
Targeted Therapies in Cancer: To Be or Not to Be, Selective


Abstract
:
1. Introduction
2. Results
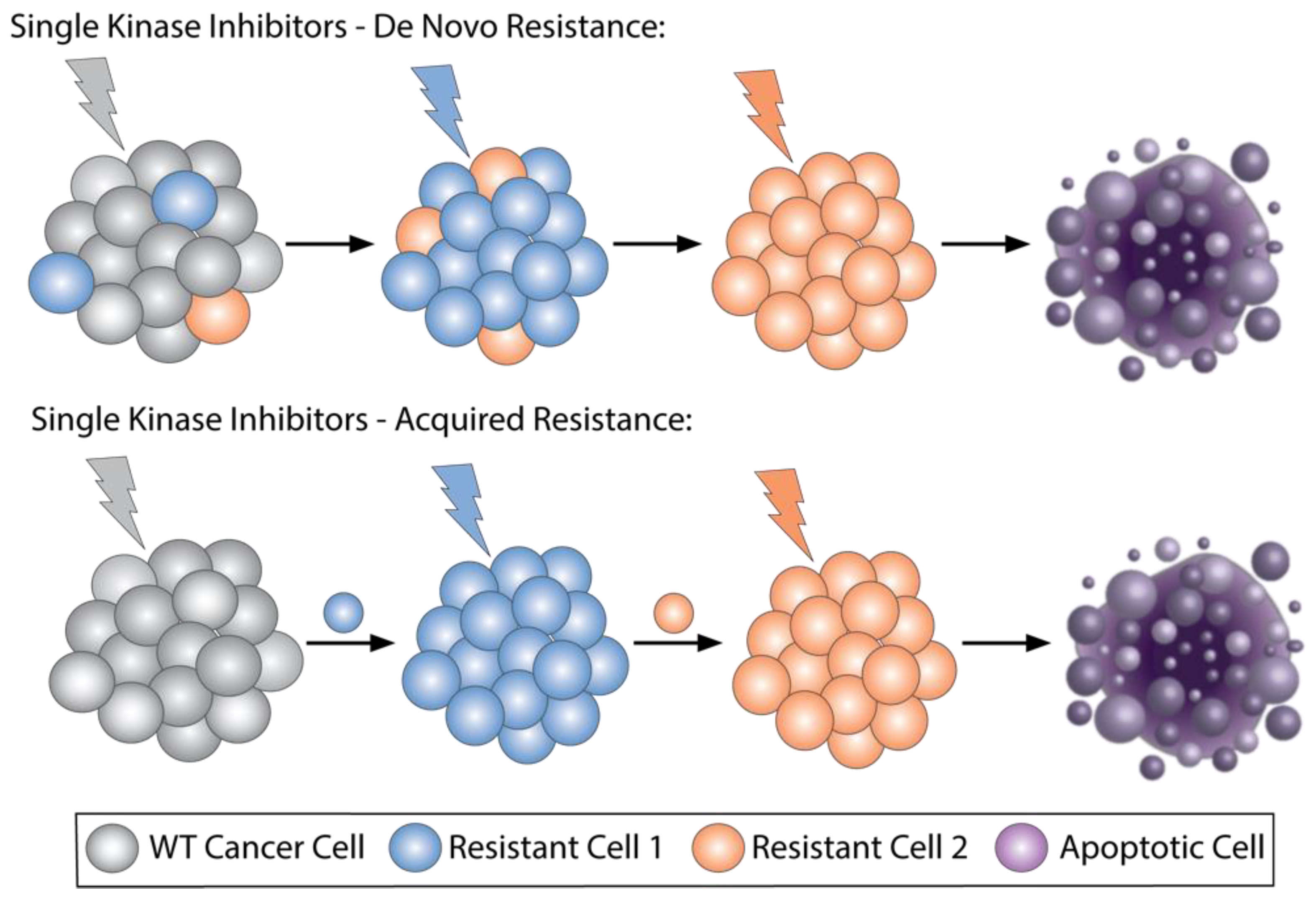
2.1. Selective Kinase Inhibitors Ipproved for Cncer, Resistance Mechanisms and How Combinations Overcome Resistance

{kind=link}
{kind=link}
{kind=link}
| Chemical Structure: | Drug Name: | Target: | Generation: | Known Resistance Mechanism: | Reference: |
|---|---|---|---|---|---|
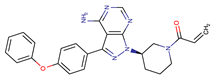 | Ibrutinib | BTK | II | BTK mutation (C418S) PLCγ2 mutations (R665W, L845F, (S707Y) | [2,7] |
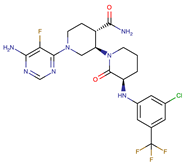 | Vecabrutinib | BTK | II | N.A. | [1,2,6] |
 | Vemurafenib | BRAF | II | Loss of NF1 or CUL3, VEGFR-1 up-regulation, CRAF up-regulation, BRAF amplification, BRAF N-terminal truncation (splice variant), MCF2 and VAV1 overexpression, EGFR–SFK–STAT3 signaling pathway increased activity, BOP1 loss, Akt signaling activation, ALK activation, COT/TPL2 expression, downregulation of the ubiquitin ligase RNF125, RTK (EGFR and c-MET) upregulation, MAPK, PI3K/AKT and SRC signaling networks activation, CD271 upregulation | [1,6,10] |
 | Dabrafenib | BRAF | II | BRAF amplification, BRAF N-terminal truncation, KRAS mutations, N-RAS or MEK1/2 activation, reactivation of MAPK/Erk, alteration to members of the RAS/RAF/MEK/Erk signaling cascade | [1,6,10] |
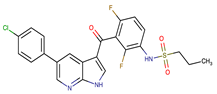
 | Encorafenib | BRAF | II | BRAF amplification, BRAF N-terminal truncation, alteration to members of the RAS/RAF/MEK/Erk signaling cascade | [1,6,10] |
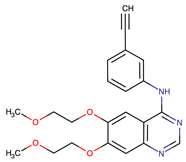 | Erlotinib | EGFR | I | EGFR T790M mutation | [12] |
 | Gefitinib | EGFR | I | EGFR T790M mutation | [12] |
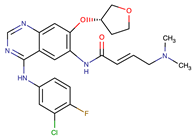 | Afatinib | EGFR | II | NRAS amplification or activating mutations KRAS amplification or activating mutations | [12,13] |
 | Dacomitinib | EGFR | II | NRAS amplification or activating mutations KRAS amplification or activating mutations | [12] |
 | Neratinib | EGFR | II | N.A. | [12] |
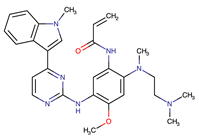 | Osimertinib | EGFR | III | EGFR C797S mutation EGFR L798Q mutation NRAS amplification or activating mutations KRAS amplification or activating mutations HER2 and MET amplification | [12] |
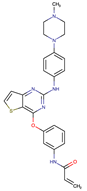 | Olmutinib | EGFR | III | EGFR C797S mutation | [12] |
 | Rociletinib | EGFR | III | EGFR L798I mutation HER2 and MET amplification | [12] |
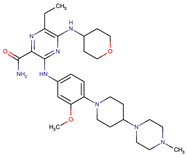 | Gilteritinib | FLT3 | IRAK1/4 hyperphosphorylation | [19,20] | |
| N.A. | EAIO45 | EGFR | IV | N.A. | [12,13] |
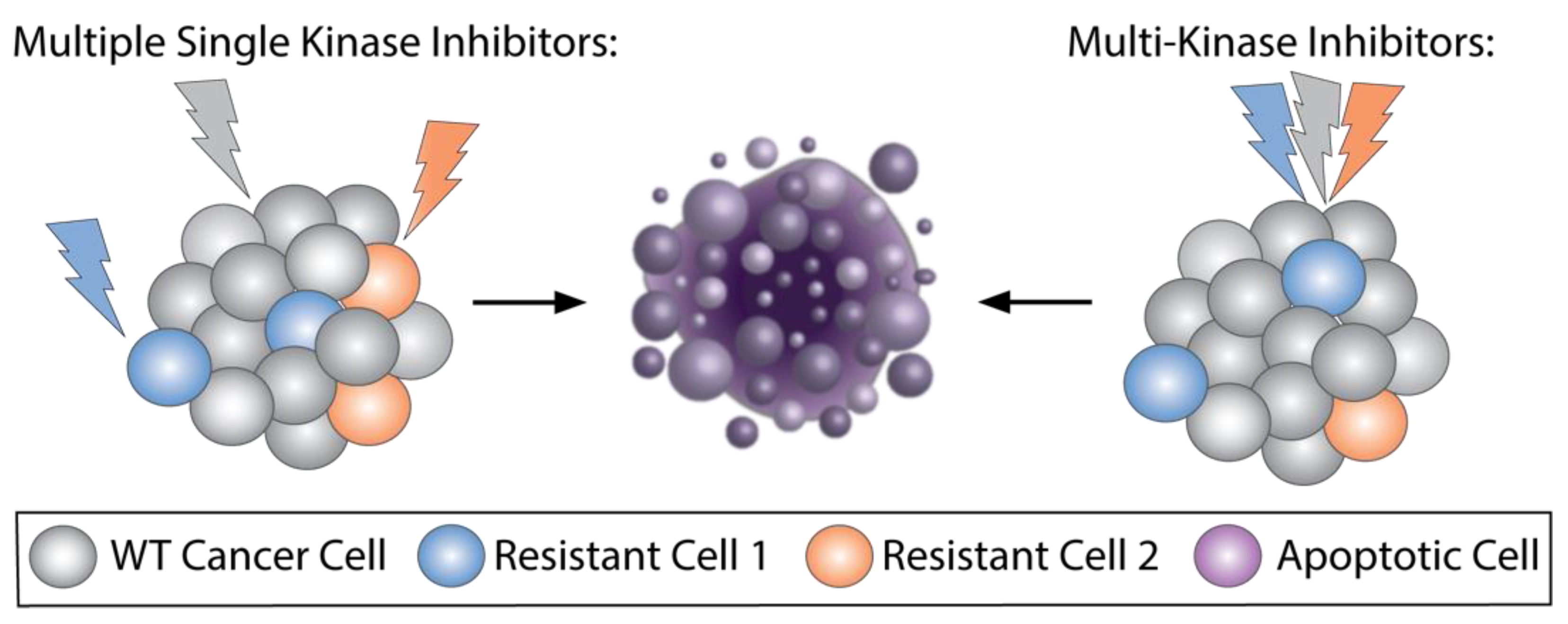
2.2. Multikinase Targeted Therapies That Are Approved in Cancer and Potential Drawbacks
2.3. Ongoing Trials of Combining Two Selective Targeted Agents, Dual-Targeted Agents, or Multikinase Inhibitors
2.4. Immunotherapies, Cancer Stem Cells, Degraders, and Drugs That Target Multiple Pathways
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 1–20. [Google Scholar] [CrossRef]
- Block, K.I.; Gyllenhaal, C.; Lowe, L.; Amedei, A.; Amin, A.R.M.R.; Amin, A.; Aquilano, K.; Arbiser, J.; Arreola, A.; Arzumanyan, A.; et al. Designing a broad-spectrum integrative approach for cancer prevention and treatment. Semin. Cancer Biol. 2015, 35, S276–S304. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Q.; Smith, S.M.; Zhang, S.Y.; Wang, Y.L. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 2015, 170, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, I.; Planchard, D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front. Med. 2017, 3, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorganic Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Wiestner, A. Ibrutinib and Venetoclax—Doubling Down on CLL. N. Engl. J. Med. 2019, 380, 2169–2171. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Mencher, S.K.; Wang, L.G. Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol. 2005, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2014, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Iorio, N.; Sawaya, R.A.; Friedenberg, F.K. Review article: The biology, diagnosis and management of gastrointestinal stromal tumours. Aliment. Pharmacol. Ther. 2014, 39, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Zhou, T.; Commodore, L.; Huang, W.-S.; Wang, Y.; Thomas, M.; Keats, J.; Xu, Q.; Rivera, V.M.; Shakespeare, W.C.; Clackson, T.; et al. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem. Biol. Drug Des. 2010, 77, 1–11. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Abbas, H.A.; Alfayez, M.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. Midostaurin in Acute Myeloid Leukemia: An Evidence-Based Review And Patient Selection. Cancer Manag. Res. 2019, 11, 8817–8828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the Treatment of Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, Y.; Wan, J.; Liu, X.; Yu, C.; Li, W. ABT-263 enhances sorafenib-induced apoptosis associated with Akt activity and the expression of Bax and p21(CIP1/WAF1) in human cancer cells. Br. J. Pharmacol. 2014, 171, 3182–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.B.; Choi, Y.; Kim, Y.S.; Lee, H.-K.; Kim, D.; Hur, D.Y. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in Sorafenib-induced apoptosis of EBV-transformed B cells. Int. J. Oncol. 2014, 44, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.; et al. Next-Generation Sequencing Reveals a Novel NSCLC ALK F1174V Mutation and Confirms ALK G1202R Mutation Confers High-Level Resistance to Alectinib (CH5424802/RO5424802) in ALK-Rearranged NSCLC Patients Who Progressed on Crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent-Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A. TRK inhibitors in TRK fusion-positive cancers. Ann. Oncol. 2019, 30, viii23–viii30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib inTRKFusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Repotrectinib Exhibits Potent Antitumor Activity in Treatment-Naïve and Solvent-Front–Mutant ROS1-Rearranged Non–Small Cell Lung Cancer|Clinical Cancer Research. Available online: https://clincancerres.aacrjournals.org/content/26/13/3287 (accessed on 31 August 2021).
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2011, 26, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yuan, Y.; Cordova, C.; Aboud, O.; Penas-Prado, M.; Theeler, B.J.; Bryla, C.; Su, Y.-T.; Grajkowska, E.; McCoy, A.; et al. Phase I trial of TG02 plus dose-dense or metronomic temozolomide for recurrent anaplastic astrocytoma and glioblastoma in adults. J. Clin. Oncol. 2019, 37, 2031. [Google Scholar] [CrossRef]
- Pallis, M.; Burrows, F.; Russell, N.H. The Multi-Kinase Inhibitor TG02 targets CD34+CD38-CD123+ AML Cells. Blood 2010, 116, 1823. [Google Scholar] [CrossRef]
- Kuykendall, A.T.; Horvat, N.P.; Pandey, G.; Komrokji, R.; Reuther, G.W. Finding a Jill for JAK: Assessing Past, Present, and Future JAK Inhibitor Combination Approaches in Myelofibrosis. Cancers 2020, 12, 2278. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Cerrano, M.; Itzykson, R. New Treatment Options for Acute Myeloid Leukemia in 2019. Curr. Oncol. Rep. 2019, 21, 16. [Google Scholar] [CrossRef]
- Kayser, S.; Levis, M.J. FLT3tyrosine kinase inhibitors in acute myeloid leukemia: Clinical implications and limitations. Leuk. Lymphoma 2013, 55, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, S.B.; Zhang, H.; Rice, W.G. A Phase 1 a/b Dose Escalation Trial to Evaluate the Safety and Tolerability of CG-806 in Patients with Relapsed or Refractory CLL/SLL or Non-Hodgkin’s Lymphomas. Blood 2019, 134, 5477. [Google Scholar] [CrossRef]
- Rastgoo, N.; Thayer, M.; Benbatoul, K.; Howell, S.; Rice, W.; Zhang, H. Abstract 4225: CG-806, a first-in-class FLT3/BTK inhibitor, and venetoclax synergize to inhibit cell proliferation and to induce apoptosis in aggressive B-cell lymphomas. Cancer Res. 2020, 80, 4225. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoebe, K.; Janssen, E.; Beutler, B. The interface between innate and adaptive immunity. Nat. Immunol. 2004, 5, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Wang, J.; Pratilas, C.A. FLT3-IRAK dual targeting: An exciting new therapeutic option guided by adaptive activation of immune response pathways. Ann. Transl. Med. 2020, 8, 511. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Melgar, K.; Bolanos, L.; Hueneman, K.; Walker, M.M.; Jiang, J.-K.; Wilson, K.M.; Zhang, X.; Shen, J.; Jiang, F.; et al. Targeting AML-associated FLT3 mutations with a type I kinase inhibitor. J. Clin. Investig. 2020, 130, 2017–2023. [Google Scholar] [CrossRef]
- Inc, M.G. A PHASE 1, DOSE ESCALATION TRIAL WITH NOVEL ORAL IRAK4 INHIBITOR... by Dr. Guillermo Garcia-Manero. Available online: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/324573/guillermo.garcia-manero.a.phase.1.dose.escalation.trial.with.novel.oral.irak4.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D1%2Asearch%3Dca-4948 (accessed on 29 September 2021).
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Mechanisms of Thalidomide and Its Derivatives. Available online: https://www.jstage.jst.go.jp/article/pjab/96/6/96_PJA9606B-01/_article/-char/ja/ (accessed on 8 September 2021).
- Koul, D.; Wang, S.; Wu, S.; Saito, N.; Zheng, S.; Gao, F.; Kaul, I.; Setoguchi, M.; Nakayama, K.; Koyama, K.; et al. Preclinical therapeutic efficacy of a novel blood-brain barrier-penetrant dual PI3K/mTOR inhibitor with preferential response in PI3K/PTEN mutant glioma. Oncotarget 2017, 8, 21741–21753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.-T.; Chen, R.; Wang, H.; Song, H.; Zhang, Q.; Chen, L.-Y.; Lappin, H.; Vasconcelos, G.; Lita, A.; Maric, D.; et al. Novel Targeting of Transcription and Metabolism in Glioblastoma. Clin. Cancer Res. 2017, 24, 1124–1137. [Google Scholar] [CrossRef] [Green Version]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging agents that target signaling pathways in cancer stem cells. J. Hematol. Oncol. 2020, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.J.; Rodriguez-Bravo, V.; Galsky, M.; Cordon-Cardo, C.; Domingo-Domenech, J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 2013, 33, 4451–4463. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Senthebane, D.; Ganz, C.; Thomford, N.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2019, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yu, C.; Xu, M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front. Oncol. 2021, 11, 678333. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, Y.; Wang, Z.; Sarkar, F.H. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins? Cancers 2011, 3, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; A Barbie, D.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2016, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Bae, T.; Hur, J.W.; Kim, D.; Hur, J.K. Recent trends in CRISPR-Cas system: Genome, epigenome, and transcriptome editing and CRISPR delivery systems. Genes Genom. 2019, 41, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, V.; Hashemi, A. Challenges of in vitro genome editing with CRISPR/Cas9 and possible solutions: A review. Gene 2020, 753, 144813. [Google Scholar] [CrossRef] [PubMed]
- Sioson, V.A.; Kim, M.; Joo, J. Challenges in delivery systems for CRISPR-based genome editing and opportunities of nanomedicine. Biomed. Eng. Lett. 2021, 11, 217–233. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.; Jura, N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 2016, 24, 7–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Study Evaluating the Safety of a New Drug (ARV-110) in Patients with MCRPC—UCLA Institute of Urologic Oncology, Los Angeles, CA. Available online: https://www.uclahealth.org/urology/iuo/a-study-evaluating-the-safety-of-a-new-drug-arv-110-in-patients-with-mcrpc (accessed on 7 September 2021).
- Perino, S.; Class, B.; Henderson, C.; Isasa, M.; Kirby, R.J.; Agafonov, R.V.; Chaturvedi, P.; Eron, S.J.; Good, A.; Hart, A.A.; et al. CFT7455: A NOVEL, IKZF1/3 DEGRADER THAT DEMONSTRATES POTENT TUMOR REGRESSION IN A SPECTRUM OF NHL XENOGRAFT MODELS. Hematol. Oncol. 2021, 39, 37–38. [Google Scholar] [CrossRef]
- Kaehler, M.; Cascorbi, I. Pharmacogenomics of Impaired Tyrosine Kinase Inhibitor Response: Lessons Learned From Chronic Myelogenous Leukemia. Front. Pharmacol. 2021, 12, 696960. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2011, 81, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontiers|Understanding the Mechanisms by Which Epigenetic Modifiers Avert Therapy Resistance in Cancer | Oncology. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2020.00992/full (accessed on 29 August 2021).
- Tyler, P.M.; Servos, M.M.; De Vries, R.C.; Klebanov, B.; Kashyap, T.; Sacham, S.; Landesman, Y.; Dougan, M.; Dougan, S.K. Clinical Dosing Regimen of Selinexor Maintains Normal Immune Homeostasis and T-cell Effector Function in Mice: Implications for Combination with Immunotherapy. Mol. Cancer Ther. 2017, 16, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2020, 18, 152–169. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.L.; Grant, S.; Shain, K.H.; Dalton, W.S.; Dai, Y.; Meads, M.; Baz, R.; Kauffman, M.; Shacham, S.; et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J. Hematol. Oncol. 2016, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Rebecca, V.W.; Smalley, K.S.M. Tumor heterogeneity and strategies to overcome kinase inhibitor resistance in cancer: Lessons from melanoma. Expert Opin. Investig. Drugs 2011, 20, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Vander Velde, R.; Yoon, N.; Marusyk, V.; Durmaz, A.; Dhawan, A.; Miroshnychenko, D.; Lozano-Peral, D.; Desai, B.; Balynska, O.; Poleszhuk, J.; et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, P.H.; Walker, R.P.; Jones, S.W.; Stephens, K.; Meurer, J.; Zajchowski, D.A.; Luke, M.M.; Eeckman, F.; Tan, Y.; Wong, L.; et al. Multiplex gene expression analysis for high-throughput drug discovery: Screening and analysis of compounds affecting genes overexpressed in cancer cells. Mol. Cancer Ther. 2002, 1, 1293–1304. [Google Scholar]
- Stathias, V.; Jermakowicz, A.M.; Maloof, M.E.; Forlin, M.; Walters, W.; Suter, R.K.; Durante, M.A.; Williams, S.L.; Harbour, J.W.; Volmar, C.-H.; et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
| Chemical Structure: | Drug Name: | Target: | Generation: | Known Resistance Mechanism: | Reference: |
|---|---|---|---|---|---|
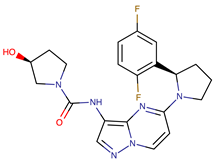 | Larotrectinib | TRKA, B and C | I | G623R, G696A, and F617L mutations in the kinase domain | [36,37,38] |
 | Entrectinib | TRKA, B and C ROS1 ALK | I | G623R, G696A, and F617L mutations in the kinase domain L1951R, S1986Y/F, F2004V, L2026M, and G2032R mutations in the kinase domain Activation of the EGFR, RAS or KIT signaling pathways C1156Y, L1196M, and G1269A (C1156Y/G1269A) mutations altering the ATP-binding pocket Activation of the EGFR, cMET, KRAS, or AXL signaling pathways | [36,37,38] |
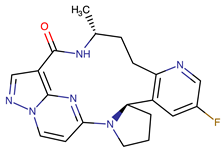 | Selitrectinib | TRKA, B and C | II | Kinase domain mutation G667C | [37,40] |
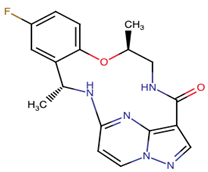 | Repotrectinib | ROS1, TRKA, B and C, ALK | II | N.A. | [37,40,41,42] |
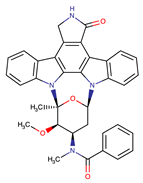 | Midostaurin | PDGFR/PKC/SYK/c-Kit/SRC/VEGFR/FLT3 | I | N676K and F691I/L mutations in FLT3 | [19,20,25,26,27,28] |
 | Quizartinib | FLT3 | II | FLT3 F691L, D835F/V/Y and Y842C/H mutation KIT D816 mutation | [19,20] |
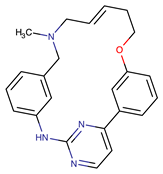 | Zotiraciclib | CDK9/JAK2/FLT3 | I | Compensatory induction of MYC expression | [43,44,45] |
 | Ruxolitinib | JAK1/JAK2 | II | Reactivation of JAK/STAT signaling via heterodimer formation between JAK2 and JAK1 or TYK2 Paracrine protective effects by cytokines (interleukin-6, fibroblast growth factor) Activation of alternative kinases not inhibited by ruxolitinib (MEK/ERK) Epigenetic mutations Mutations in JAK2 Y931C, G935R, R938L, I960V and E985K (in vitro only) | [46,47] |
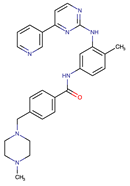 | Imatinib | ABL1/PDGFR/cKIT | I | Mutations within the Abl kinase domain/Overexpression of Bcr-Abl/Src activation | [21,22,23] |
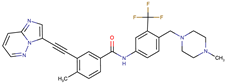 | Ponatinib | ABL1,FGFR, PDGFR, SRC, RET, KIT, and FLT1 | III | BCR/ABL compound mutations: T315I/F359V, E255V/T315I, T315I/F359C, T315I/E453K | [21,22] |
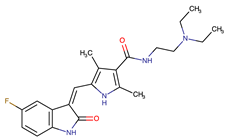 | Sunitinib | VEGF/KIT/PDGFR | I | Androgen receptor (AR) expression EIF3D or EZH2 overexpression EGFR activation | [25] |
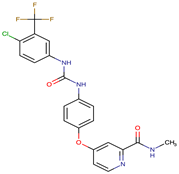 | Sorafenib | c-KIT, VEGFR -1/2/3, FLT3, RET, RAF, and PDGFR | I | Overexpression of both αB-Crystallin and 14-3-3ζ GRP78 overexpression | [29,30,31,32,33,34,35] |
| Combination Drugs/Ongoing Trials: | Targets: | Trial Phase | ClinicalTrials.gov Identifier: | Cancer Treated |
|---|---|---|---|---|
| Ibrutinib + Venetoclax | EGFR + BCL2 | 2 | NCT03045328 | Refractory chronic lymphocytic leukemia (CLL) and small lymphocytic leukemia (SLL) |
| Dabrafenib + Trametinib | EGFR + MEK | 1/2 | NCT01767454 NCT02296996 | Melanoma |
| Zotiraciclib | CDK, JAK2, FLT3 | 1/2 | NCT02942264 | Gliomas |
| Ruxolitonib + Thalidomide | JAK1/2, CRBN | 2 | NCT03069326 | PMF, post-PV MF, or post-ET MF |
| CA-4948 | FLT3, IRAK | 1/2 | NCT04278768 | AML |
| CA-4948 + Ibrutinib | FLT3, IRAK BTK | 1/2 | NCT03328078 | NHL, CLL, and Waldenstrom’s macroglobulinemia (WM) |
| CG-806 | BTK, FLT3 | 1a/b | NCT04477291 NCT03893682 | AML, CLL/SLL, NHL |
| Repotrectinib | ROS1, TRK, ALK | 1/2 | NCT03093116 | NTRK fusion tumors |
| Selective Inhibition | Non-Selective Inhibition | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montoya, S.; Soong, D.; Nguyen, N.; Affer, M.; Munamarty, S.P.; Taylor, J. Targeted Therapies in Cancer: To Be or Not to Be, Selective. Biomedicines 2021, 9, 1591. https://doi.org/10.3390/biomedicines9111591
Montoya S, Soong D, Nguyen N, Affer M, Munamarty SP, Taylor J. Targeted Therapies in Cancer: To Be or Not to Be, Selective. Biomedicines. 2021; 9(11):1591. https://doi.org/10.3390/biomedicines9111591
Chicago/Turabian StyleMontoya, Skye, Deborah Soong, Nina Nguyen, Maurizio Affer, Sailasya P. Munamarty, and Justin Taylor. 2021. "Targeted Therapies in Cancer: To Be or Not to Be, Selective" Biomedicines 9, no. 11: 1591. https://doi.org/10.3390/biomedicines9111591
APA StyleMontoya, S., Soong, D., Nguyen, N., Affer, M., Munamarty, S. P., & Taylor, J. (2021). Targeted Therapies in Cancer: To Be or Not to Be, Selective. Biomedicines, 9(11), 1591. https://doi.org/10.3390/biomedicines9111591