
Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- The curation of blood candidate set of disease-related genes (DRGs);
- (2)
- The selection of DRG sets with high prediction performance;
- (3)
- The selection of DRG sets having convergent results with single-cell RNA seq-based findings;
- (4)
- The identification of upstream genes via network analysis.
2. Methods
2.1. Retrieval of Blood, Brain, Heart, Fat, and Vessel Transcriptomic Datasets
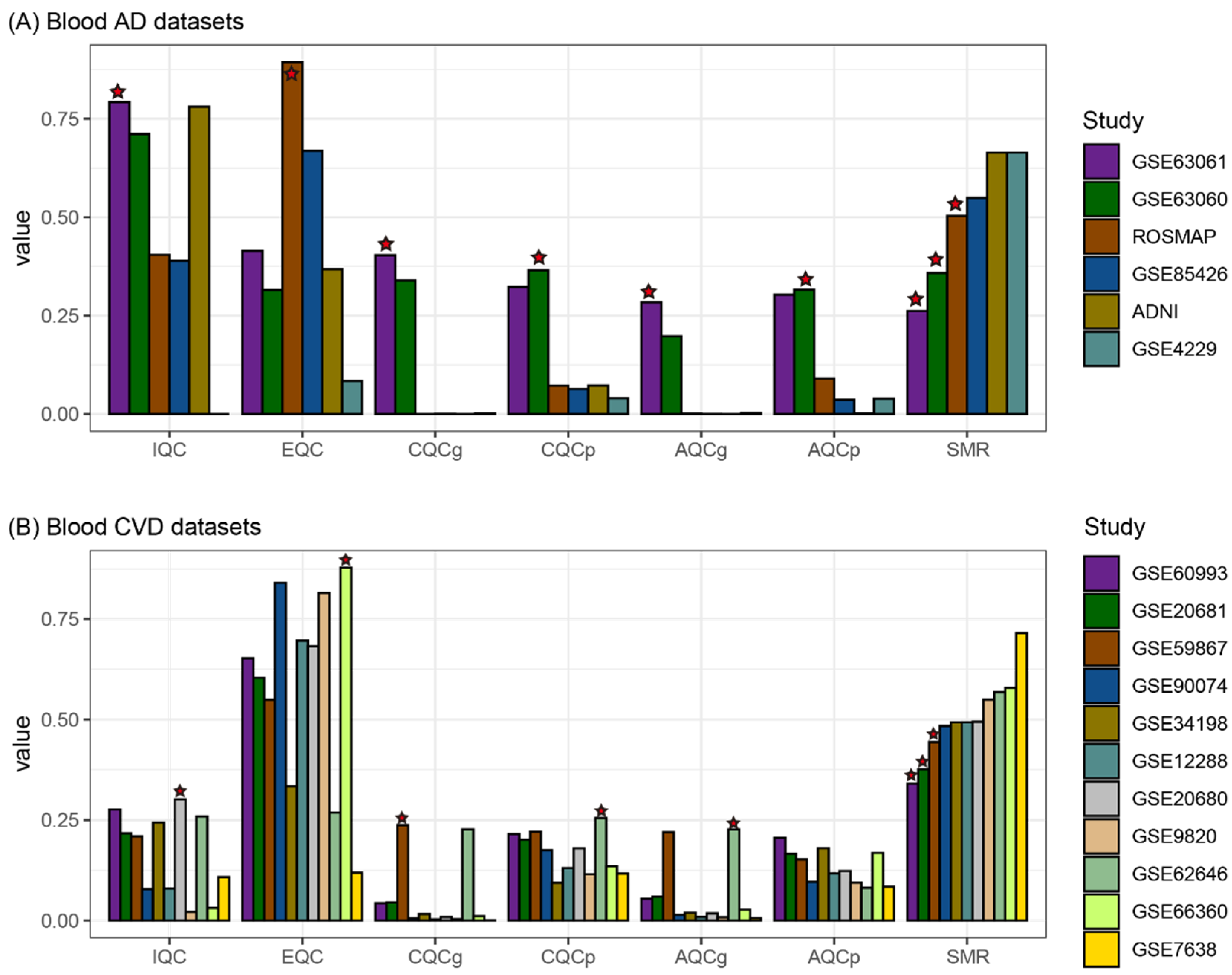
2.2. Selection of High Quality Datasets for Feature Selection
2.3. Differential Gene Expression Analysis
2.4. Identification of Blood AD-Related Genes
2.5. Identification of Blood CVD-Related Genes
2.6. Evaluation of the Blood DRGs Based on Disease Classification Performance
2.7. Comparison of the DRGs Obtained from the Blood and Single-Cell Datasets
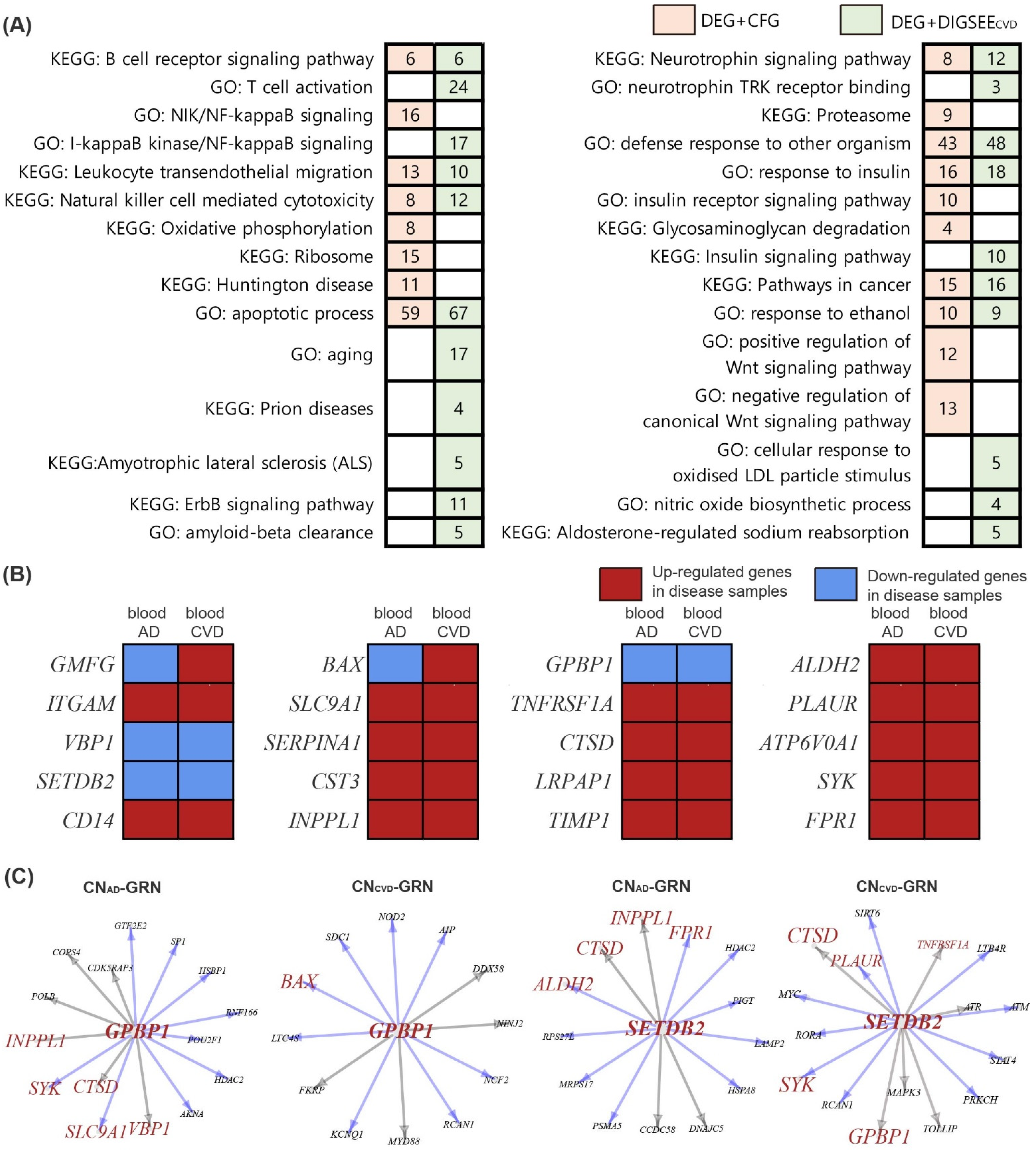
2.8. Pathway Analysis
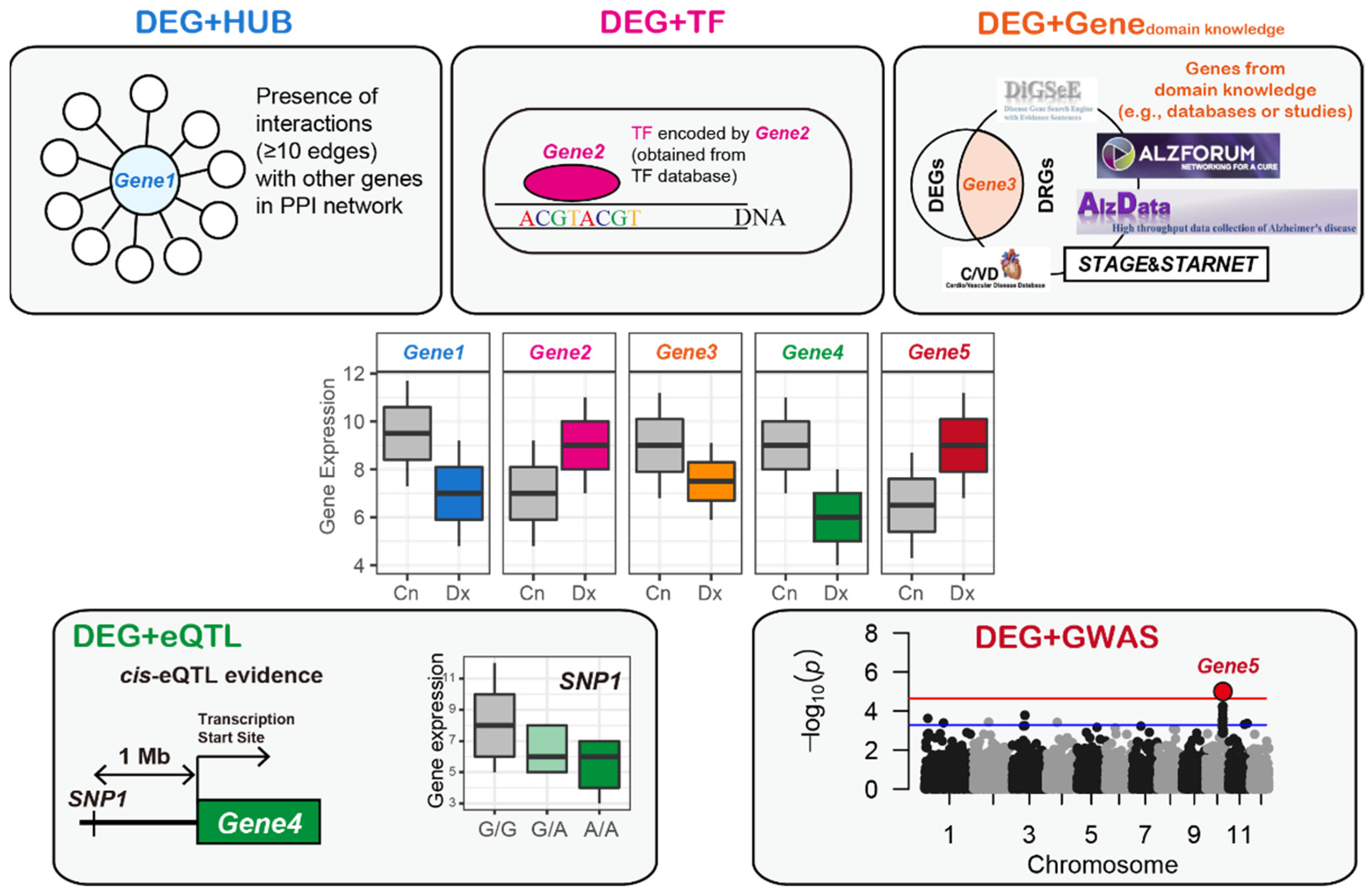
2.9. Establishment of a Gene Regulatory Network
- (1)
- To reduce the false-positive edges, we selected edges between the genes with weight values in which the degree of interaction strength calculated by GENIE3 was greater than the mean plus two standard deviations of the weight values.
- (2)
- Similar to a study by Zhang et al. [49], we excluded cases (i.e., interactions or edges) in which the genes without any cis-eSNPs were parents of genes with one or more cis-eSNPs. There were some cases in which the parent and child genes both had cis-eSNPs, which is referred to as bi-directional edges. Kirsten et al. [50] suggested that genes are not only regulated by the most significant cis-eSNP but also by a considerable number of other possible cis-regulations. Jansen et al. [51] hypothesized that a cis-eSNP with an independent association after adjusting for other cis-eSNPs might be likely to regulate gene expression and found that the possibility of the presence of a gene with an independent cis-eSNP is positively correlated with the number of cis-eSNPs in the gene. Based on these studies, a gene with a greater number of eSNPs was assigned as the parent of other genes with fewer eSNPs.
- (3)
- If two genes had the same number of eSNPs and were bi-directional, a directed edge with a higher weight value was selected.
- (4)
- If two genes did not have eSNPs and were bi-directional, a directed edge with a higher weight value was selected.
3. Results
3.1. Blood Datasets and High Quality Dataset Selection
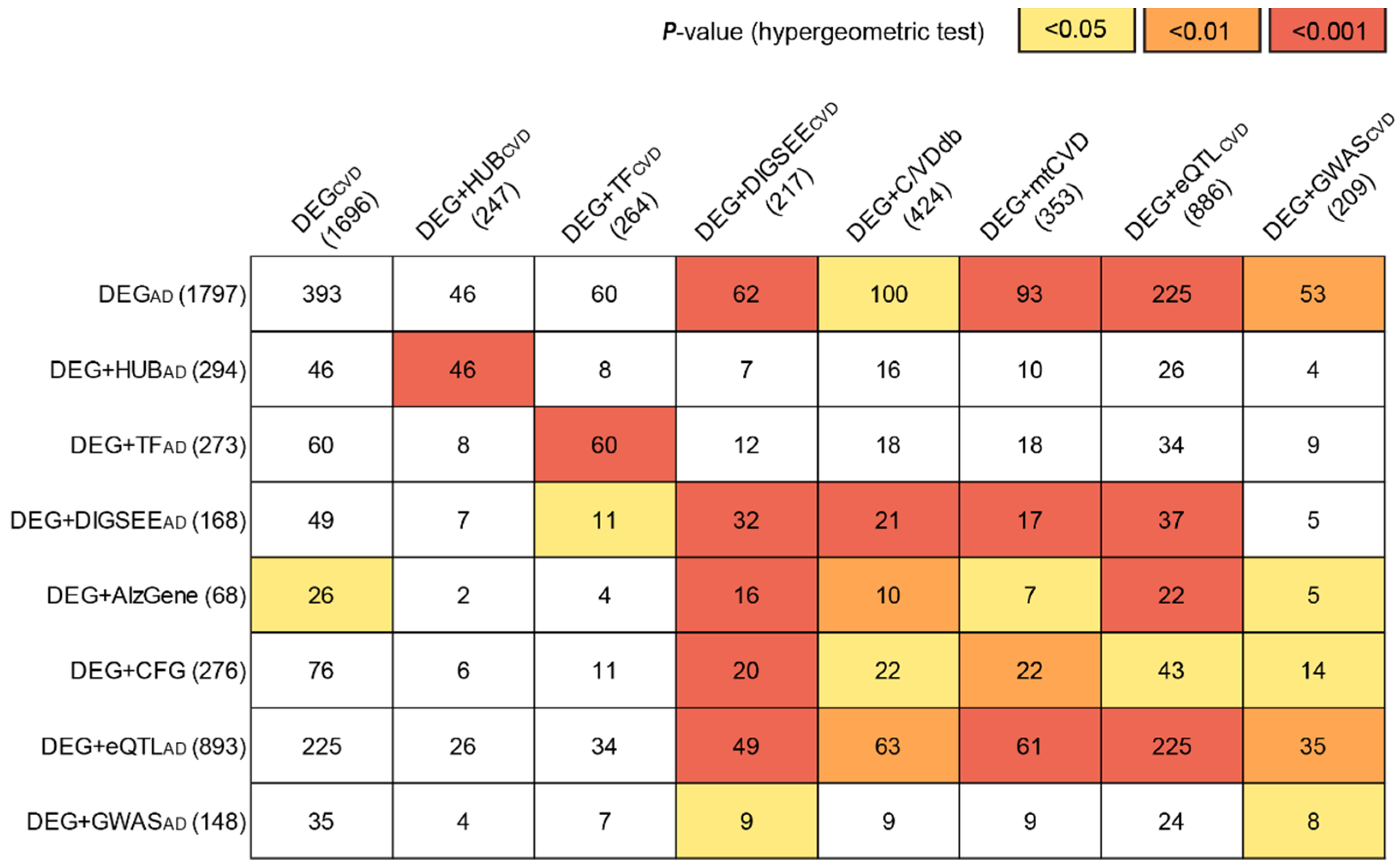
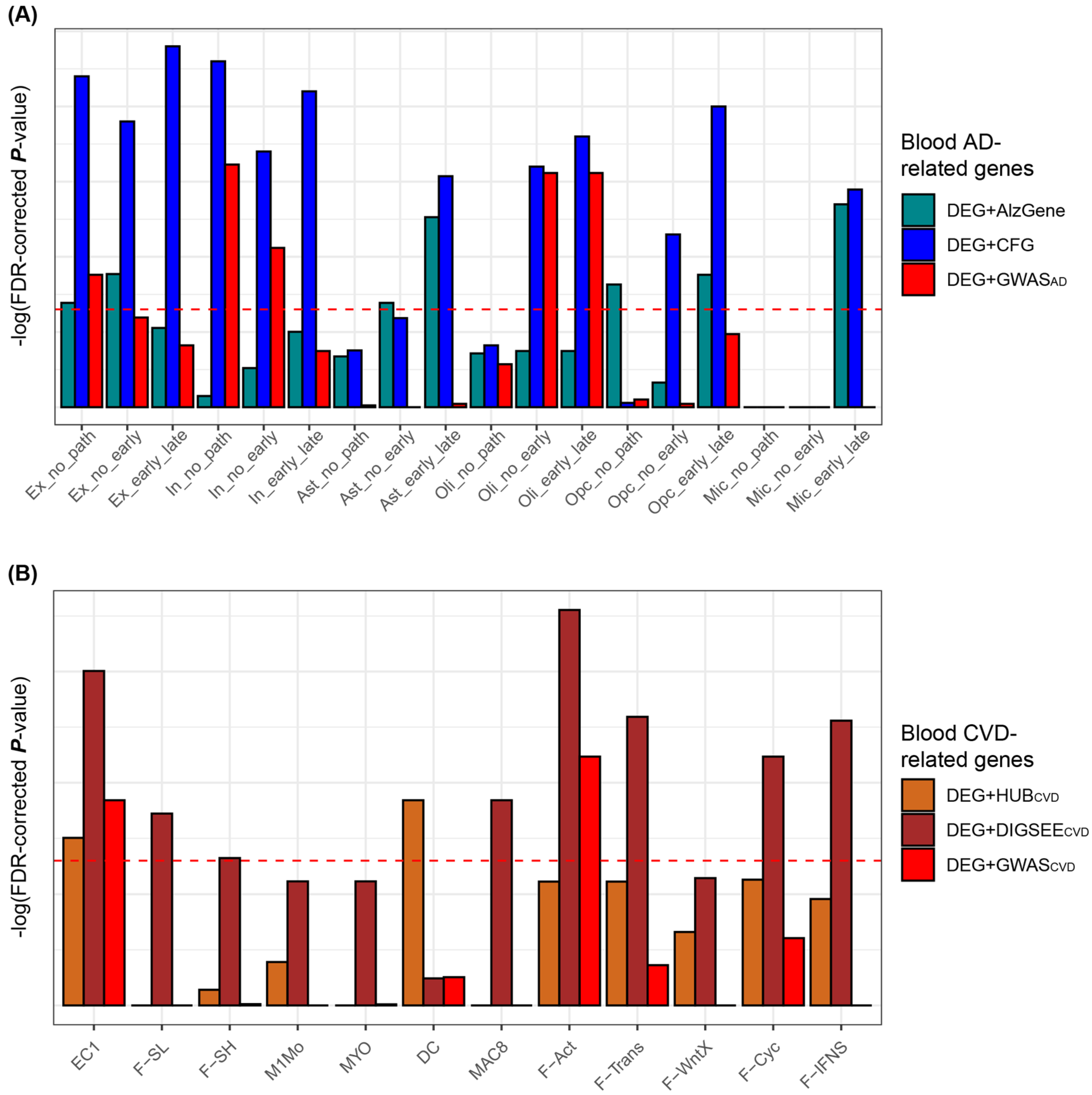
3.2. Identification of the Blood AD-Related Genes
3.3. Identification of the Blood CVD-Related Genes
3.4. Blood AD-Related Genes for Brain AD and Blood CVD Prediction
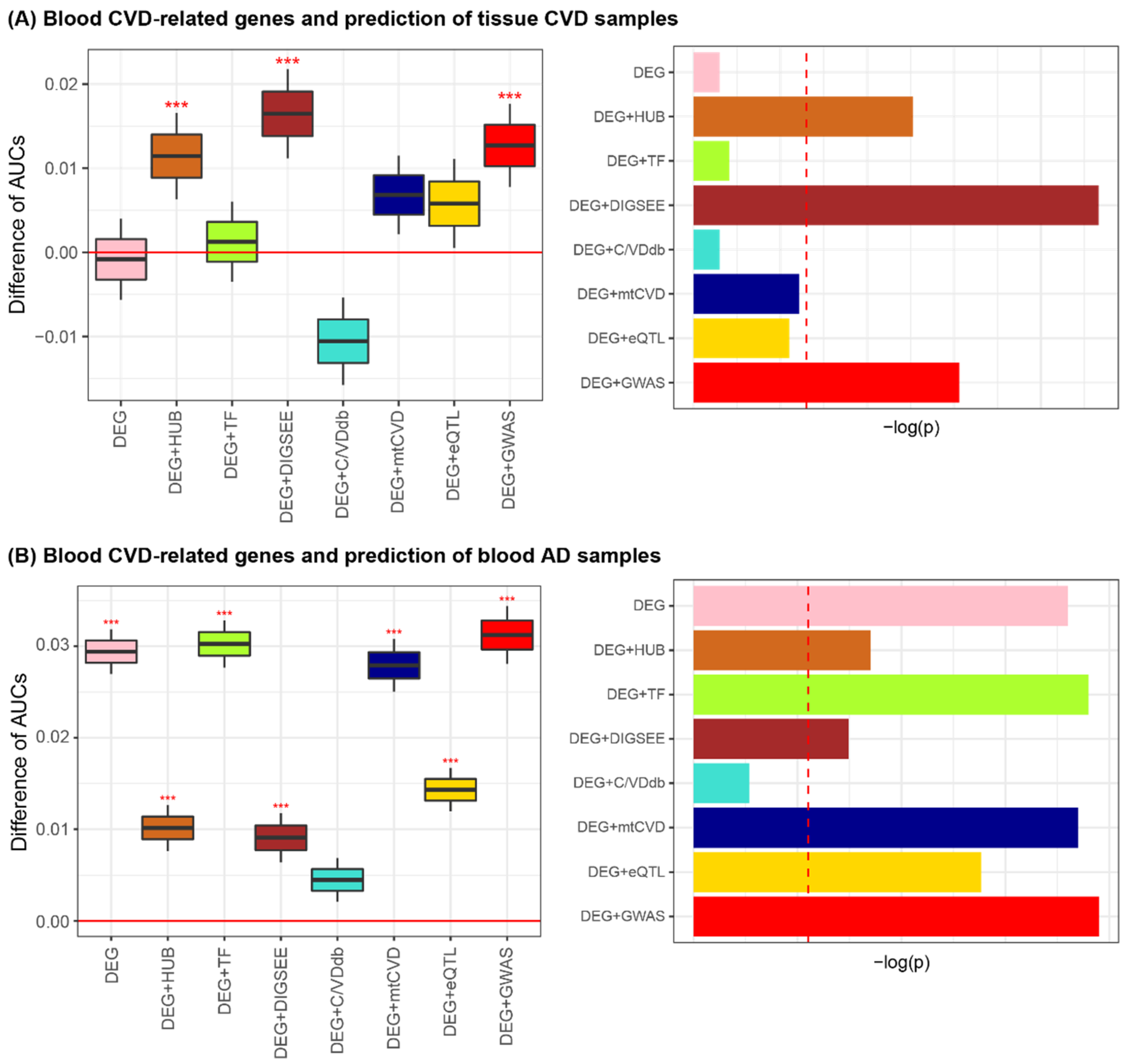
3.5. Blood CVD-Related Genes for Tissue CVD and Blood AD Prediction
3.6. Comparison of DRGs Obtained from the Blood Microarrays and Tissue (Brain or Heart) Single Cell RNA-Sequencing Datasets
3.7. Gene Regulatory Network and Identification of Altered Genes in the Disease Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018; pp. 32–36. [Google Scholar]
- Ritchie, K.; Lovestone, S. The dementias. Lancet 2002, 360, 1759–1766. [Google Scholar] [CrossRef]
- Wolters, F.J.; Ikram, M.A. Epidemiology of Vascular Dementia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Custodio, N.; Montesinos, R.; Lira, D.; Herrera-Pérez, E.; Bardales, Y.; Valeriano-Lorenzo, L. Mixed dementia: A review of the evidence. Dement. Neuropsychol. 2017, 11, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breteler, M.M.; Claus, J.J.; Grobbee, D.E.; Hofman, A. Cardiovascular disease and distribution of cognitive function in elderly people: The Rotterdam Study. BMJ 1994, 308, 1604–1608. [Google Scholar] [CrossRef] [Green Version]
- Bleckwenn, M.; Kleineidam, L.; Wagner, M.; Jessen, F.; Weyerer, S.; Werle, J.; Wiese, B.; Luhmann, D.; Posselt, T.; Konig, H.H.; et al. Impact of coronary heart disease on cognitive decline in Alzheimer’s disease: A prospective longitudinal cohort study in primary care. Br. J. Gen. Pract. J. R. Coll. Gen. Pract. 2017, 67, e111–e117. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.B.; Fitzpatrick, A.L.; Lopez, O.; Jackson, S.; Lyketsos, C.; Jagust, W.; Ives, D.; Dekosky, S.T.; Kuller, L.H. Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J. Am. Geriatr. Soc. 2005, 53, 1101–1107. [Google Scholar] [CrossRef]
- Tini, G.; Scagliola, R.; Monacelli, F.; La Malfa, G.; Porto, I.; Brunelli, C.; Rosa, G.M. Alzheimer’s Disease and Cardiovascular Disease: A Particular Association. Cardiol. Res. Pract. 2020, 2020, 2617970. [Google Scholar] [CrossRef]
- Beeri, M.S.; Rapp, M.; Silverman, J.M.; Schmeidler, J.; Grossman, H.T.; Fallon, J.T.; Purohit, D.P.; Perl, D.P.; Siddiqui, A.; Lesser, G.; et al. Coronary artery disease is associated with Alzheimer disease neuropathology in APOE4 carriers. Neurology 2006, 66, 1399–1404. [Google Scholar] [CrossRef]
- Nho, K.; Nudelman, K.; Allen, M.; Hodges, A.; Kim, S.; Risacher, S.L.; Apostolova, L.G.; Lin, K.; Lunnon, K.; Wang, X.; et al. Genome-wide transcriptome analysis identifies novel dysregulated genes implicated in Alzheimer’s pathology. Alzheimers Dement. 2020, 16, 1213–1223. [Google Scholar] [CrossRef]
- Maciejak, A.; Kiliszek, M.; Michalak, M.; Tulacz, D.; Opolski, G.; Matlak, K.; Dobrzycki, S.; Segiet, A.; Gora, M.; Burzynska, B. Gene expression profiling reveals potential prognostic biomarkers associated with the progression of heart failure. Genome Med. 2015, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Michiels, S.; Koscielny, S.; Hill, C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 2005, 365, 488–492. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R. On testing the significance of sets of genes. Ann. Appl. Stat. 2007, 1, 107–129. [Google Scholar] [CrossRef] [Green Version]
- Zander, T.; Hofmann, A.; Staratschek-Jox, A.; Classen, S.; Debey-Pascher, S.; Maisel, D.; Ansén, S.; Hahn, M.; Beyer, M.; Thomas, R.K.; et al. Blood-based gene expression signatures in non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 3360–3367. [Google Scholar] [CrossRef] [Green Version]
- Venet, D.; Dumont, J.E.; Detours, V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, Y. Association between expression of random gene sets and survival is evident in multiple cancer types and may be explained by sub-classification. PLoS Comput. Biol. 2018, 14, e1006026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. Limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.K.; Montgomery, K.S.; Kauer, N.; Woo, K.H.; Leanza, Z.J.; Poehlman, W.L.; Gockley, J.; Sieberts, S.K.; Bradic, L.; Logsdon, B.A.; et al. The AD Knowledge Portal: A Repository for Multi-Omic Data on Alzheimer’s Disease and Aging. Curr. Protoc. Hum. Genet. 2020, 108, e105. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Lee, H. Shared Blood Transcriptomic Signatures between Alzheimer’s Disease and Diabetes Mellitus. Biomedicines 2021, 9, 34. [Google Scholar] [CrossRef]
- Kang, D.D.; Sibille, E.; Kaminski, N.; Tseng, G.C. MetaQC: Objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 2012, 40, e15. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Lee, H. Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep. 2020, 10, 3485. [Google Scholar] [CrossRef] [PubMed]
- Hägg, S.; Skogsberg, J.; Lundström, J.; Noori, P.; Nilsson, R.; Zhong, H.; Maleki, S.; Shang, M.M.; Brinne, B.; Bradshaw, M.; et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: The Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009, 5, e1000754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed] [Green Version]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, J.J.; Lee, H. An analysis of disease-gene relationship from Medline abstracts by DigSee. Sci. Rep. 2017, 7, 40154. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, D.-F.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.-L.; Bi, R.; Yao, Y.-G. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Fernandes, M.; Patel, A.; Husi, H. C/VDdb: A multi-omics expression profiling database for a knowledge-driven approach in cardiovascular disease (CVD). PLoS ONE 2018, 13, e0207371. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, H.A.; Foroughi Asl, H.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Franzén, O.; Kidd, B.A.; Readhead, B.; Giannarelli, C.; Kovacic, J.C.; et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016, 2, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Talukdar, H.A.; Koplev, S.; Giannarelli, C.; Ivert, T.; Gan, L.M.; Ruusalepp, A.; Schadt, E.E.; Kovacic, J.C.; Lusis, A.J.; et al. Contribution of Gene Regulatory Networks to Heritability of Coronary Artery Disease. J. Am. Coll. Cardiol. 2019, 73, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, e43882. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies. genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Kirsten, H.; Al-Hasani, H.; Holdt, L.; Gross, A.; Beutner, F.; Krohn, K.; Horn, K.; Ahnert, P.; Burkhardt, R.; Reiche, K.; et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci†. Hum. Mol. Genet. 2015, 24, 4746–4763. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Hottenga, J.J.; Nivard, M.G.; Abdellaoui, A.; Laport, B.; de Geus, E.J.; Wright, F.A.; Penninx, B.; Boomsma, D.I. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum. Mol. Genet. 2017, 26, 1444–1451. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, J.; Jung, E.S.; Kim, M.-H.; Kim, I.B.; Son, H.; Kim, S.; Kim, S.; Park, Y.M.; Mook-Jung, I.; et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun. 2019, 10, 3090. [Google Scholar] [CrossRef]
- Sang, Q.; Liu, X.; Wang, L.; Qi, L.; Sun, W.; Wang, W.; Sun, Y.; Zhang, H. CircSNCA downregulation by pramipexole treatment mediates cell apoptosis and autophagy in Parkinson’s disease by targeting miR-7. Aging (Albany NY) 2018, 10, 1281–1293. [Google Scholar] [CrossRef]
- Bijnens, A.P.; Gils, A.; Jutten, B.; Faber, B.C.; Heeneman, S.; Kitslaar, P.J.; Tordoir, J.H.; de Vries, C.J.; Kroon, A.A.; Daemen, M.J.; et al. Vasculin, a novel vascular protein differentially expressed in human atherogenesis. Blood 2003, 102, 2803–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.Y.; Ng, M.P.; Loke, S.Y.; Jin, S.; Wu, Y.J.; Tanaka, K.; Wong, P.T. Comprehensive gene expression profiling reveals synergistic functional networks in cerebral vessels after hypertension or hypercholesterolemia. PLoS ONE 2013, 8, e68335. [Google Scholar] [CrossRef] [PubMed]
- Roqueta-Rivera, M.; Esquejo, R.M.; Phelan, P.E.; Sandor, K.; Daniel, B.; Foufelle, F.; Ding, J.; Li, X.; Khorasanizadeh, S.; Osborne, T.F. SETDB2 Links Glucocorticoid to Lipid Metabolism through Insig2a Regulation. Cell Metab. 2016, 24, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schliehe, C.; Flynn, E.K.; Vilagos, B.; Richson, U.; Swaminathan, S.; Bosnjak, B.; Bauer, L.; Kandasamy, R.K.; Griesshammer, I.M.; Kosack, L.; et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat. Immunol. 2015, 16, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kroetz, D.N.; Allen, R.M.; Schaller, M.A.; Cavallaro, C.; Ito, T.; Kunkel, S.L. Type I Interferon Induced Epigenetic Regulation of Macrophages Suppresses Innate and Adaptive Immunity in Acute Respiratory Viral Infection. PLoS Pathog. 2015, 11, e1005338. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Kim, S.H.; Arifuzzaman, S.; Yoon, T.; Chai, J.C.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome sequencing reveals that LPS-triggered transcriptional responses in established microglia BV2 cell lines are poorly representative of primary microglia. J. Neuroinflammation 2016, 13, 182. [Google Scholar] [CrossRef] [Green Version]
- Eyster, K.M.; Appt, S.E.; Mark-Kappeler, C.J.; Chalpe, A.; Register, T.C.; Clarkson, T.B. Gene expression signatures differ with extent of atherosclerosis in monkey iliac artery. Menopause 2011, 18, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef]
- Paris, D.; Ait-Ghezala, G.; Bachmeier, C.; Laco, G.; Beaulieu-Abdelahad, D.; Lin, Y.; Jin, C.; Crawford, F.; Mullan, M. The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-β production and Tau hyperphosphorylation. J. Biol. Chem. 2014, 289, 33927–33944. [Google Scholar] [CrossRef] [Green Version]
- van Eeuwijk, J.M.; Stegner, D.; Lamb, D.J.; Kraft, P.; Beck, S.; Thielmann, I.; Kiefer, F.; Walzog, B.; Stoll, G.; Nieswandt, B. The Novel Oral Syk Inhibitor, Bl1002494, Protects Mice From Arterial Thrombosis and Thromboinflammatory Brain Infarction. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Hamano, T.; Sada, K.; Kanaan, N.M.; Sasaki, H.; Yen, S.H.; Asano, R.; Enomoto, S.; Shirafuji, N.; Ueno, A. Syk inhibitor reduces oligomeric tau associated with GSK3β inactivation and autophagy activation: Molecular and cell biology/tau. Alzheimer’s Dement. 2020, 16, e042633. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Sumsuzzman, D.M.; Ashraf, G.M.; Perveen, A.; Bungau, S.G.; Mousa, S.A.; El-Seedi, H.R.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Emerging Promise of Cannabinoids for the Management of Pain and Associated Neuropathological Alterations in Alzhei.imer’s Disease. Front. Pharmacol. 2020, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef]
- Santos, C.Y.; Snyder, P.J.; Wu, W.-C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement. (Amst) 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, T.; Kaur, I.; Sehgal, A.; Kumar, A.; Uddin, M.S.; Bungau, S. The Interplay of ABC Transporters in Aβ Translocation and Cholesterol Metabolism: Implicating Their Roles in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1564–1582. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.; Lee, H.; the Alzheimer’s Disease Neuroimaging Initiative. Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis. Biomedicines 2021, 9, 1525. https://doi.org/10.3390/biomedicines9111525
Lee T, Lee H, the Alzheimer’s Disease Neuroimaging Initiative. Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis. Biomedicines. 2021; 9(11):1525. https://doi.org/10.3390/biomedicines9111525
Chicago/Turabian StyleLee, Taesic, Hyunju Lee, and the Alzheimer’s Disease Neuroimaging Initiative. 2021. "Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis" Biomedicines 9, no. 11: 1525. https://doi.org/10.3390/biomedicines9111525
APA StyleLee, T., Lee, H., & the Alzheimer’s Disease Neuroimaging Initiative. (2021). Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis. Biomedicines, 9(11), 1525. https://doi.org/10.3390/biomedicines9111525