli, M.; Favia, P.; D’Agostino, R.; Gristina, R. Behaviour of SH-SY5Y neuroblastoma cell line grown in different media and on different chemically modified substrates. Biomaterials 2007, 28, 2932–2945. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A primary neural cell culture model to study neuron, astrocyte, and microglia interactions in neuroinflammation. J. Neuroinflamm. 2020, 17, 1–16. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lee, Y.I. Differential expressions of synaptogenic markers between primary cultured cortical and hippocampal neurons. Exp. Neurobiol. 2012, 21, 61–67. [Google Scholar] [CrossRef]
- Tönges, L.; Frank, T.; Tatenhorst, L.; Saal, K.A.; Koch, J.C.; Szegő, É.M.; Bähr, M.; Weishaupt, J.H.; Lingor, P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain 2012, 135 Pt 11, 3355–3370. [Google Scholar] [CrossRef]
- Ito, C.; Wakamori, M.; Akaike, N. Dual effect of glycine on isolated rat suprachiasmatic neurons. Am. J. Physiol. Content 1991, 260 Pt 1, 213–218. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Eichert, N.; Robinson, E.C.; Bryant, K.L.; Jbabdi, S.; Jenkinson, M.; Li, L.; Krug, K.; E Watkins, K.; Mars, R.B. Cross-species cortical alignment identifies different types of anatomical reorganization in the primate temporal lobe. Elife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nat. Cell Biol. 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Pang, Z.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nat. Cell Biol. 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Liu, M.L.; Zang, T.; Zou, Y.; Chang, J.C.; Gibson, J.R.; Huber, K.M.; Zhang, C.-L. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 2013, 4, 2183. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nat. Cell Biol. 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Karow, M.; Sánchez, R.; Schichor, C.; Masserdotti, G.; Ortega, F.; Heinrich, C.; Gascón, S.; Khan, M.A.; Lie, D.C.; Dellavalle, A.; et al. Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 2012, 11, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Pang, Z.; Yang, N.; Tsai, M.C.; Qu, K.; Chang, H.Y.; Südhof, T.C.; Wernig, M. Direct lineage conversion of terminally differentiated hepatocytes to functional neurons. Cell Stem Cell 2011, 9, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Niu, W.; Liu, M.-L.; Zou, Y.; Zhang, C.-L. In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chouchane, M.; de Farias, A.R.M.; Moura, D.M.D.S.; Hilscher, M.M.; Schroeder, T.; Leao, R.; Costa, M.R. Lineage reprogramming of astroglial cells from different origins into distinct neuronal subtypes. Stem Cell Rep. 2017, 9, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Ang, C.E.; Chanda, S.; Olmos, V.; Haag, D.; Levinson, D.F.; Südhof, T.C.; Wernig, M. Transdifferentiation of human adult peripheral blood T cells into neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 6470–6475. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Reid, D.; Lau, S.; Kim, Y.; Gage, F.H. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 2018, 52, 271–293. [Google Scholar] [CrossRef]
- Traxler, L.; Edenhofer, F.; Mertens, J. Next-generation disease modeling with direct conversion: A new path to old neurons. FEBS Lett. 2019, 593, 3316–3337. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Wilson, S.I.; Edlund, T. Neural induction: Toward a unifying mechanism. Nat. Neurosci. 2001, 4, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.; Berliner, A.J.; Papanayotou, C.; Sirulnik, A.; Stern, C. Initiation of neural induction by FGF signalling before gastrulation. Nat. Cell Biol. 2000, 406, 74–78. [Google Scholar] [CrossRef]
- Briscoe, J.; Ericson, J. Specification of neuronal fates in the ventral neural tube. Curr. Opin. Neurobiol. 2001, 11, 43–49. [Google Scholar] [CrossRef]
- Lee, K.J.; Jessell, T.M. The specification of dorsal cell fates in the vertebrate central nervous system. Annu. Rev. Neurosci. 1999, 22, 261–294. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.K.; Inokuma, M.S.; Denham, J.; Mujtaba, T.; Chiu, C.-P.; Rao, M.S. Enrichment of neurons and neural precursors from human embryonic stem cells. Exp. Neurol. 2001, 172, 383–397. [Google Scholar] [CrossRef]
- Dhara, S.K.; Hasneen, K.; Machacek, D.W.; Boyd, N.L.; Rao, R.R.; Stice, S.L. Human neural progenitor cells derived from embryonic stem cells in feeder-free cultures. Differentiation 2008, 76, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef]
- Shin, S.; Mitalipova, M.; Noggle, S.; Tibbitts, D.; Venable, A.; Rao, R.; Stice, S.L. Long-term proliferation of human embryonic stem cell-derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 2006, 24, 125–138. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Baharvand, H.; Mehrjardi, N.Z.; Hatami, M.; Kiani, S.; Rao, M.; Haghighi, M.M. Neural differentiation from human embryonic stem cells in a defined adherent culture condition. Int. J. Dev. Biol. 2003, 51, 371–378. [Google Scholar] [CrossRef]
- Erceg, S.; Laínez, S.; Ronaghi, M.; Stojkovic, P.; Pérez-Aragó, M.A.; Moreno-Manzano, V.; Moreno-Palanques, R.; Planells-Cases, R.; Stojkovic, M. Differentiation of human embryonic stem cells to regional specific neural precursors in chemically defined medium conditions. PLoS ONE 2008, 3, e2122. [Google Scholar] [CrossRef]
- Levenberg, S.; Huang, N.; Lavik, E.; Rogers, A.B.; Itskovitz-Eldor, J.; Langer, R. Differentiation of human embryonic stem cells on three-dimensional polymer scaffolds. Proc. Natl. Acad. Sci. USA 2003, 100, 12741–12746. [Google Scholar] [CrossRef]
- Reubinoff, B.; Itsykson, P.; Turetsky, T.; Pera, M.; Reinhartz, E.; Itzik, A.; Ben-Hur, T. Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1134–1140. [Google Scholar] [CrossRef]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The cellular and molecular landscapes of the developing human central nervous system. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Son, E.Y.; Ichida, J.K.; Wainger, B.J.; Toma, J.S.; Rafuse, V.F.; Woolf, C.J.; Eggan, K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 2011, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jiang, H.; Zhong, P.; Yan, Z.; Chen, S.; Feng, J. Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 2015, 21, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nat. Cell Biol. 2011, 476, 224–227. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.; Phatnani, H.; et al. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Yang, N.; Zuchero, J.B.; Ahlenius, H.; Marro, S.; Ng, Y.H.; Vierbuchen, T.; Hawkins, J.S.; Geissler, R.; Barres, B.A.; Wernig, M. Generation of oligodendroglial cells by direct lineage conversion. Nat. Biotechnol. 2013, 31, 434–439. [Google Scholar] [CrossRef]
- Chen, S.W.; Hung, Y.S.; Fuh, J.L.; Chen, N.J.; Chu, Y.S.; Chen, S.C.; Fann, M.J.; Wong, Y.H. Efficient conversion of human induced pluripotent stem cells into microglia by defined transcription factors. Stem Cell Rep. 2021, 16, 1363–1380. [Google Scholar] [CrossRef]
- Studer, L.; Vera, E.; Cornacchia, D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 2015, 16, 591–600. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Xu, L.; Brunet, A. Turning back time with emerging rejuvenation strategies. Nat. Cell Biol. 2019, 21, 32–43. [Google Scholar] [CrossRef]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial rejuvenation after induced pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; DE Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Olova, N.N.; Chandra, T. Cellular reprogramming and epigenetic rejuvenation. Clin. Epigenet. 2021, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.M.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.C.; Fann, M.J.; Tran, T.T.; Chen, S.C.; Devina, T.; Cheng, I.H.J.; Lien, C.C.; Kao, L.S.; Wang, S.J.; Fuh, J.L.; et al. Assessing the therapeutic potential of Graptopetalum paraguayense on Alzheimer’s disease using patient iPSC-derived neurons. Sci. Rep. 2019, 9, 19301. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.-G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Tesson, L.; Usal, C.; Ménoret, S.; Leung, E.; Niles, B.J.; Remy, S.; Santiago, Y.; I Vincent, A.; Meng, X.; Zhang, L.; et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 2011, 29, 695–696. [Google Scholar] [CrossRef]
- Hale, C.R.; Zhao, P.; Olson, S.; Duff, M.O.; Graveley, B.R.; Wells, L.; Terns, R.M.; Terns, M.P. RNA-guided RNA cleavage by a CRISPR RNA-cas protein complex. Cell 2009, 139, 945–956. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Güell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Gilbert, L.; Wang, X.; A Lim, W.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Gilbert, L.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.D.; Pruitt, B.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Abarientos, A.; Hong, J.; Hashemi, S.H.; Yan, R.; Dräger, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1–15. [Google Scholar] [CrossRef]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 2019, 104, 239–255. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, C.; Daley, T.P.; Wang, F.; Cao, W.S.; Bhate, S.; Lin, X.; Still, C.; Liu, H.; Zhao, D.; et al. CRISPR activation screens systematically identify factors that drive neuronal fate and reprogramming. Cell Stem Cell 2018, 23, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, Y.; Sun, S.; Li, W.; Song, M.; Ji, Q.; Wu, Z.; Liu, Z.; Fan, Y.; Liu, F.; et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Kallunki, T.; Barisic, M.; Jäättelä, M.; Liu, B. How to choose the right inducible gene expression system for mammalian studies? Cells 2019, 8, 796. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, L.; Ji, W.; Chen, Z.; Wang, D.; Chen, C.; Tong, H.; Han, Z.; Niu, C.; Chu, M.; et al. Generation of a human induced pluripotent stem cell line with Cas9 driven by Tet-on operator via AAVS1 safe harbor gene-editing. Stem Cell Res. 2020, 49, 102064. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.-M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Meng, X.; Liu, Y.; Song, D.; Jiang, C.; Cai, J. Applications of brain organoids in neurodevelopment and neurological diseases. J. Biomed. Sci. 2021, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line [Ref] | Source | Method of Generating Neurons | Characterization of Neurons | Method of Inducing Cellular Aging (Reagent/Time) | Phenotypes of Cellular Aging | |
|---|---|---|---|---|---|---|
| Immortalized cell lines | SH-SY5Y (ATCC CRL-2266) [187,188,189] | Human neuroblastoma |
| Marker expression:
characterization:
| H2O2/24 h D-galactose/2~3 days | H2O2
|
| Neuro-2a (ATCC CCL-131) [190,191] | Mouse neuroblastoma |
| ||||
| PC-12 (ATCC CRL-1721) [192] | Rat adrenal gland |
| ||||
| P19 (ATCC CRL-1825) [193] | Mouse embryonic carcinoma |
| ||||
| Primary neuronal cultures | Primary neurons [178,189,194,195,196,197,198,199,200,201,202,203,204,205,206,207] | Rodent (mouse or rat) hippocampus or cortex |
| D-galactose/~12 DIV Long term culture/ 6~35 DIV | Long-term culture
| |
| Human ibroblast (hFib)- converted neurons | hFib- converted neurons [208,209,210,211] | Human fibroblasts from donor individuals |
| Preserve age-associated features | Epigenetic modification | |
| Human induced pluripotent stem cell (hiPSC) -derived neurons | hiPSC- derived neurons [212,213,214] | Human iPSCs derived from donor individuals |
|
|
|
| Benefits | Drawbacks | |
|---|---|---|
| Immortalized cell lines |
|
|
| Primary neuronal cultures |
|
|
| Human fibroblast (hFib)-converted neurons |
|
|
| Human induced pluripotent stem cell (hiPSC) -derived neurons |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, C.-C.; Shen, P.-W.; Tzeng, T.-Y.; Kung, H.-J.; Tsai, T.-F.; Wong, Y.-H. Human iPSC-Derived Neurons as A Platform for Deciphering the Mechanisms behind Brain Aging. Biomedicines 2021, 9, 1635. https://doi.org/10.3390/biomedicines9111635
Chao C-C, Shen P-W, Tzeng T-Y, Kung H-J, Tsai T-F, Wong Y-H. Human iPSC-Derived Neurons as A Platform for Deciphering the Mechanisms behind Brain Aging. Biomedicines. 2021; 9(11):1635. https://doi.org/10.3390/biomedicines9111635
Chicago/Turabian StyleChao, Chuan-Chuan, Po-Wen Shen, Tsai-Yu Tzeng, Hsing-Jien Kung, Ting-Fen Tsai, and Yu-Hui Wong. 2021. "Human iPSC-Derived Neurons as A Platform for Deciphering the Mechanisms behind Brain Aging" Biomedicines 9, no. 11: 1635. https://doi.org/10.3390/biomedicines9111635
APA StyleChao, C.-C., Shen, P.-W., Tzeng, T.-Y., Kung, H.-J., Tsai, T.-F., & Wong, Y.-H. (2021). Human iPSC-Derived Neurons as A Platform for Deciphering the Mechanisms behind Brain Aging. Biomedicines, 9(11), 1635. https://doi.org/10.3390/biomedicines9111635