
Pathway-Based Personalized Analysis of Pan-Cancer Transcriptomic Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
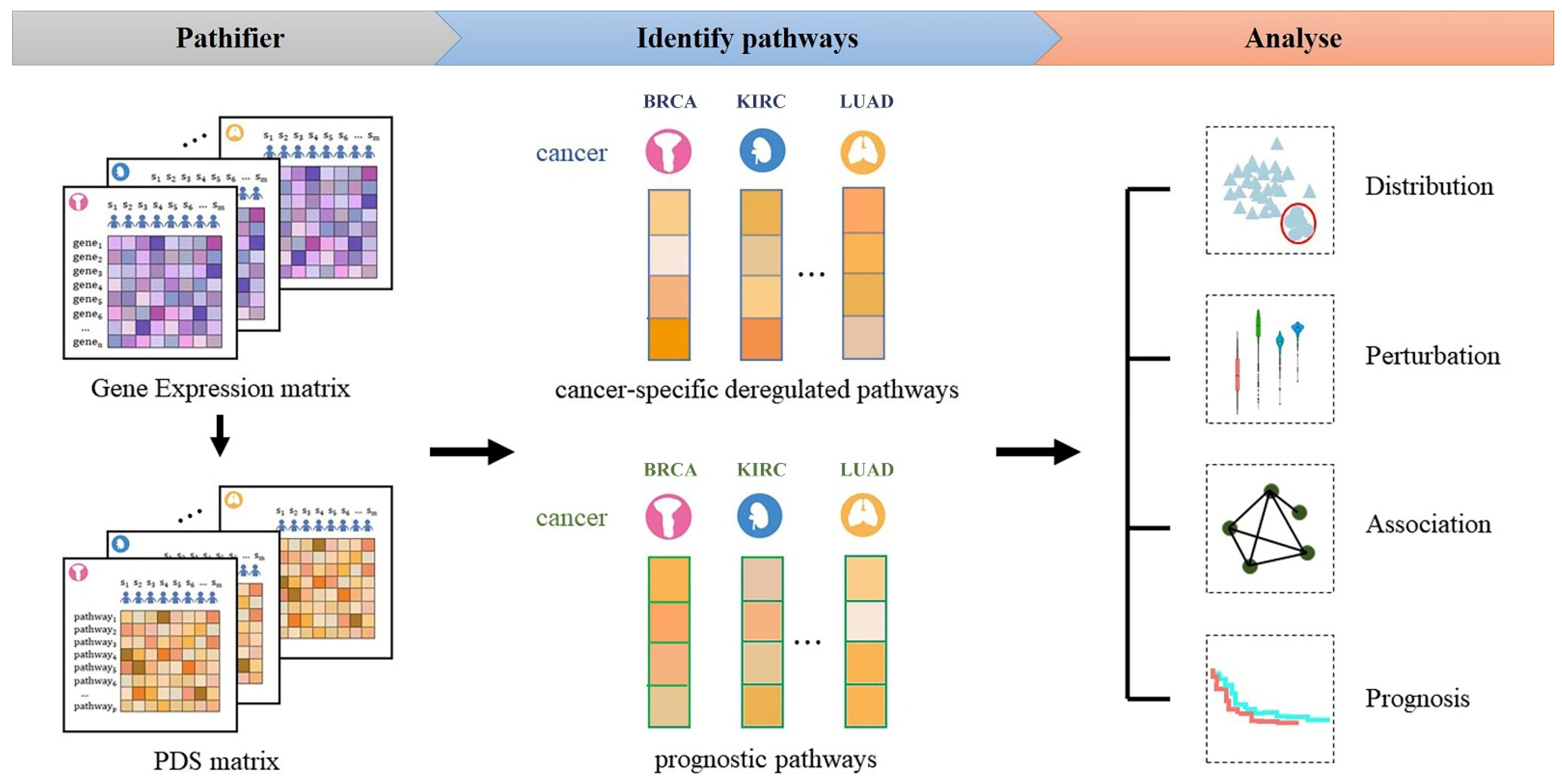
2.2. Overview of the Approach
2.3. Calculating the PDS
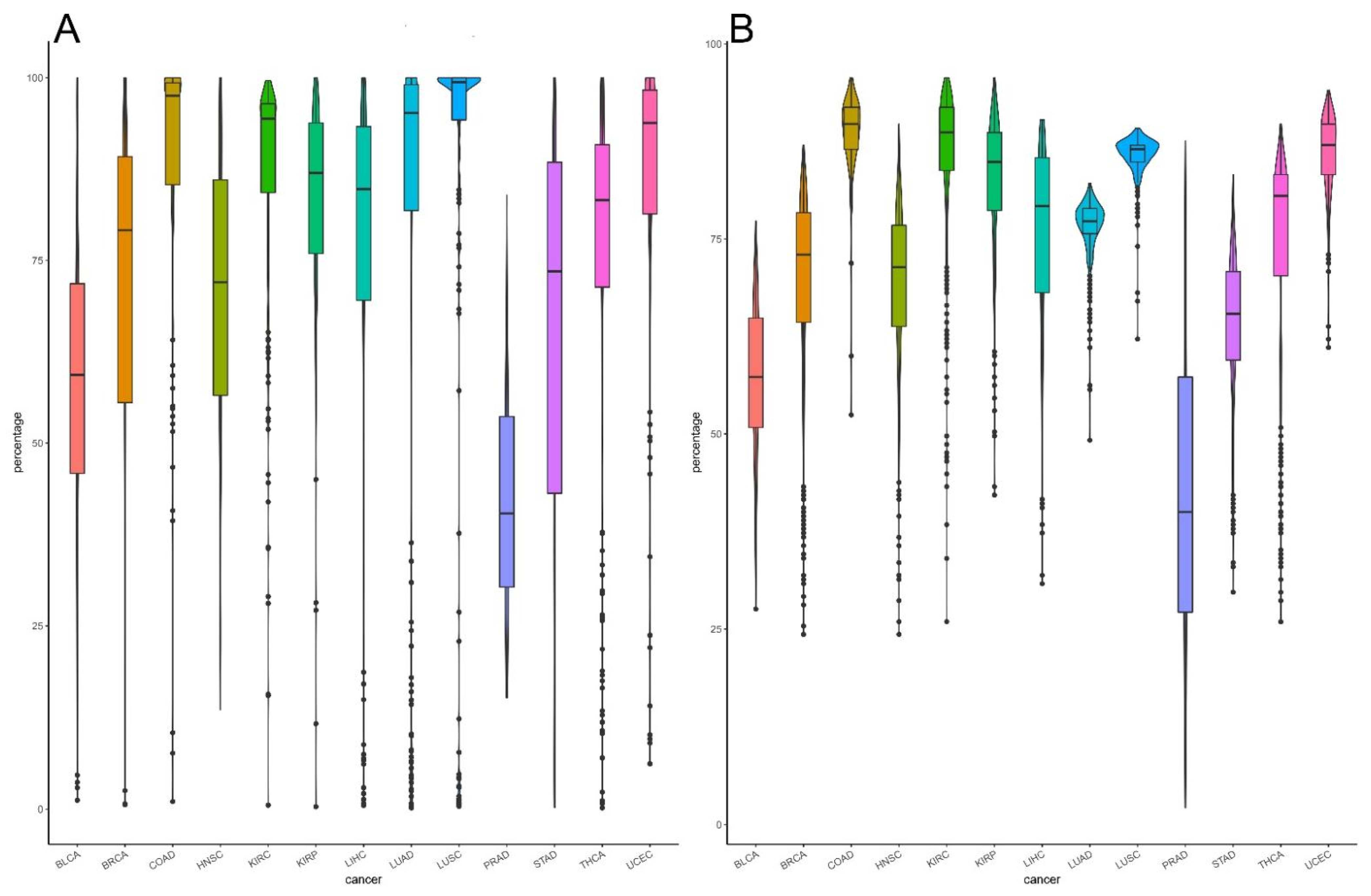
2.4. Constructing Classifier to Distinguish Cancers from Normal
2.5. Identifying Deregulated Cancer-Specific Deregulated Pathways
2.6. Identifying Prognostic Pathways
3. Results
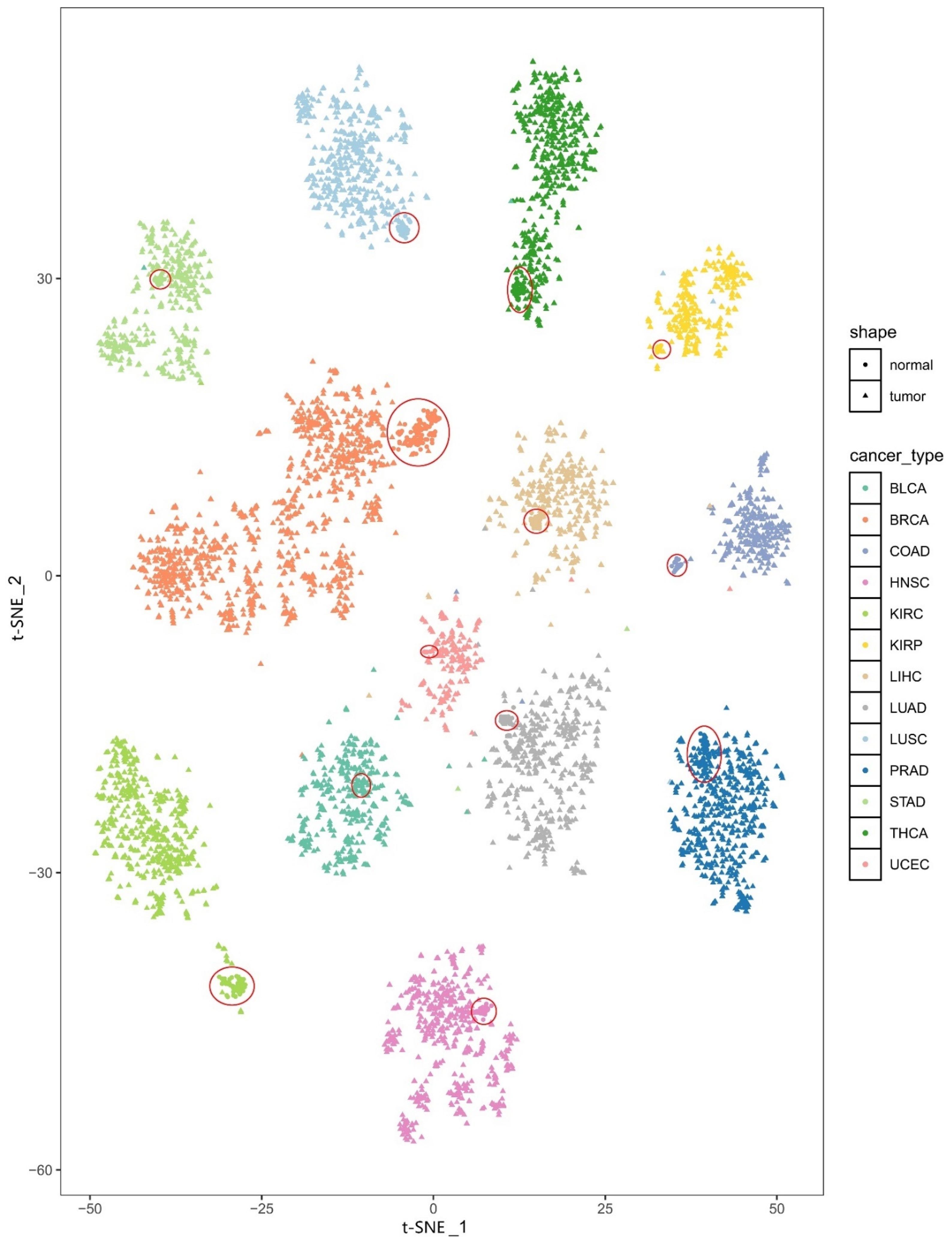
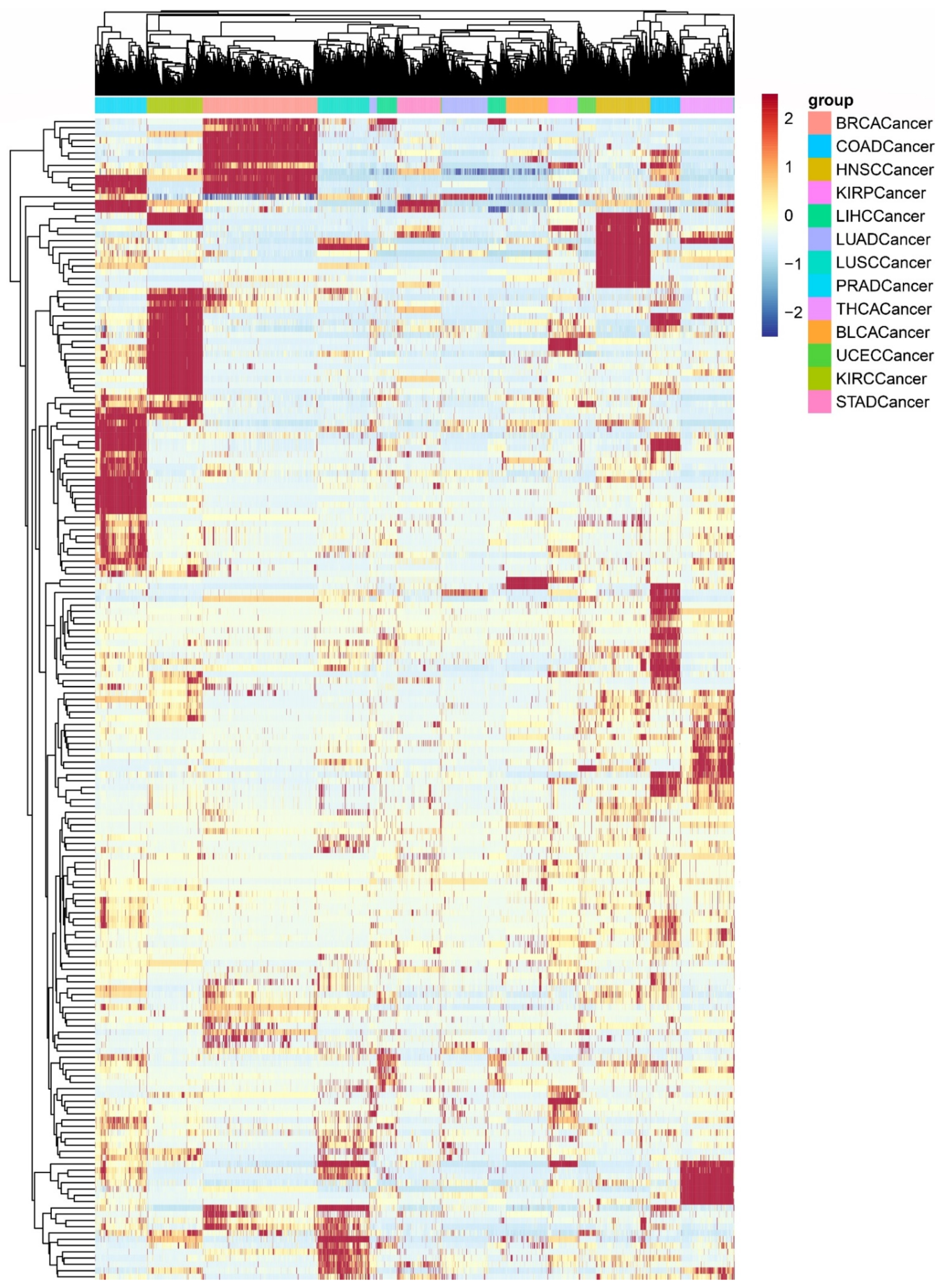
3.1. The Heterogeneity of the PDS
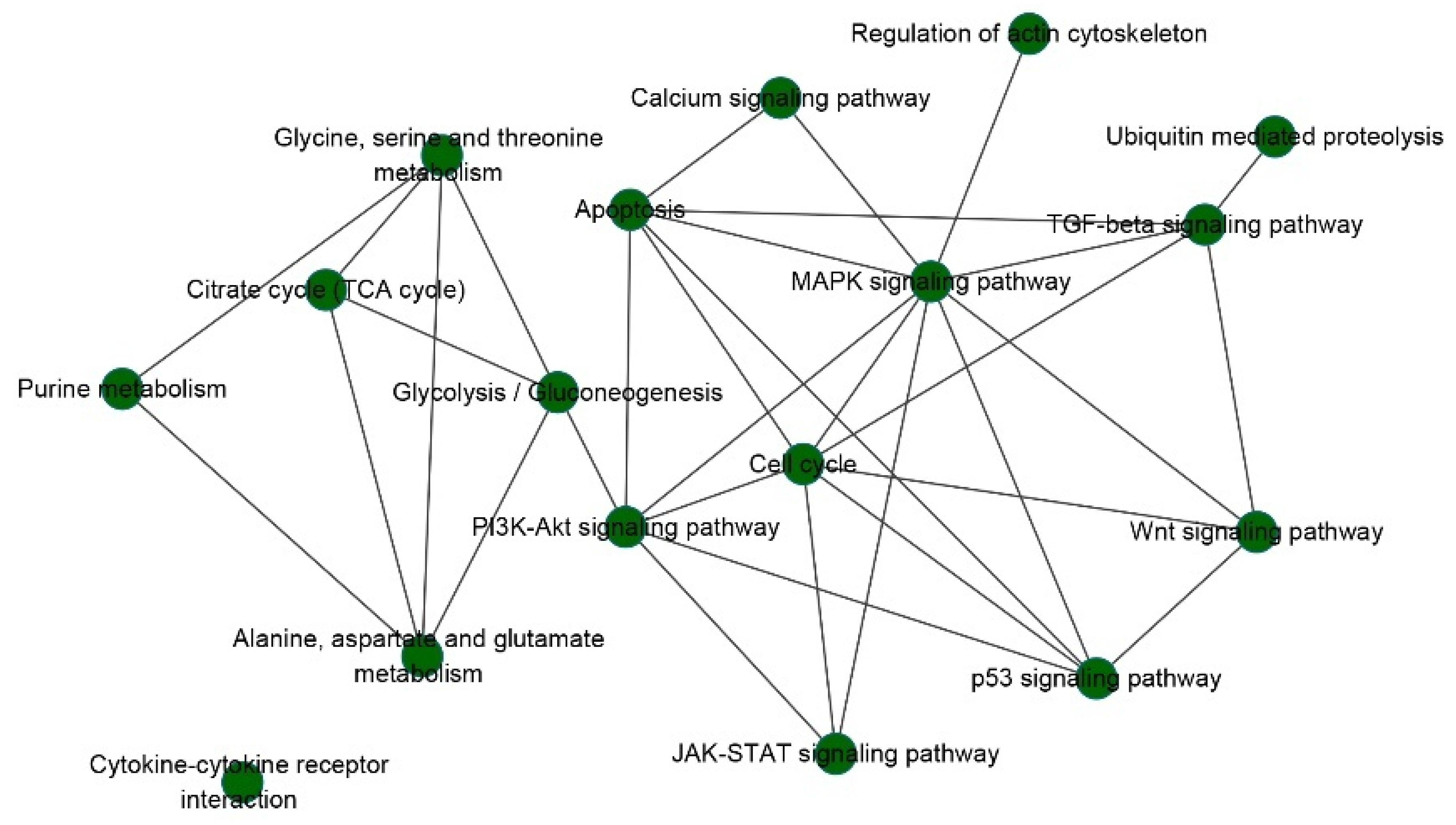
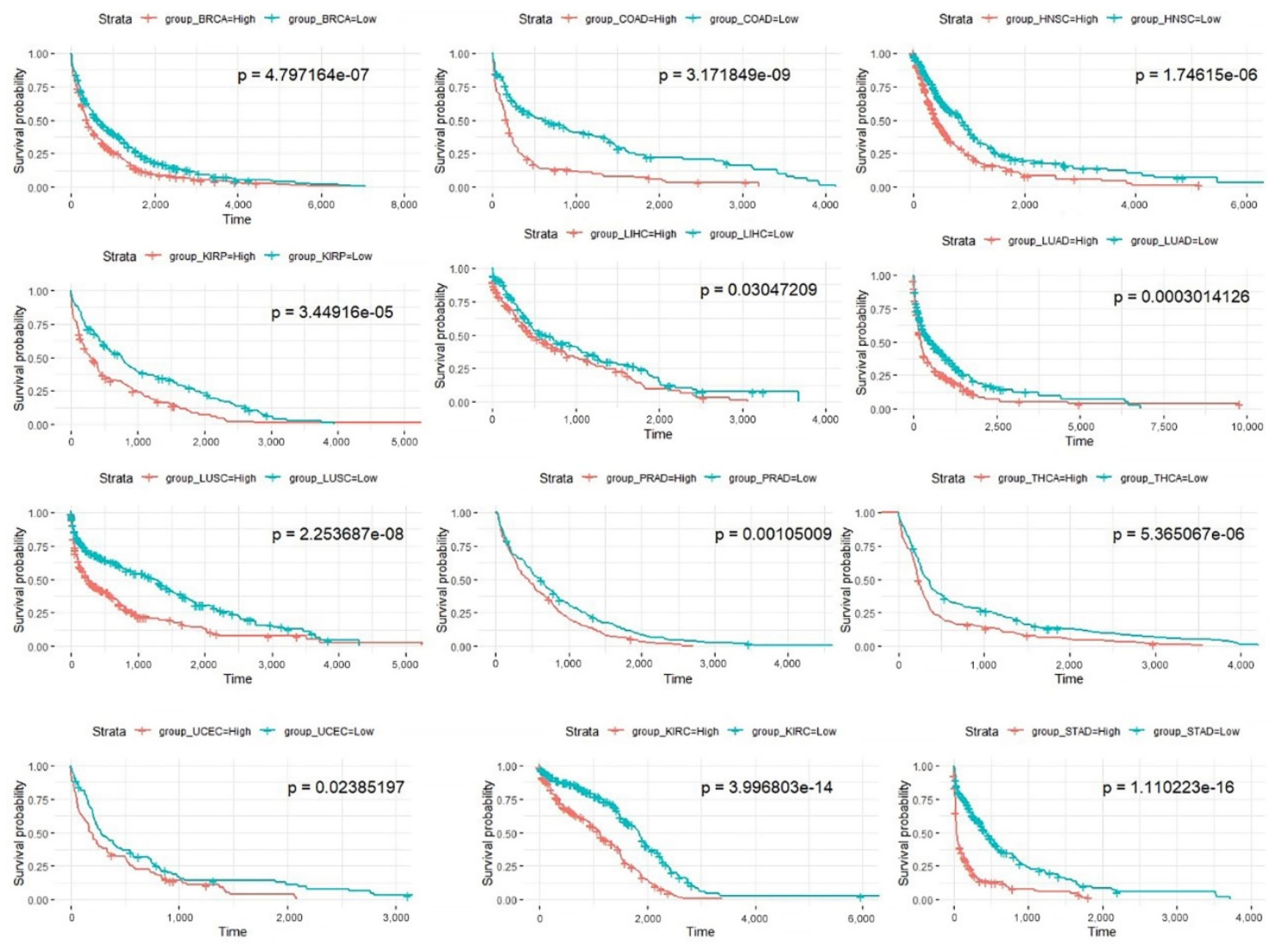
3.2. Prognostic Pathways
3.3. Prognostic Models Based on Pathways
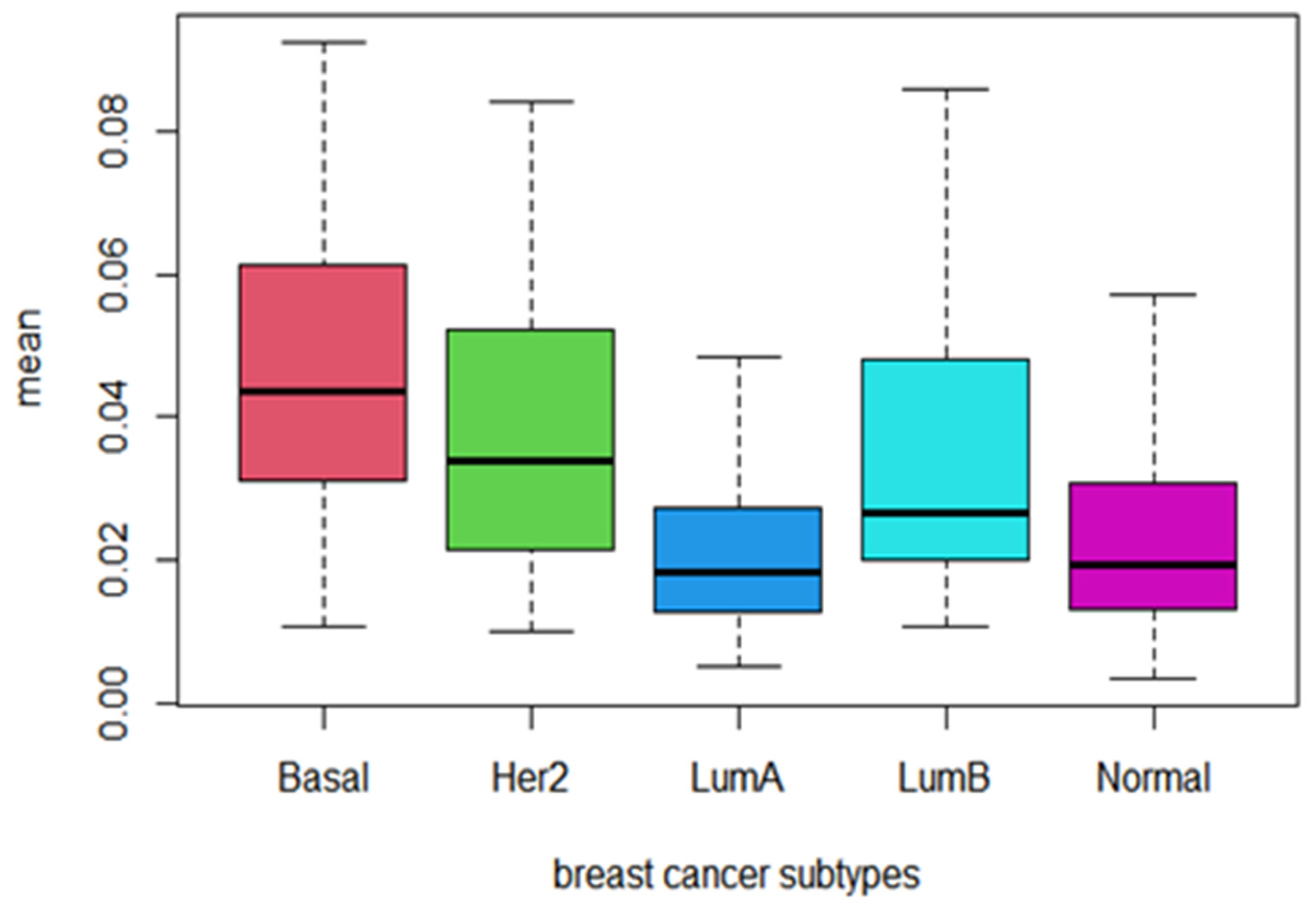
3.4. Behavior of Prognostic Pathways among Cancer Subtypes
3.5. Genes in Prognostic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Efroni, S.; Schaefer, C.F.; Buetow, K.H. Identification of Key Processes Underlying Cancer Phenotypes Using Biologic Pathway Analysis. PLoS ONE 2007, 2, e425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmert-Streib, F.; Glazko, G.V. Pathway Analysis of Expression Data: Deciphering Functional Building Blocks of Complex Diseases. PLoS Comput. Biol. 2011, 7, e1002053. [Google Scholar] [CrossRef] [Green Version]
- Drier, Y.; Sheffer, M.; Domany, E. Pathway-based personalized analysis of cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 6388–6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaske, C.; Benz, S.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Huang, S.; Yee, C.; Ching, T.; Yu, H.; Garmire, L.X. A Novel Model to Combine Clinical and Pathway-Based Transcriptomic Information for the Prognosis Prediction of Breast Cancer. PLoS Comput. Biol. 2014, 10, e1003851. [Google Scholar] [CrossRef]
- Livshits, A.; Git, A.; Fuks, G.; Caldas, C.; Domany, E. Pathway-based personalized analysis of breast cancer expression data. Mol. Oncol. 2015, 9, 1471–1483. [Google Scholar] [CrossRef]
- Fa, B.; Luo, C.; Tang, Z.; Yan, Y.; Zhang, Y.; Yu, Z. Pathway-based biomarker identification with crosstalk analysis for robust prognosis prediction in hepatocellular carcinoma. EBioMedicine 2019, 44, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nat. Cell Biol. 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; METABRIC Group; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Stuetzle, W. Principal Curves. J. Am. Stat. Assoc. 1989, 84, 502–516. [Google Scholar] [CrossRef]
- Gill, R. Multistate life-tables and regression models. Math. Popul. Stud. 1992, 3, 259–276. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Khanna, P.; Chua, P.J.; Wong, B.S.E.; Yin, C.; Thike, A.A.; Wan, W.K.; Tan, P.H.; Baeg, G.H. GRAM domain-containing protein 1B (GRAMD1B), a novel component of the JAK/STAT signaling pathway, functions in gastric carcinogenesis. Oncotarget 2017, 8, 115370–115383. [Google Scholar] [CrossRef]
- Pronobis, M.; Deuitch, N.; Posham, V.; Mimori-Kiyosue, Y.; Peifer, M. Reconstituting regulation of the canonical Wnt pathway by engineering a minimal β-catenin destruction machine. Mol. Biol. Cell 2017, 28, 41–53. [Google Scholar] [CrossRef]
- Miller, K.D.; Sauer, A.G.; Ortiz, A.P.; Fedewa, S.A.; Pinheiro, P.S.; Tortolero-Luna, G.; Martinez-Tyson, D.; Jemal, A.; Siegel, R.L. Cancer Statistics for Hispanics/Latinos. CA A Cancer J. Clin. 2018, 68, 425–445. [Google Scholar] [CrossRef] [Green Version]
- Brawley, O.W. Donna P Ankerst; Ian M Thompson. Screening for prostate cancer. CA Cancer J. Clin. 2009, 59, 264–273. [Google Scholar] [CrossRef]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell Survival Promoted by the Ras-MAPK Signaling Pathway by Transcription-Dependent and -Independent Mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradi-Marjaneh, R.; Hassanian, S.M.; Fiuji, H.; Soleimanpour, S.; Ferns, G.A.; Avan, A.; Khazaei, M. Toll like receptor signaling pathway as a potential therapeutic target in colorectal cancer. J. Cell. Physiol. 2018, 233, 5613–5622. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, T.O.P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF Therapies in the Clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef] [Green Version]
- Poultsides, G.A.; Servais, E.L.; Saltz, L.; Patil, S.; Kemeny, N.E.; Guillem, J.G.; Weiser, M.; Temple, L.K.; Wong, W.D.; Paty, P.B. Outcome of Primary Tumor in Patients With Synchronous Stage IV Colorectal Cancer Receiving Combination Chemotherapy Without Surgery As Initial Treatment. J. Clin. Oncol. 2009, 27, 3379–3384. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.-Y.; Hwang, J.-S.; Son, H.; Park, H.-I.; Oh, E.-S.; Kim, H.; Kim, D.K.; Choi, W.S.; Lee, B.-J.; Kim, H.-R.C.; et al. Identification of H-Ras-Specific Motif for the Activation of Invasive Signaling Program in Human Breast Epithelial Cells. Neoplasia 2011, 13, 98–107, IN1. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Simon, I.; Moreno, V.; Roepman, P.; Tabernero, J.; Snel, M.; Veer, L.V.; Salazar, R.; Bernards, R.; Capella, G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 2012, 62, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Cardone, R.A.; Bagorda, A.; Bellizzi, A.; Busco, G.; Guerra, L.; Paradiso, A.; Casavola, V.; Zaccolo, M.; Reshkin, S.J. Protein Kinase A Gating of a Pseudopodial-located RhoA/ROCK/p38/NHE1 Signal Module Regulates Invasion in Breast Cancer Cell Lines. Mol. Biol. Cell 2005, 16, 3117–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β an excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.; Tian, Y.; Ye, Q.; Li, J.; Ding, K.; Li, J. Improving the Prognosis of Colon Cancer through Knowledge-Based Clinical-Molecular Integrated Analysis. BioMed Res. Int. 2021, 2021, 9987819. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | TCGA RNA-Seq | METABRIC RNA-Seq | |
|---|---|---|---|
| Tumour Samples | Normal Samples | Tumour Samples | |
| Breast invasive carcinoma(BRCA) | 1102 | 113 | 1904 |
| Colon adenocarcinoma(COAD) | 287 | 41 | |
| Head and Neck squamous cell carcinoma(HNSC) | 522 | 44 | |
| Kidney renal papillary cell carcinoma(KIRP) | 291 | 32 | |
| Liver hepatocellular carcinoma(LIHC) | 374 | 50 | |
| Lung adenocarcinoma(LUAD) | 517 | 59 | |
| Lung squamous cell carcinoma(LUSC) | 502 | 51 | |
| Prostate adenocarcinoma(PRAD) | 498 | 52 | |
| Thyroid carcinoma(THCA) | 513 | 59 | |
| Bladder Urothelial Carcinoma(BLCA) | 408 | 19 | |
| Uterine Corpus Endometrial Carcinoma(UCEC) | 177 | 24 | |
| Kidney renal clear cell carcinoma(KIRC) | 534 | 72 | |
| Stomach adenocarcinoma(STAD) | 415 | 35 | |
| Total | 6140 | 651 | 1904 |
| Cancer | BRCA | COAD | HNSC | KIRP | LIHC | LUAD | LUSC | PRAD | THCA | BLCA | UCEC | KIRC | STAD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn | 0.966 | 0.986 | 0.972 | 0.973 | 0.943 | 0.987 | 0.992 | 0.904 | 0.973 | 0.985 | 0.969 | 0.968 | 0.965 |
| Sp | 0.998 | 1 | 0.991 | 0.992 | 0.999 | 1 | 1 | 0.928 | 0.969 | 0.82 | 0.995 | 0.984 | 0.999 |
| Acc | 0.968 | 0.988 | 0.973 | 0.975 | 0.949 | 0.988 | 0.993 | 0.906 | 0.972 | 0.977 | 0.971 | 0.984 | 0.967 |
| Starch and sucrose metabolism |
| Riboflavin metabolism |
| TGF-beta signaling pathway |
| Prostate cancer |
| Thyroid cancer |
| Small cell lung cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pian, C.; He, M.; Chen, Y. Pathway-Based Personalized Analysis of Pan-Cancer Transcriptomic Data. Biomedicines 2021, 9, 1502. https://doi.org/10.3390/biomedicines9111502
Pian C, He M, Chen Y. Pathway-Based Personalized Analysis of Pan-Cancer Transcriptomic Data. Biomedicines. 2021; 9(11):1502. https://doi.org/10.3390/biomedicines9111502
Chicago/Turabian StylePian, Cong, Mengyuan He, and Yuanyuan Chen. 2021. "Pathway-Based Personalized Analysis of Pan-Cancer Transcriptomic Data" Biomedicines 9, no. 11: 1502. https://doi.org/10.3390/biomedicines9111502
APA StylePian, C., He, M., & Chen, Y. (2021). Pathway-Based Personalized Analysis of Pan-Cancer Transcriptomic Data. Biomedicines, 9(11), 1502. https://doi.org/10.3390/biomedicines9111502