
A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies

Abstract
:
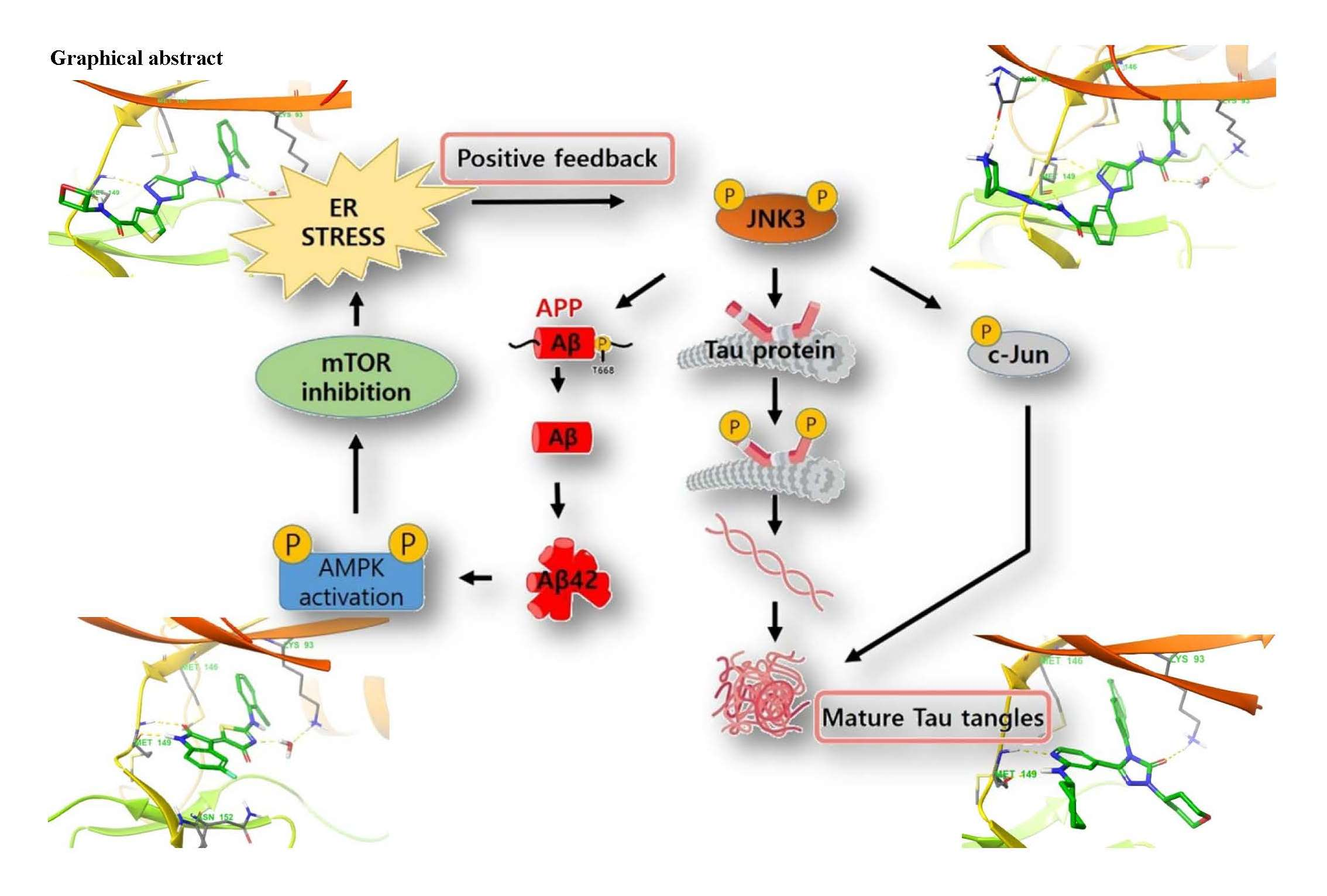{kind=link}
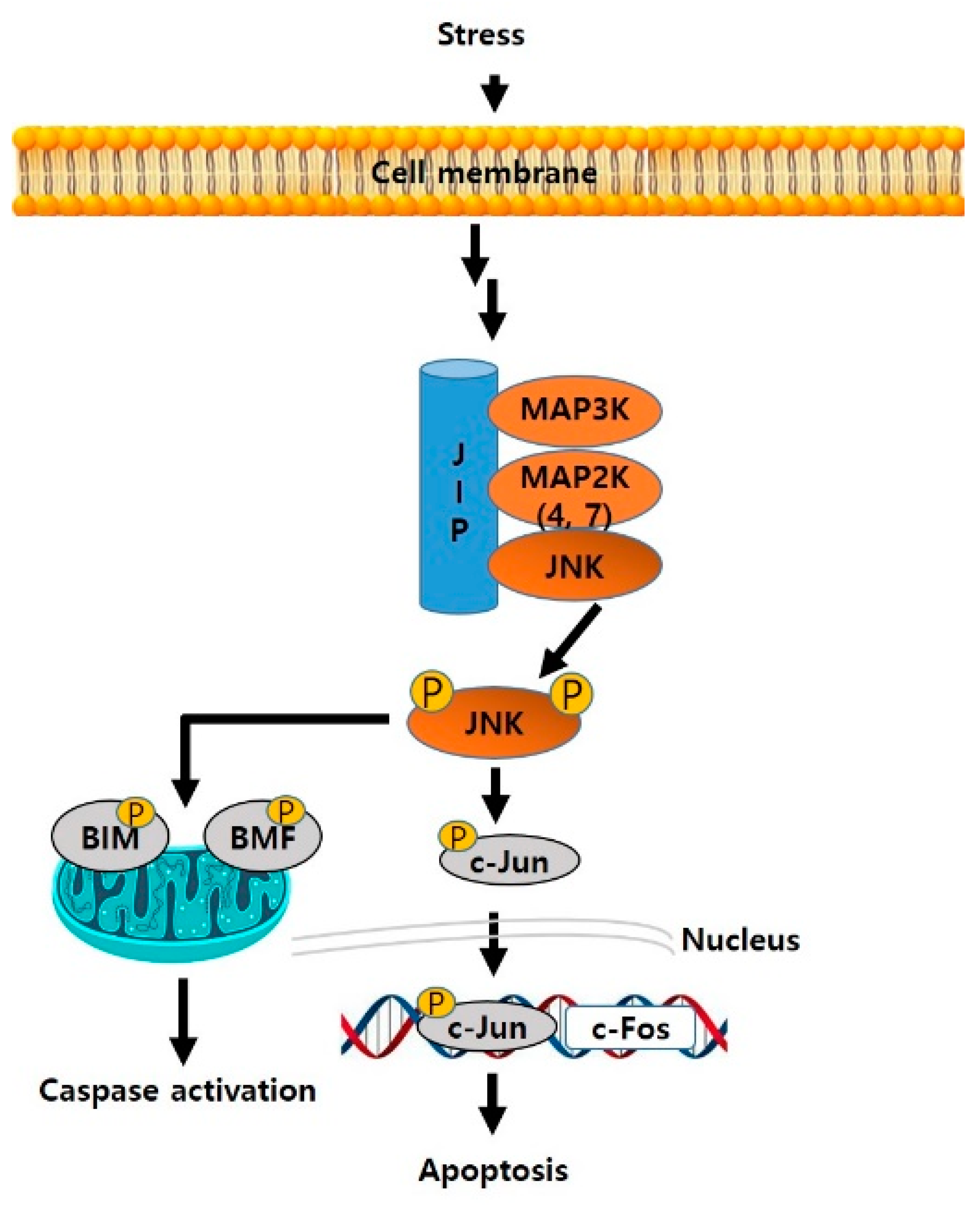{kind=link}
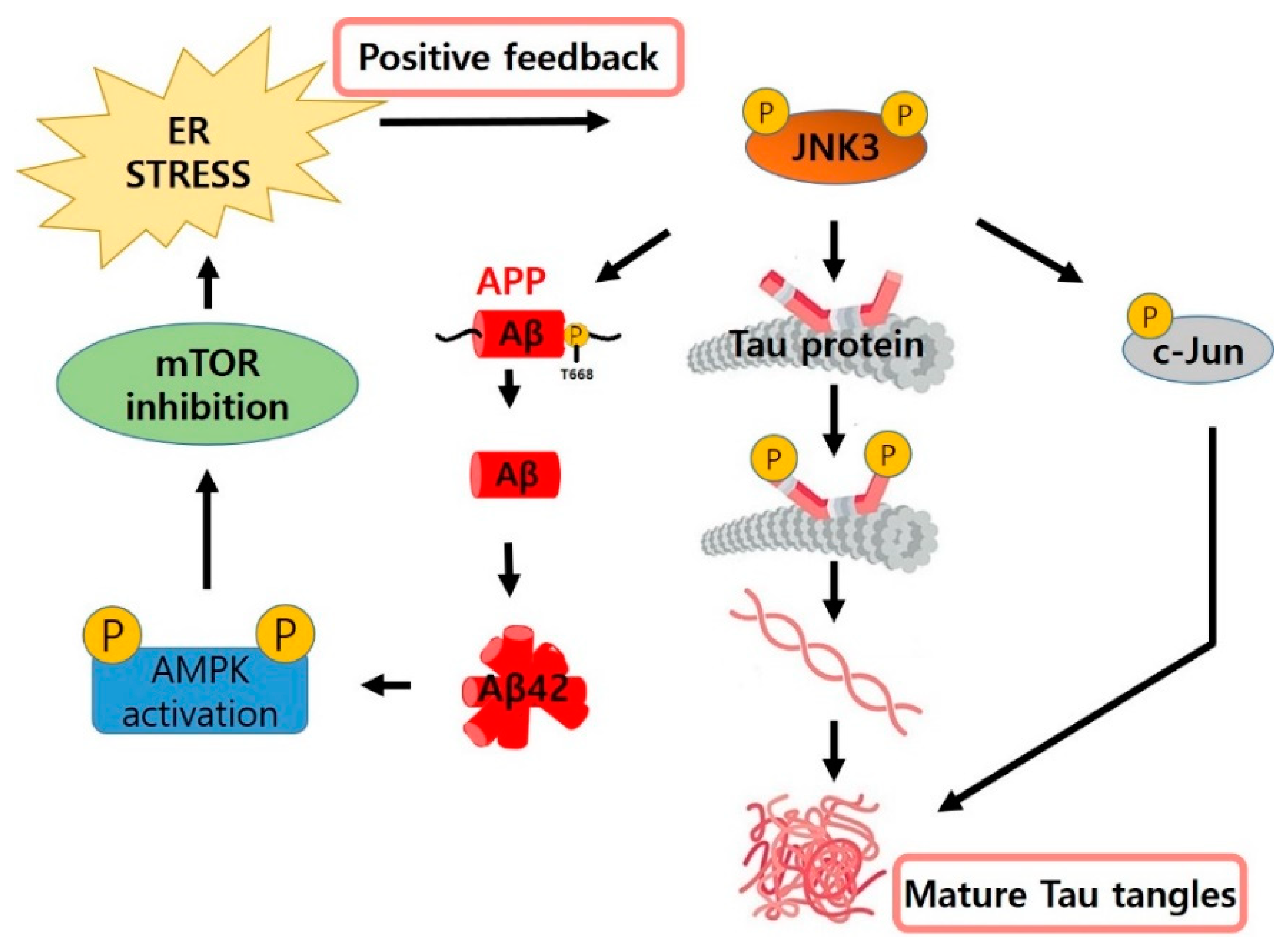{kind=link}
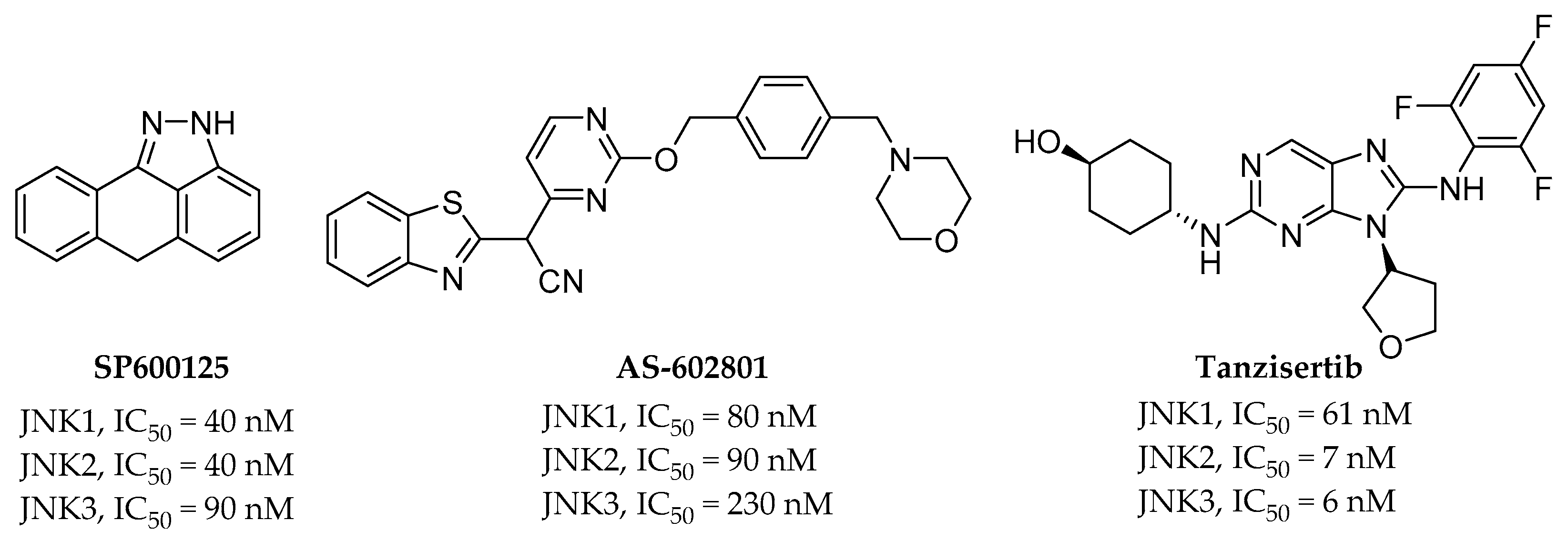{kind=link}
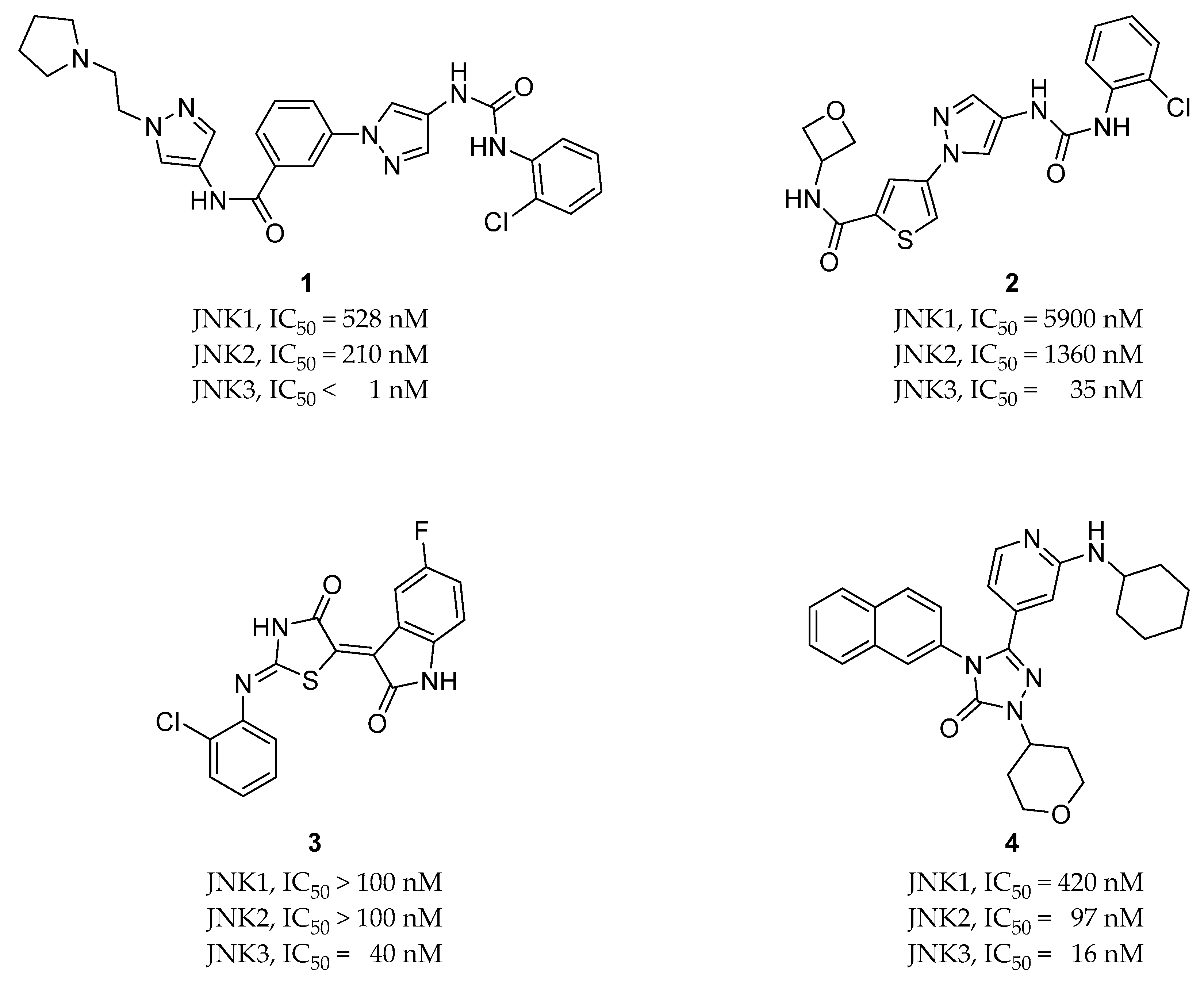{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
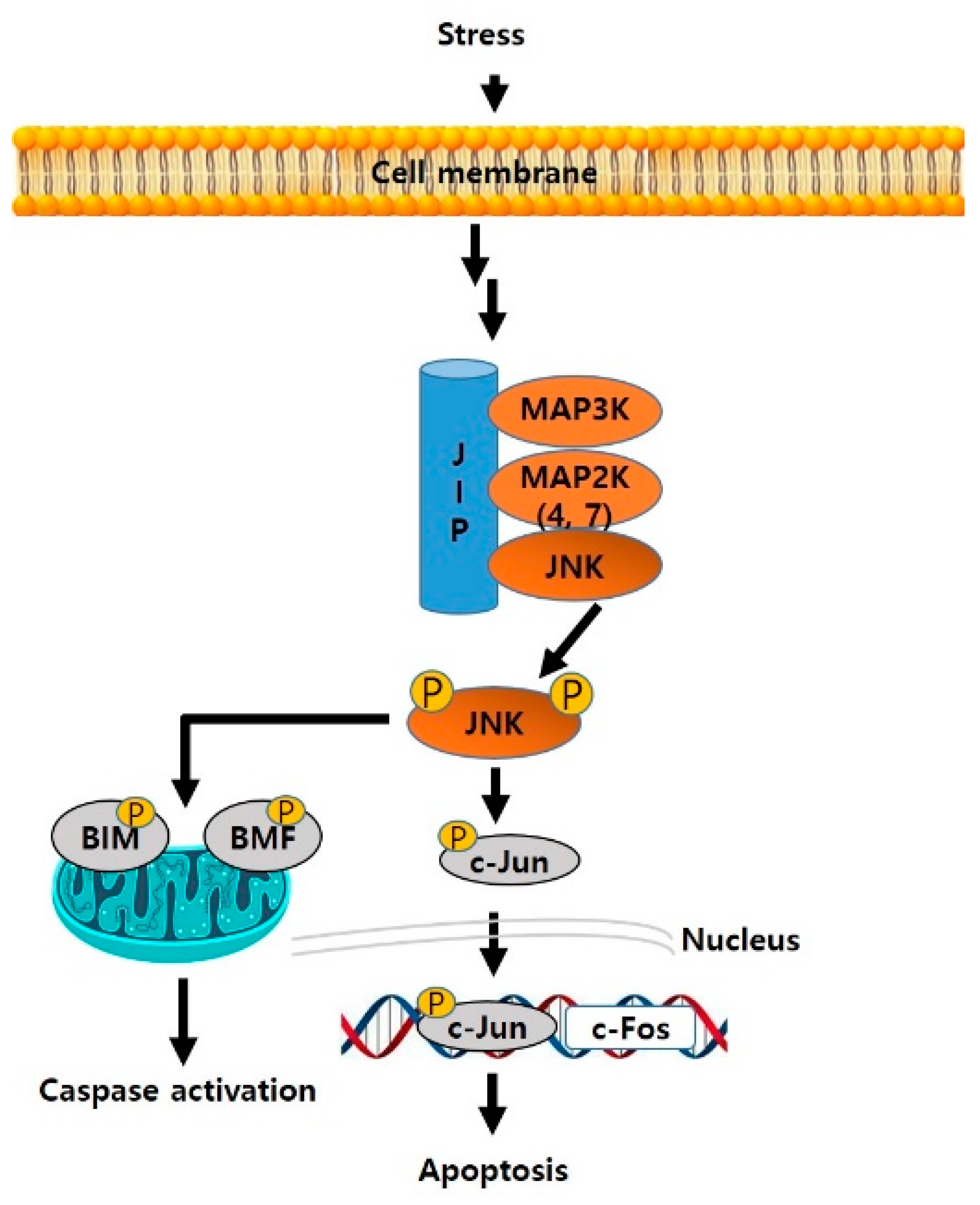
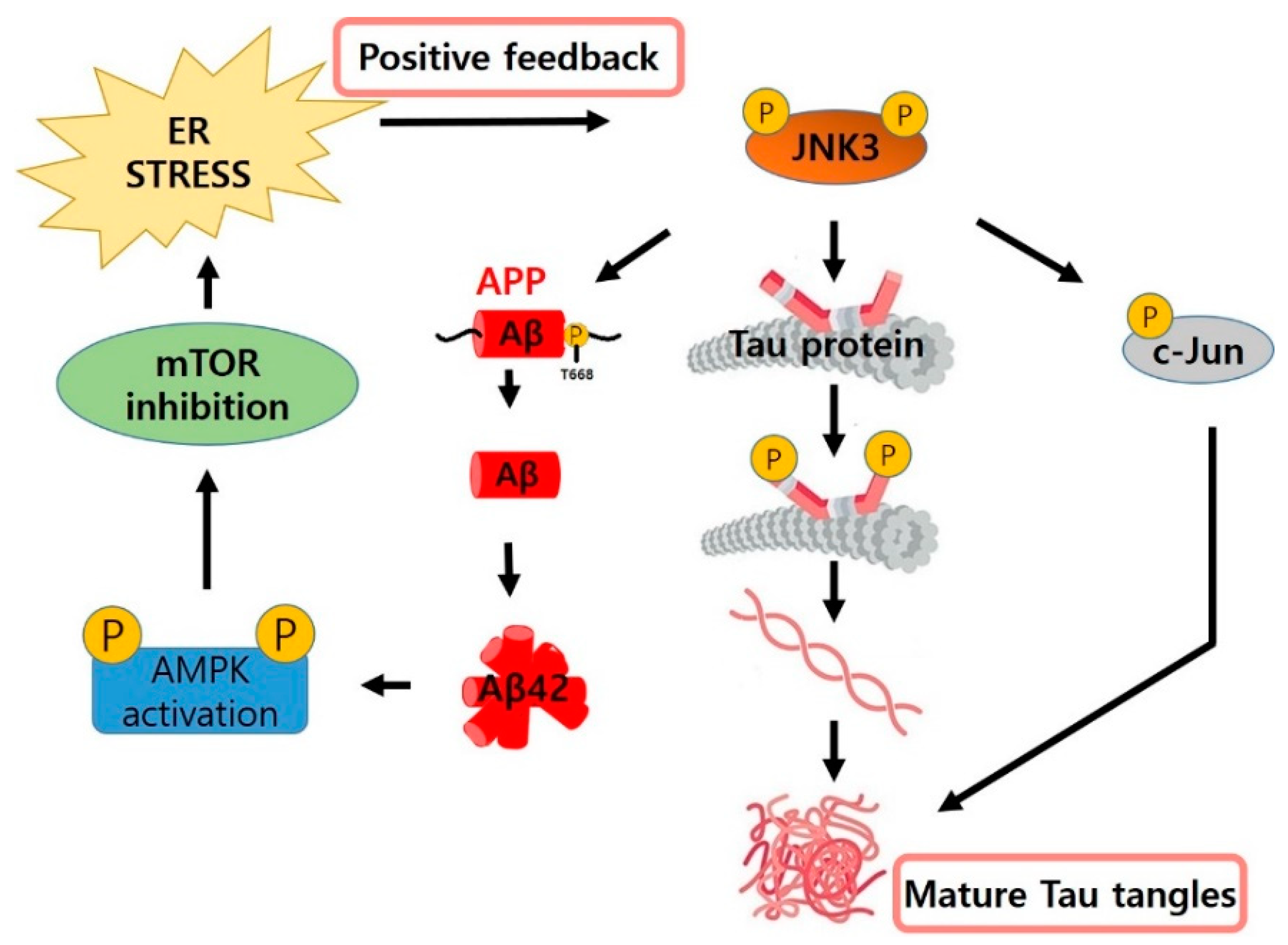
1. Introduction
2. Current Development Status of JNK Inhibitors
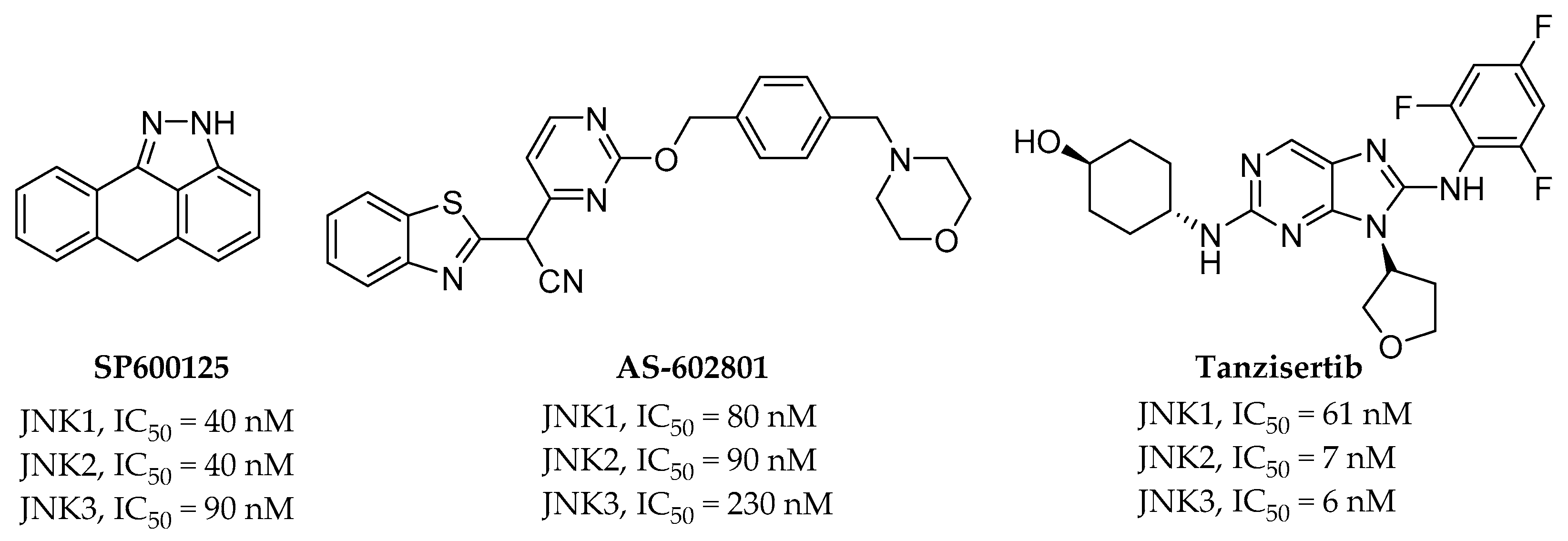
2.1. Pan-JNK Inhibitors
2.2. Selective JNK Inhibitors
3. Structural Perspective Analysis of JNK’s Active Site
3.1. Superimposition of JNK1, JNK2, and JNK3 Crystal Structures
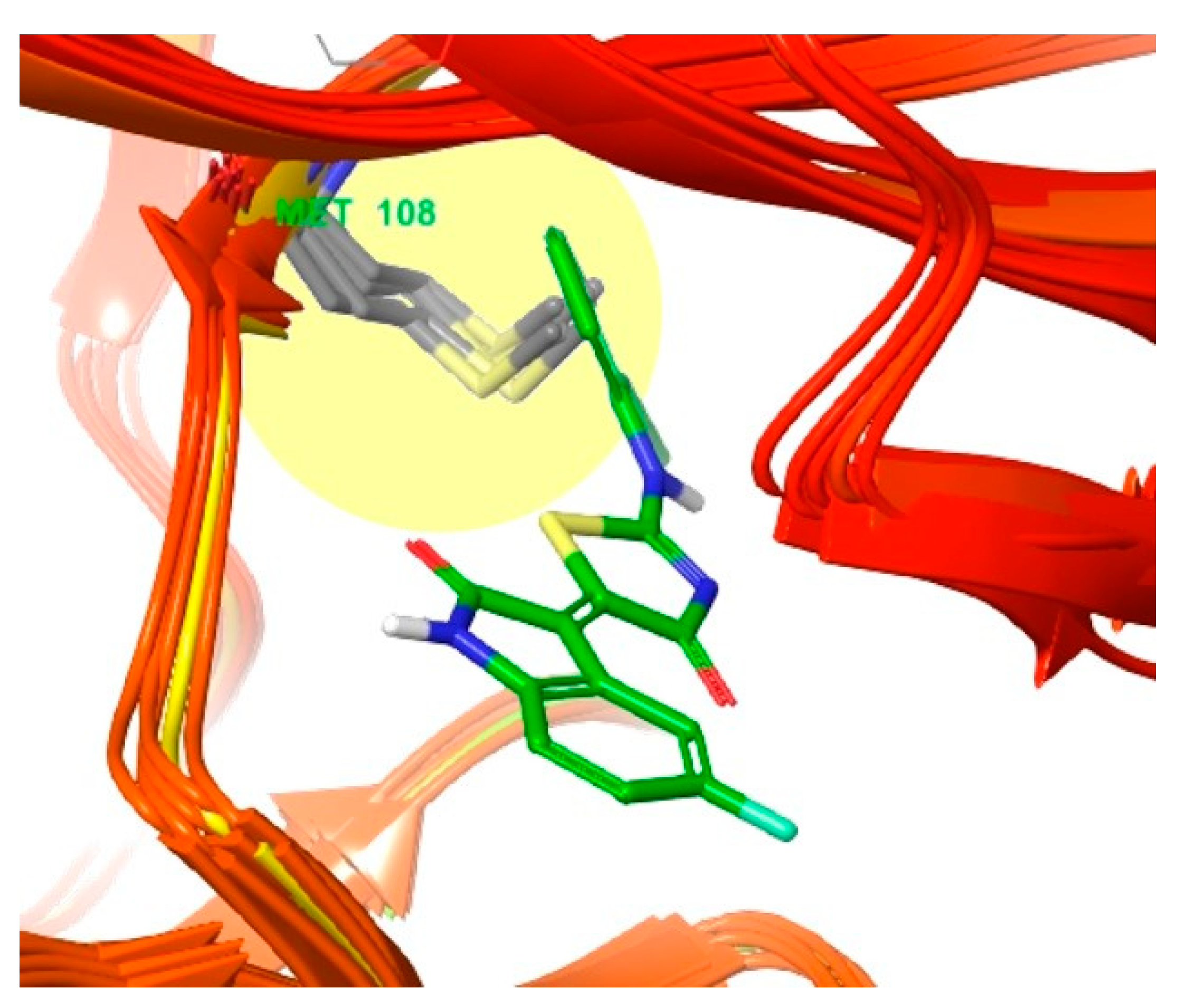
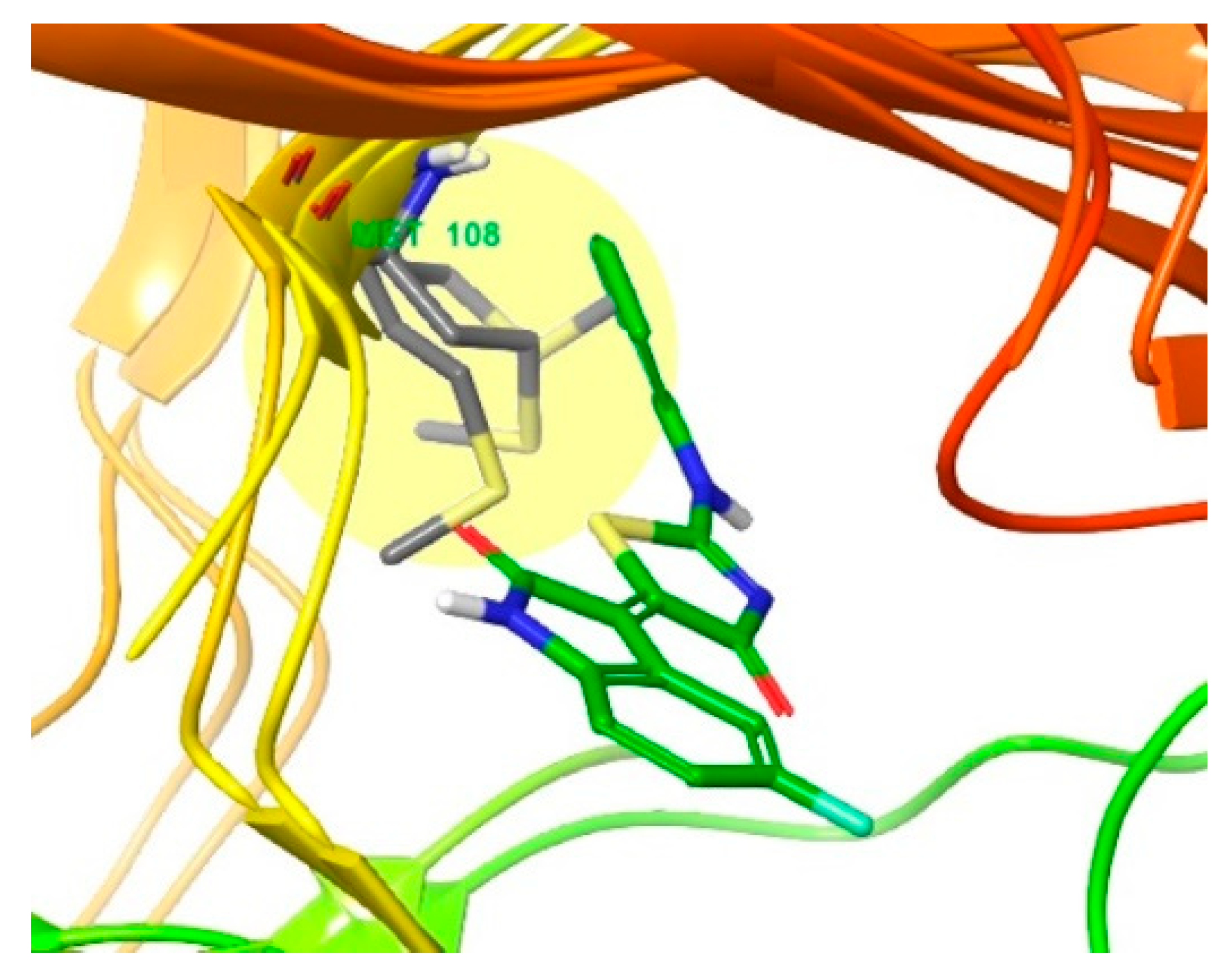
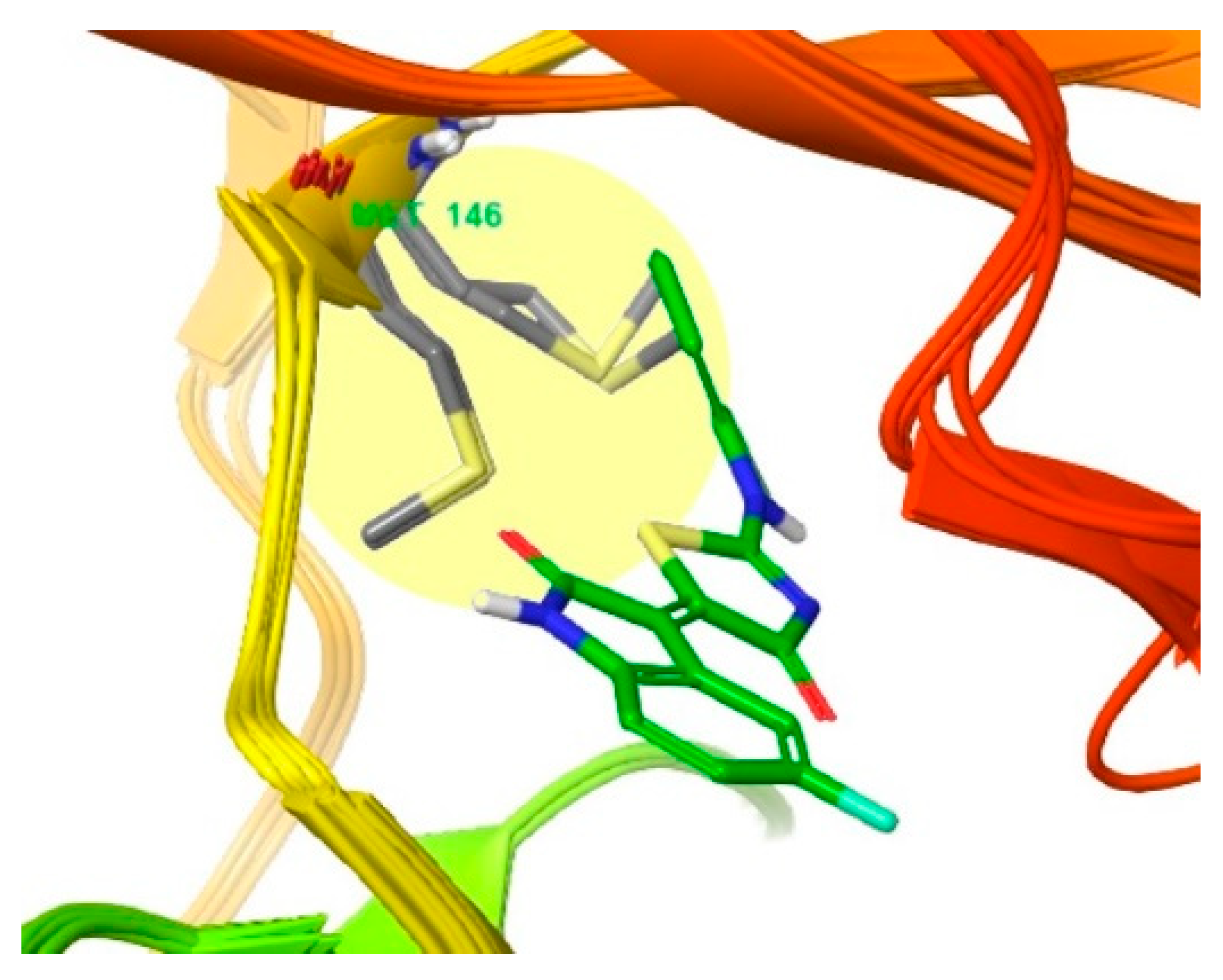
3.2. Different Residues between JNK1 and JNK3
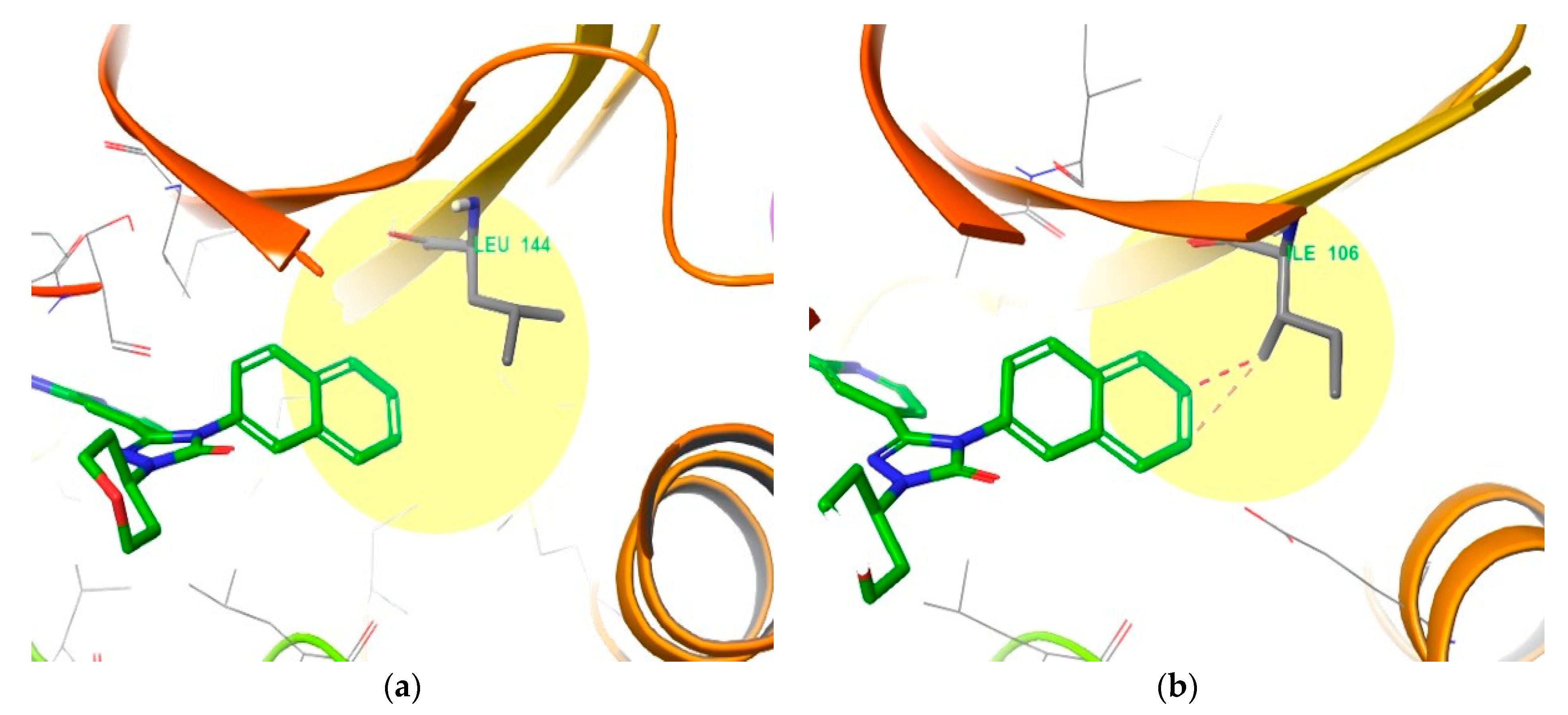
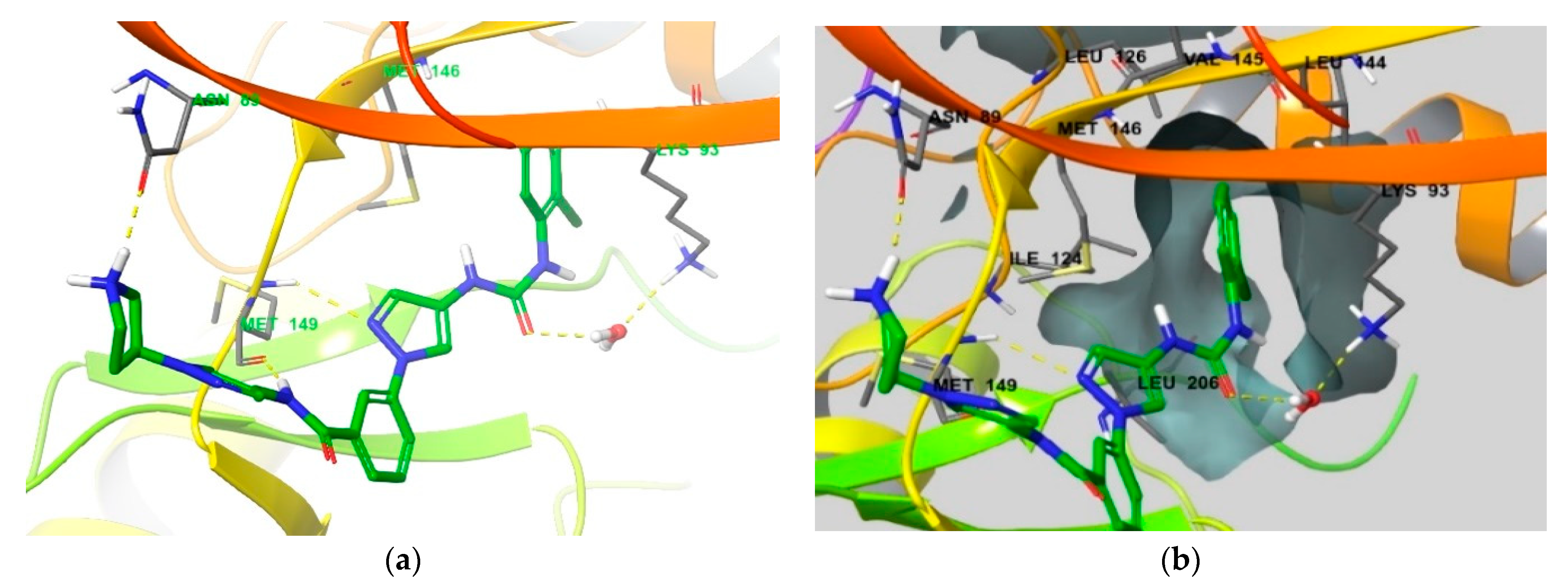
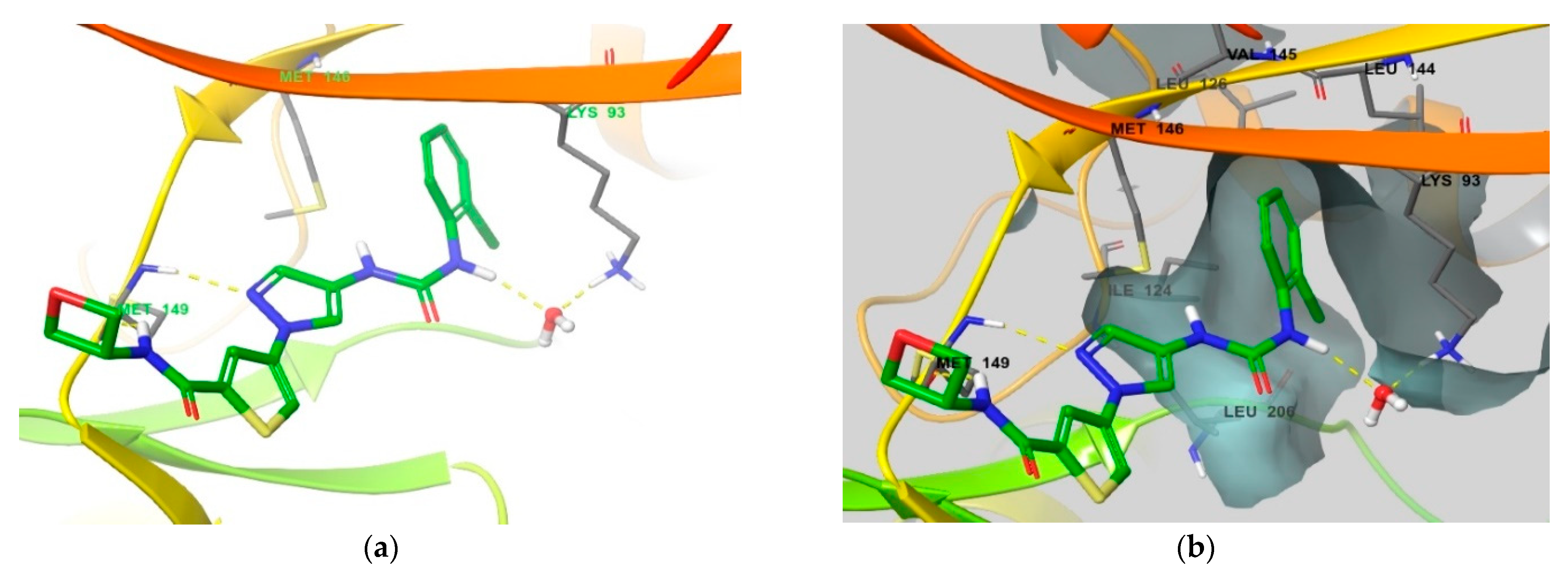
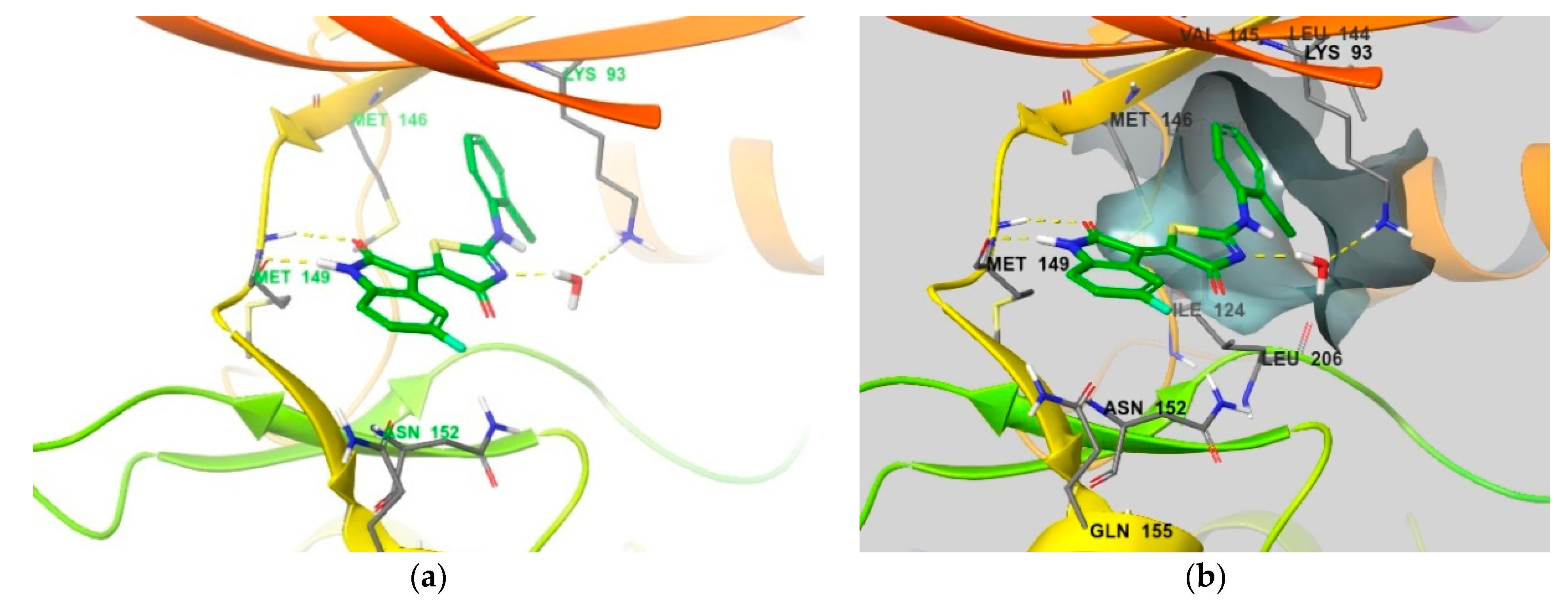
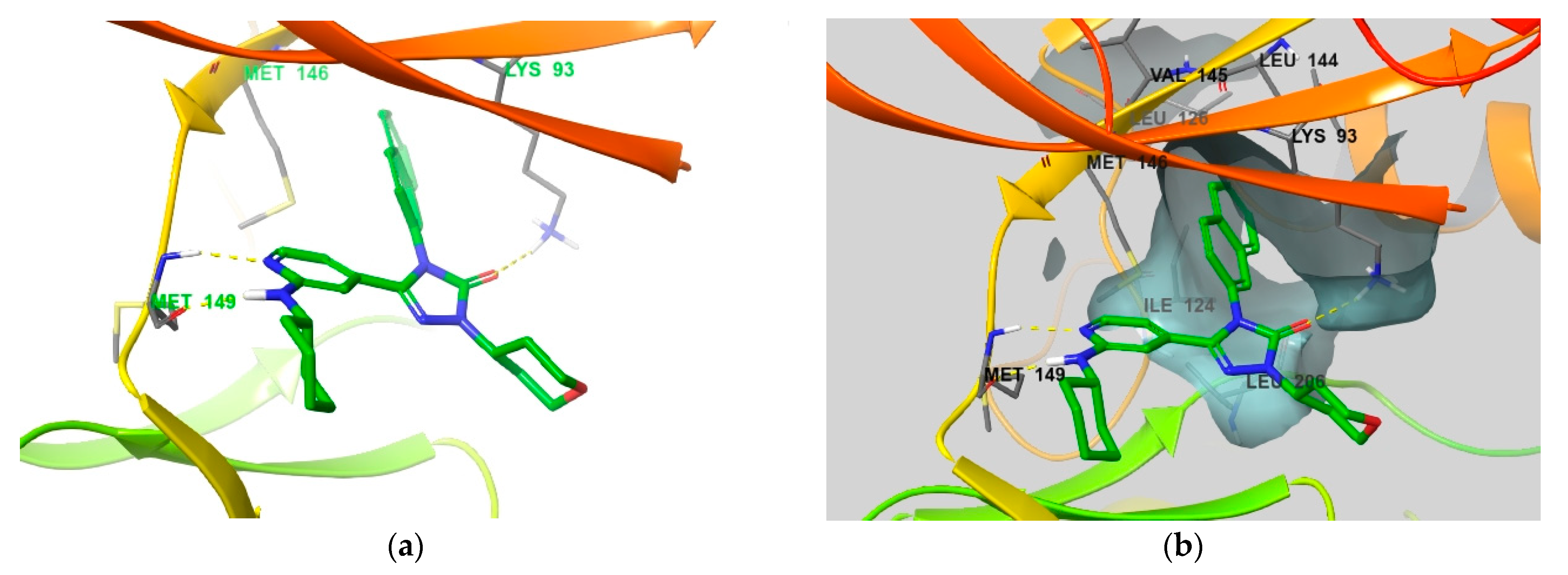
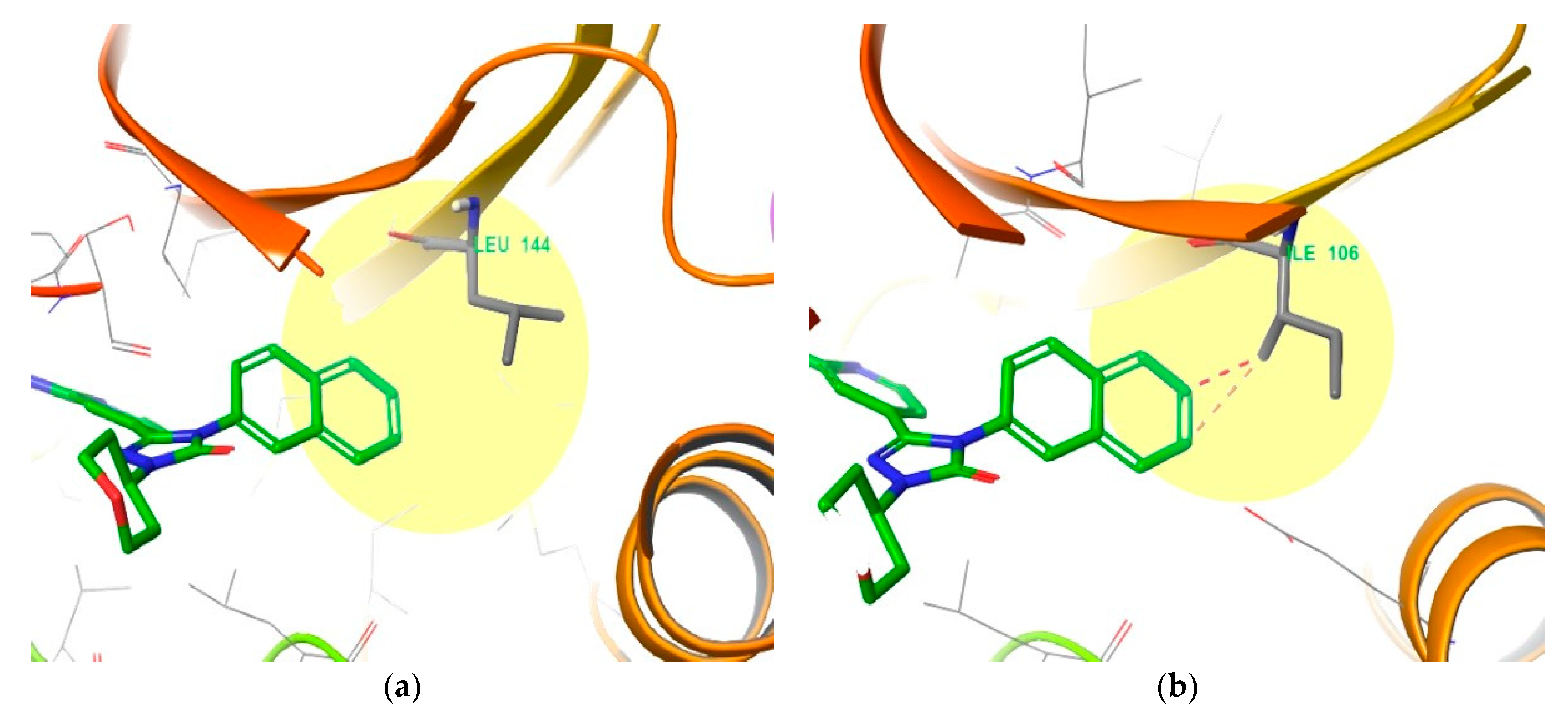
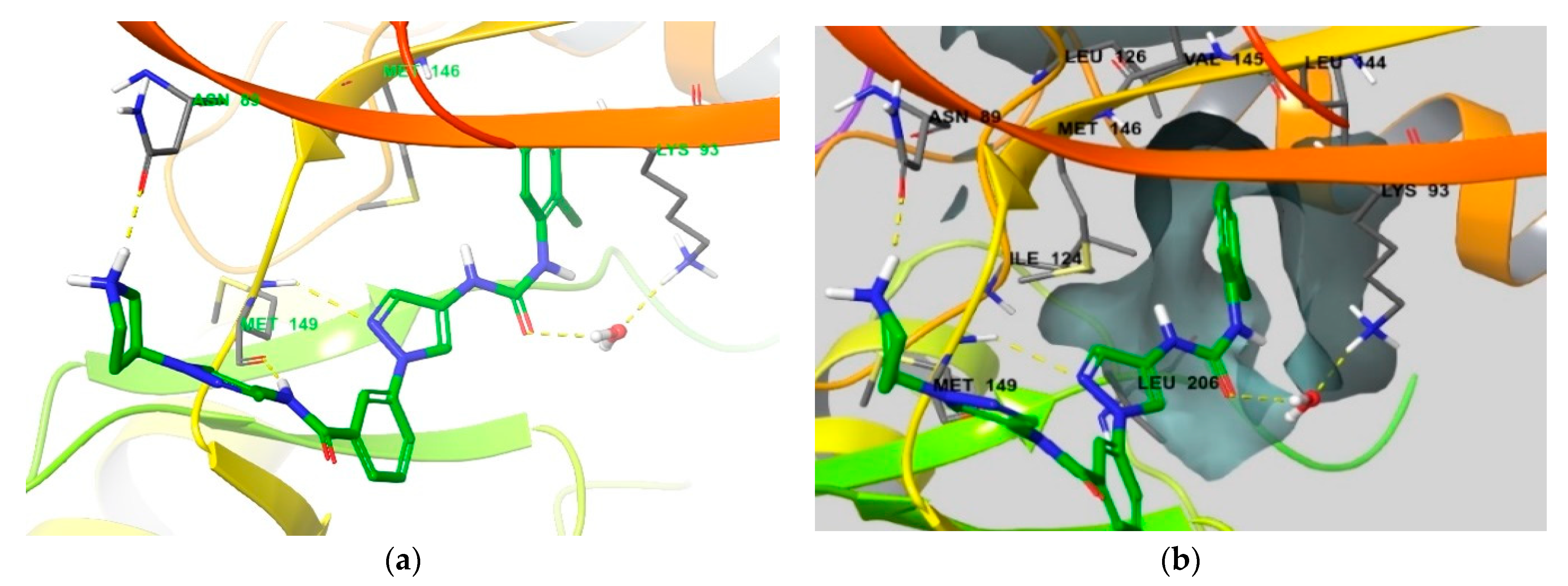
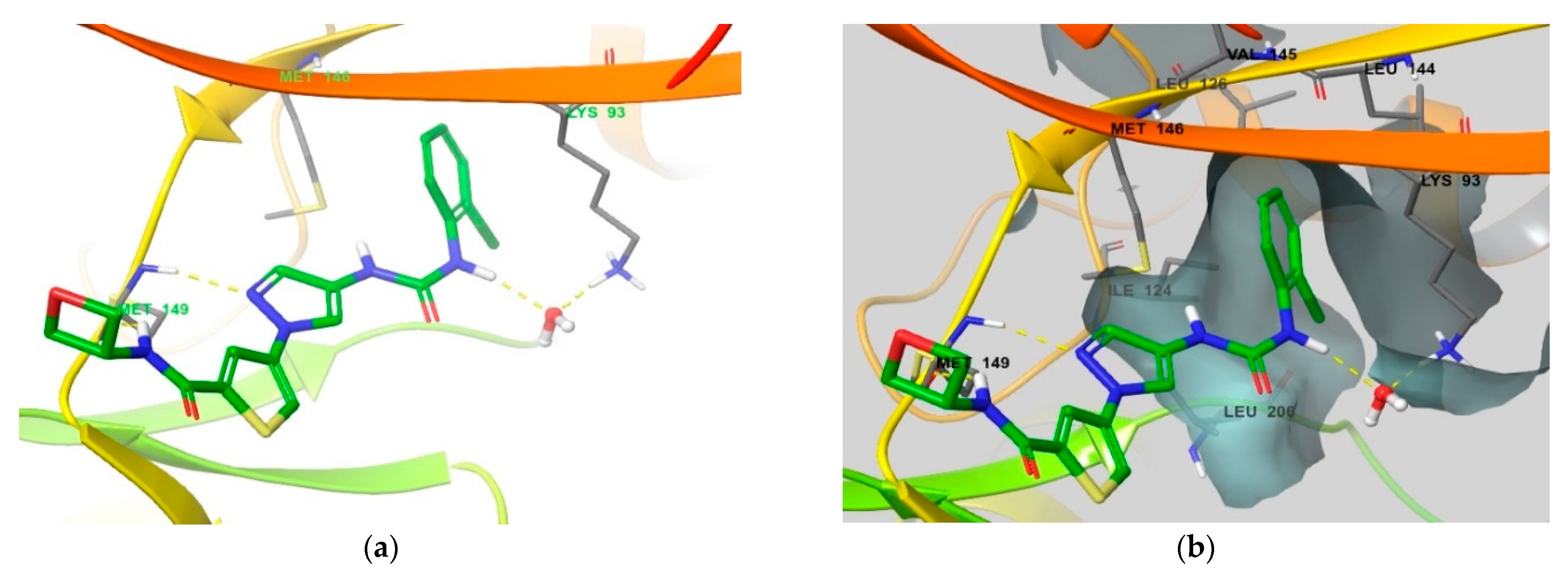
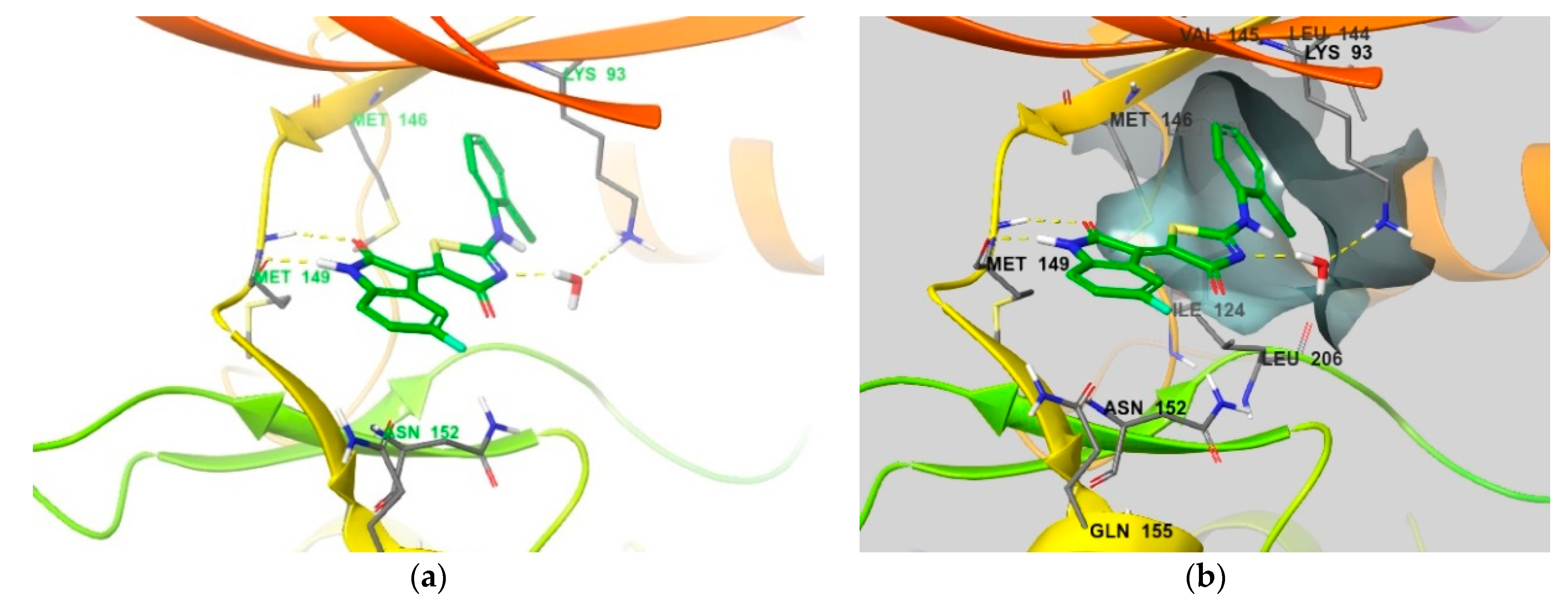
4. Functional Groups in Each Compound Contributing to Isoform Selectivity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/compilation-cder-new-molecular-entity-nme-drug-and-new-biologic-approvals (accessed on 14 September 2021).
- Ficarro, S.B.; McCleland, M.L.; Stukenberg, P.T.; Burke, D.J.; Ross, M.M.; Shabanowitz, J.; Hunt, D.F.; White, F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002, 20, 301–305. [Google Scholar] [CrossRef]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Roskoski, R.J. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Muller, S.; Chaikuad, A.; Gray, N.S.; Knapp, S. The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol. 2015, 11, 818–821. [Google Scholar] [CrossRef]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Small, D.H.; Cappai, R. Alois Alzheimer and Alzheimer’s disease: A centennial perspective. J. Neurochem. 2006, 99, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1341–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazawa, H.; Estus, S. The JNK/c-Jun cascade and Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2002, 17, 79–88. [Google Scholar] [CrossRef]
- Bruckner, S.R.; Tammariello, S.P.; Kuan, C.Y.; Flavell, R.A.; Rakic, P.; Estus, S. JNK3 contributes to c-Jun activation and apoptosis but not oxidative stress in nerve growth factor-deprived sympathetic neurons. J. Neurochem. 2001, 78, 298–303. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 Perpetuates Metabolic Stress Induced by Aβ Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef]
- Wang, W.; Shi, L.; Xie, Y.; Ma, C.; Li, W.; Su, X.; Huang, S.; Chen, R.; Zhu, Z.; Mao, Z.; et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 2004, 48, 195–202. [Google Scholar] [CrossRef]
- Hussein, M.; Chai, D.C.; Kyama, C.M.; Mwenda, J.M.; Palmer, S.S.; Gotteland, J.P.; D’Hooghe, T.M. c-Jun NH2-terminal kinase inhibitor bentamapimod reduces induced endometriosis in baboons: An assessor-blind placebo-controlled randomized study. Fertil. Steril. 2016, 105, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.S.; Altan, M.; Denis, D.; Tos, E.G.; Gotteland, J.P.; Osteen, K.G.; Bruner-Tran, K.L.; Nataraja, S.G. Bentamapimod (JNK inhibitor AS602801) induces regression of endometriotic lesions in animal models. Reprod. Sci. 2016, 23, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Plantevin, K.V.; Nadolny, L.; Delgado, M.; Ayala, L.; Clareen, S.S.; Hilgraf, R.; Albers, R.; Hegde, S.; D’Sidocky, N.; Sapienza, J.; et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 1433–1438. [Google Scholar] [CrossRef]
- Zheng, K.; Iqbal, S.; Hernandez, P.; Park, H.J.; LoGrasso, P.V.; Feng, Y. Design and Synthesis of Highly Potent and Isoform Selective JNK3 Inhibitors: SAR Studies on Aminopyrazole Derivatives. J. Med. Chem. 2014, 57, 10013–10030. [Google Scholar] [CrossRef]
- Feng, Y.; Park, H.J.; Bauer, L.; Ryu, J.C.; Yoon, S.O. Thiophene-Pyrazolourea Derivatives as Potent, Orally Bioavailable, and Isoform-Selective JNK3 Inhibitors. ACS Med. Chem. Lett. 2021, 12, 24–29. [Google Scholar] [CrossRef]
- Dou, X.; Huang, H.; Li, Y.; Jiang, L.; Wang, Y.; Jin, H.; Jiao, N.; Zhang, L.; Zhang, L.; Liu, Z. Multistage Screening Reveals 3-Substituted Indolin-2-one Derivatives as Novel and Isoform-Selective c-Jun N-terminal Kinase 3 (JNK3) Inhibitors: Implications to Drug Discovery for Potential Treatment of Neurodegenerative Diseases. J. Med. Chem. 2019, 62, 6645–6664. [Google Scholar] [CrossRef]
- Probst, G.D.; Bowers, S.; Sealy, J.M.; Truong, A.P.; Hom, R.K.; Galemmo, R.A.J.; Konradi, A.W.; Sham, H.L.; Quincy, D.A.; Pan, H.; et al. Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg. Med. Chem. Lett. 2011, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.; Ashwell, S.; Almeida, L.; Brassil, P.; Breed, J.; Deng, C.; Gero, T.; Grondine, M.; Horn, C.; Ioannidis, S.; et al. Discovery of Checkpoint Kinase Inhibitor (S)-5-(3-Fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by Structure-Based Design and Optimization of Thiophenecarboxamide Ureas. J. Med. Chem. 2012, 55, 5130–5142. [Google Scholar] [CrossRef] [PubMed]
- Garai, A.; Zeke, A.; Gogl, G.; Toro, I.; Fordos, F.; Blankenburg, H.; Barkai, T.; Varga, J.; Alexa, A.; Emig, D.; et al. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Eliana, M.C.T.; Zimmer, J.; Liang, Y.; Gray, N.S.; Tarsounas, M.; Knapp, S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, S.D.; Redman, A.M.; Wilson, J.W.; Deanda, F.; Shotwell, J.B.; Gerding, R.; Lei, H.; Yang, B.; Stevens, K.L.; Hassell, A.M.; et al. Optimization of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine IGF-1R tyrosine kinase inhibitors towards JNK selectivity. Bioorg. Med. Chem. Lett. 2009, 19, 360–364. [Google Scholar] [CrossRef]
- Liddle, J.; Bamborough, P.; Barker, M.D.; Campos, S.; Chung, C.W.; Cousins, R.P.C.; Faulder, P.; Heathcote, M.L.; Hobbs, H.; Holmes, D.S.; et al. 4-Phenyl-7-azaindoles as potent, selective and bioavailable IKK2 inhibitors demonstrating good in vivo efficacy. Bioorg. Med. Chem. Lett. 2012, 22, 5222–5226. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cociorva, O.M.; Nomanbhoy, T.; Weissig, H.; Li, Q.; Nakamura, K.; Liyanage, M.; Zhang, M.C.; Shih, A.Y.; Aban, A.; et al. Hit-to-lead optimization and kinase selectivity of imidazo[1,2-a]quinoxalin-4-amine derived JNK1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5217–5222. [Google Scholar] [CrossRef]
- Shaw, D.; Wang, S.M.; Villasenor, A.G.; Tsing, S.; Walter, D.; Browner, M.F.; Barnett, J.; Kuglstatter, A. The Crystal Structure of JNK2 Reveals Conformational Flexibility in the MAP Kinase Insert and Indicates Its Involvement in the Regulation of Catalytic Activity. J. Mol. Biol. 2008, 383, 885–893. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB. Available online: https://www.rcsb.org/structure/7CML (accessed on 14 September 2021).
- Kuglstatter, A.; Ghate, M.; Tsing, S.; Villasenor, A.G.; Shaw, D.; Barnett, J.W.; Browner, M.F. X-ray crystal structure of JNK2 complexed with the p38α inhibitor BIRB 796: Insights into the rational design of DFG-out binding MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5217–5220. [Google Scholar] [CrossRef]
- Park, H.J.; Iqbal, S.; Hernandez, P.; Mora, R.; Zheng, K.; Feng, Y.; LoGrasso, P. Structural Basis and Biological Consequences for JNK2/3 Isoform Selective Aminopyrazoles. Sci. Rep. 2015, 5, 8047. [Google Scholar] [CrossRef] [Green Version]
- Ansideri, F.; Macedo, J.T.; Eitel, M.; El-Gokha, A.; Zinad, D.S.; Scarpellini, C.; Kudolo, M.; Schollmeyer, D.; Boeckler, F.M.; Blaum, B.S.; et al. Structural Optimization of a Pyridinylimidazole Scaffold: Shifting the Selectivity from p38α Mitogen-Activated Protein Kinase to c-Jun N-Terminal Kinase 3. ACS Omega 2018, 3, 7809–7831. [Google Scholar] [CrossRef] [Green Version]
- Christopher, J.A.; Atkinson, F.L.; Bax, B.D.; Brown, M.J.B.; Champigny, A.C.; Chuang, T.T.; Jones, E.J.; Mosley, J.E.; Musgrave, J.R. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorg. Med. Chem. Lett. 2009, 19, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.; Hah, J.-M. A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies. Biomedicines 2021, 9, 1431. https://doi.org/10.3390/biomedicines9101431
Cho H, Hah J-M. A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies. Biomedicines. 2021; 9(10):1431. https://doi.org/10.3390/biomedicines9101431
Chicago/Turabian StyleCho, Hyunwook, and Jung-Mi Hah. 2021. "A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies" Biomedicines 9, no. 10: 1431. https://doi.org/10.3390/biomedicines9101431
APA StyleCho, H., & Hah, J.-M. (2021). A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies. Biomedicines, 9(10), 1431. https://doi.org/10.3390/biomedicines9101431