
In Vitro Disease Models of the Endocrine Pancreas


 , ,
, ,  and
and 
Abstract
1. Introduction—The Need for In Vitro Models of the Pancreas
2. The Pancreas
2.1. Anatomy and Physiology of the Pancreas
2.2. Pathophysiology of Type 1 and Type 2 Diabetes Mellitus
3. Cell Sources for Pancreatic In Vitro Models
3.1. Transformed Cell Lines
3.2. Primary Cell Lines
3.3. Stem Cells
3.4. Other Cell Sources
4. Types of In Vitro Models of the Pancreas
4.1. 2D pancreatic Cell Culture Models
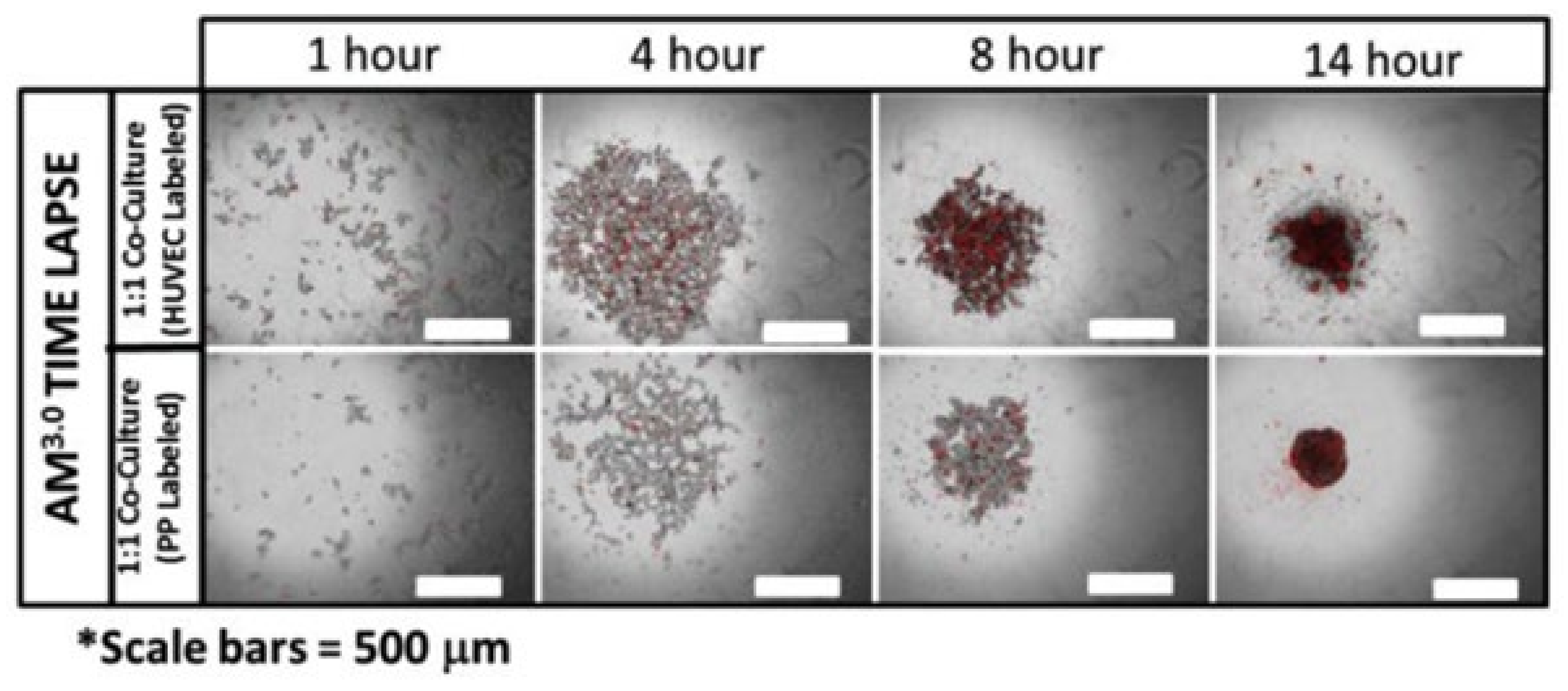
4.2. Pancreatic Co-Culture Models
4.3. The Need for 3D In vitro Models
4.3.1. Organoids
4.3.2. In vitro Modelling with 3D Biomimetic Scaffolds
4.3.3. Pancreatic Tissue-Derived Scaffolds
4.3.4. Biomimetic Scaffolds from Synthetic Polymers
4.3.5. Biomimetic Scaffolds from Naturally Derived Polymers
4.3.6. 3D Bioprinting of Biomimetic Scaffolds
4.4. Bioreactors
4.5. Pancreas-on-a-Chip
4.6. Tissue Slices
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Ramos, T.; Moroni, L. Tissue engineering and regenerative medicine 2019: The role of biofabrication—A year in review. Tissue Eng. Part C Methods 2019, 26, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Tavakol, D.N.; Fleischer, S.; Vunjak-Novakovic, G. Harnessing organs-on-a-chip to model tissue regeneration. Cell Stem Cell 2021, 28, 993–1015. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2021, 20, 345–361. [Google Scholar] [CrossRef]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; del Nery, E.; Ebner, D.; Montoya, M.C.; Östling, P.; Pietiäinen, V.; Price, L.S. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R. A decade of progress in tissue engineering. Nat. Protoc. 2016, 11, 1775–1781. [Google Scholar] [CrossRef]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.-J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R. Engineered in vitro disease models. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 195–262. [Google Scholar] [CrossRef] [PubMed]
- Trapecar, M.; Wogram, E.; Svoboda, D.; Communal, C.; Omer, A.; Lungjangwa, T.; Sphabmixay, P.; Velazquez, J.; Schneider, K.; Wright, C.W.; et al. Human physiomimetic model integrating microphysiological systems of the gut, liver, and brain for studies of neurodegenerative diseases. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Mattei, G.; Giusti, S.; Ahluwalia, A. Design criteria for generating physiologically relevant in vitro models in bioreactors. Processes 2014, 2, 548–569. [Google Scholar] [CrossRef]
- Rees, D.; Alcolado, J. Animal models of diabetes mellitus. Diabet. Med. 2005, 22, 359–370. [Google Scholar]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-cell electrical activity and insulin secretion: Of mice and men. Physiol Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef]
- Moreira, L.; Bakir, B.; Chatterji, P.; Dantes, Z.; Reichert, M.; Rustgi, A.K. Pancreas 3D organoids: Current and future aspects as a research platform for personalized medicine in pancreatic cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 289–298. [Google Scholar] [CrossRef]
- Chen, H.; Zhuo, Q.; Ye, Z.; Xu, X.; Ji, S. Organoid model: A new hope for pancreatic cancer treatment? Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188466. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Plenker, D.; Baker, L.A.; Tuveson, D.A. Organoid models for translational pancreatic cancer research. Curr. Opin. Genet. Dev. 2019, 54, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of acute and chronic pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef]
- Geron, E.; Schejter, E.D.; Shilo, B.-Z. Assessing the secretory capacity of pancreatic acinar cells. J. Vis. Exp. 2014, 51799. [Google Scholar] [CrossRef]
- Hegyi, P.; Maléth, J.; Venglovecz, V.; Rakonczay, Z. Pancreatic ductal bicarbonate secretion: Challenge of the acinar acid load. Front. Physiol. 2011, 2. [Google Scholar] [CrossRef]
- Reichert, M.; Rustgi, A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Investig. 2011, 121, 4572–4578. [Google Scholar] [CrossRef]
- Leung, P.S.; Ip, S.P. Pancreatic acinar cell: Its role in acute pancreatitis. Int. J. Biochem. Cell Biol. 2006, 38, 1024–1030. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Campbell-Thompson, M.; Kusmartseva, I.; Kaestner, K.H. Organisation of the human pancreas in health and in diabetes. Diabetologia 2020, 63, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, N.; Petersen, O.H. Pancreatic acinar cells: Localization of acetylcholine receptors and the importance of chloride and calcium for acetylcholine-evoked depolarization. J. Physiol. 1977, 269, 723–733. [Google Scholar] [CrossRef]
- Yule, D.I.; Lawrie, A.M.; Gallacher, D.V. Acetylcholine and cholecystokinin induce different patterns of oscillating calcium signals in pancreatic acinar cells. Cell Calcium 1991, 12, 145–151. [Google Scholar] [CrossRef]
- Williams, J.A. Cholecystokinin (CCK) regulation of pancreatic acinar cells: Physiological actions and signal transduction mechanisms. Compr. Physiol. 2019, 9, 535–564. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634. [Google Scholar] [CrossRef]
- Saito, K.; Iwama, N.; Takahashi, T. Morphometrical analysis on topographical difference in size distribution, number and volume of islets in the human pancreas. Tohoku J. Exp. Med. 1978, 124, 177–186. [Google Scholar] [CrossRef]
- Dolenšek, J.; Rupnik, M.S.; Stožer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef]
- Lammert, E.; Thorn, P. The role of the islet niche on beta cell structure and function. J. Mol. Biol. 2020, 432, 1407–1418. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695–709. [Google Scholar] [CrossRef]
- Stozer, A.; Dolensek, J.; Rupnik, M.S. Glucose-stimulated calcium dynamics in islets of Langerhans in acute mouse pancreas tissue slices. PLoS ONE 2013, 8, e54638. [Google Scholar] [CrossRef]
- Olofsson, C.S.; Göpel, S.O.; Barg, S.; Galvanovskis, J.; Ma, X.; Salehi, A.; Rorsman, P.; Eliasson, L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflug. Arch. 2002, 444, 43–51. [Google Scholar] [CrossRef]
- Dhatariya, K.K.; Glaser, N.S.; Codner, E.; Umpierrez, G.E. Diabetic ketoacidosis. Nat. Rev. Dis. Primers 2020, 6, 40. [Google Scholar] [CrossRef]
- Grotzke, M.; Jones, R.E. CHAPTER 2—Acute and chronic complications of diabetes. In Endocrine Secrets, 6th ed.; McDermott, M.T., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2013; pp. 16–27. [Google Scholar]
- Mathew, P.; Thoppil, D. Hypoglycemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- King, A.; Bowe, J. Animal models for diabetes: Understanding the pathogenesis and finding new treatments. Biochem. Pharmacol. 2016, 99, 1–10. [Google Scholar] [CrossRef]
- Harcourt, B.E.; Penfold, S.A.; Forbes, J.M. Coming full circle in diabetes mellitus: From complications to initiation. Nat. Rev. Endocrinol. 2013, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.M.; Johnson, R.K.; Stene, L.C. Type 1 diabetes-early life origins and changing epidemiology. Lancet Diabetes Endocrinol. 2020, 8, 226–238. [Google Scholar] [CrossRef]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar] [CrossRef]
- Craig, M.E.; Kim, K.W.; Isaacs, S.R.; Penno, M.A.; Hamilton-Williams, E.E.; Couper, J.J.; Rawlinson, W.D. Early-life factors contributing to type 1 diabetes. Diabetologia 2019, 62, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; Redondo, M.J. New insights on the genetics of type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 181–187. [Google Scholar] [CrossRef] [PubMed]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Type 2 diabetes and remission: Practical management guided by pathophysiology. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Mezza, T.; Cinti, F.; Cefalo, C.M.A.; Pontecorvi, A.; Kulkarni, R.N.; Giaccari, A. β-cell fate in human insulin resistance and type 2 diabetes: A perspective on islet plasticity. Diabetes 2019, 68, 1121–1129. [Google Scholar] [CrossRef]
- Wortham, M.; Sander, M. Mechanisms of β-cell functional adaptation to changes in workload. Diabetes Obes. Metab. 2016, 18, 78–86. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Madiraju, S.R.M. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, β-cells, and diabetes: The emperor has no clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Al-Mrabeh, A.; Sattar, N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 726–736. [Google Scholar] [CrossRef]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 11. [Google Scholar] [CrossRef]
- Skelin, M.; Rupnik, M.; Cencic, A. Pancreatic beta cell lines and their applications in diabetes mellitus research. Altex 2010, 27, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knospel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S.; Weir, G.C. New sources of pancreatic β-cells. Nat. Biotechnol. 2005, 23, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Gradisnik, L.; Maver, U.; Bosnjak, R.; Velnar, T. Optimised isolation and characterisation of adult human astrocytes from neurotrauma patients. J. Neurosci. Methods 2020, 341, 108796. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, J.F.; Stocker, C.J. Isolation and purification of rodent pancreatic islets of langerhans. Methods Mol. Biol. 2020, 2076, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.-R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Komatsu, H.; Kang, D.; Medrano, L.; Barriga, A.; Mendez, D.; Rawson, J.; Omori, K.; Ferreri, K.; Tai, Y.C.; Kandeel, F.; et al. Isolated human islets require hyperoxia to maintain islet mass, metabolism, and function. Biochem. Biophys. Res. Commun. 2016, 470, 534–538. [Google Scholar] [CrossRef]
- Komatsu, H.; Cook, C.; Wang, C.-H.; Medrano, L.; Lin, H.; Kandeel, F.; Tai, Y.-C.; Mullen, Y. Oxygen environment and islet size are the primary limiting factors of isolated pancreatic islet survival. PLoS ONE 2017, 12, e0183780. [Google Scholar] [CrossRef]
- Rosenberg, L.; Wang, R.; Paraskevas, S.; Maysinger, D. Structural and functional changes resulting from islet isolation lead to islet cell death. Surgery 1999, 126, 393–398. [Google Scholar] [CrossRef]
- Ilieva, A.; Yuan, S.; Wang, R.; Agapitos, D.; Hill, D.; Rosenberg, L. Pancreatic islet cell survival following islet isolation: The role of cellular interactions in the pancreas. J. Endocrinol. 1999, 161, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Rosenberg, L. Maintenance of beta-cell function and survival following islet isolation requires re-establishment of the islet-matrix relationship. J. Endocrinol. 1999, 163, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gosak, M.; Markovič, R.; Dolenšek, J.; Rupnik, M.S.; Marhl, M.; Stožer, A.; Perc, M. Network science of biological systems at different scales: A review. Phys. Life Rev. 2017, 24, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Green, B.J.; Altamentova, S.M.; Rocheleau, J.V. A microfluidic device designed to induce media flow throughout pancreatic islets while limiting shear-induced damage. Lab Chip 2013, 13, 4374–4384. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kumar, N.; Kalsan, M.; Saini, A.; Chandra, R. Mechanism of induction: Induced pluripotent stem cells (iPSCs). J. Stem Cells 2015, 10, 43. [Google Scholar]
- Kondo, Y.; Toyoda, T.; Inagaki, N.; Osafune, K. iPSC technology-based regenerative therapy for diabetes. J. Diabetes Investig. 2018, 9, 234–243. [Google Scholar] [CrossRef]
- Toyoda, T.; Mae, S.; Tanaka, H.; Kondo, Y.; Funato, M.; Hosokawa, Y.; Sudo, T.; Kawaguchi, Y.; Osafune, K. Cell aggregation optimizes the differentiation of human ESCs and iPSCs into pancreatic bud-like progenitor cells. Stem Cell Res. 2015, 14, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 2007, 25, 1940–1953. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ying, L.; Lu, L.; Galvão, A.M.; Mills, J.A.; Lin, H.C.; Kotton, D.N.; Shen, S.S.; Nostro, M.C.; Choi, J.K. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell 2012, 10, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Hrvatin, S.; O’Donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, β cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012, 30, 261. [Google Scholar] [CrossRef]
- Merkle, F.T.; Eggan, K. Modeling human disease with pluripotent stem cells: From genome association to function. Cell Stem Cell 2013, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.-R.; Harb, G.; Poh, Y.-C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Millman, J.R.; Xie, C.; van Dervort, A.; Gürtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Melton, D.A. How to make a functional β-cell. Development 2013, 140, 2472–2483. [Google Scholar] [CrossRef]
- Kerr-Conte, J.; Pattou, F.; Lecomte-Houcke, M.; Xia, Y.; Boilly, B.; Proye, C.; Lefebvre, J. Ductal cyst formation in collagen-embedded adult human islet preparations: A means to the reproduction of nesidioblastosis in vitro. Diabetes 1996, 45, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Prabakar, K.R.; Domínguez-Bendala, J.; Molano, R.D.; Pileggi, A.; Villate, S.; Ricordi, C.; Inverardi, L. Generation of glucose-responsive, insulin-producing cells from human umbilical cord blood-derived mesenchymal stem cells. Cell Transplant. 2012, 21, 1321–1339. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tableros, V.; Gai, C.; Gomez, Y.; Giunti, S.; Pasquino, C.; Deregibus, M.C.; Tapparo, M.; Pitino, A.; Tetta, C.; Brizzi, M.F.; et al. Islet-like structures generated in vitro from adult human liver stem cells revert hyperglycemia in diabetic SCID mice. Stem Cell Rev. Rep. 2019, 15, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Romo-Morales, A.; Przyborski, S. An introduction to the third dimension for routine cell culture. In Technology Platforms for 3D Cell Culture A User’s Guide; Wiley: Chichester, UK, 2017; Volume 1. [Google Scholar]
- Knight, E.; Przyborski, S. Advances in 3D cell culture technologies enabling tissue-like structures to be created in vitro. J. Anat. 2015, 227, 746–756. [Google Scholar] [CrossRef]
- Phelps, E.A.; Cianciaruso, C.; Santo-Domingo, J.; Pasquier, M.; Galliverti, G.; Piemonti, L.; Berishvili, E.; Burri, O.; Wiederkehr, A.; Hubbell, J.A.; et al. Advances in pancreatic islet monolayer culture on glass surfaces enable super-resolution microscopy and insights into beta cell ciliogenesis and proliferation. Sci. Rep. 2017, 7, 45961. [Google Scholar] [CrossRef]
- Nagata, N.; Gu, Y.; Hori, H.; Balamurugan, A.; Touma, M.; Kawakami, Y.; Wang, W.; Baba, T.T.; Satake, A.; Nozawa, M. Evaluation of insulin secretion of isolated rat islets cultured in extracellular matrix. Cell Transplant. 2001, 10, 447–451. [Google Scholar] [CrossRef]
- Hammar, E.B.; Irminger, J.-C.; Rickenbach, K.; Parnaud, G.; Ribaux, P.; Bosco, D.; Rouiller, D.G.; Halban, P.A. Activation of NF-κB by extracellular matrix is involved in spreading and glucose-stimulated insulin secretion of pancreatic beta cells. J. Biol. Chem. 2005, 280, 30630–30637. [Google Scholar] [CrossRef]
- Pinkse, G.G.; Bouwman, W.P.; Jiawan-Lalai, R.; Terpstra, O.; Bruijn, J.A.; de Heer, E. Integrin signaling via RGD peptides and anti-β1 antibodies confers resistance to apoptosis in islets of Langerhans. Diabetes 2006, 55, 312–317. [Google Scholar] [CrossRef]
- Andersen, P.L.; Vermette, P. Biomimetic Surfaces Supporting Dissociated Pancreatic Islet Cultures. Colloids Surf. B Biointerfaces 2017, 159, 166–173. [Google Scholar] [CrossRef] [PubMed]
- De Souza, B.M.; Bouças, A.P.; Oliveira, F.D.; Reis, K.P.; Ziegelmann, P.; Bauer, A.C.; Crispim, D. Effect of co-culture of mesenchymal stem/stromal cells with pancreatic islets on viability and function outcomes: A systematic review and meta-analysis. Islets 2017, 9, 30–42. [Google Scholar] [CrossRef]
- Jun, Y.; Kang, A.R.; Lee, J.S.; Park, S.J.; Lee, D.Y.; Moon, S.H.; Lee, S.H. Microchip-based engineering of super-pancreatic islets supported by adipose-derived stem cells. Biomaterials 2014, 35, 4815–4826. [Google Scholar] [CrossRef]
- Kaufman-Francis, K.; Koffler, J.; Weinberg, N.; Dor, Y.; Levenberg, S. Engineered vascular beds provide key signals to pancreatic hormone-producing cells. PLoS ONE 2012, 7, e40741. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, G.; Nicolas, V.; Matsusaki, M.; Akashi, M.; Couvreur, P.; Mura, S. Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 2018, 78, 296–307. [Google Scholar] [CrossRef]
- Jun, Y.; Kang, A.R.; Lee, J.S.; Jeong, G.S.; Ju, J.; Lee, D.Y.; Lee, S.-H. 3D co-culturing model of primary pancreatic islets and hepatocytes in hybrid spheroid to overcome pancreatic cell shortage. Biomaterials 2013, 34, 3784–3794. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Collins, J.W.; Cruz Walma, D.A.; Doyle, A.D.; Morales, S.G.; Lu, J.; Matsumoto, K.; Nazari, S.S.; Sekiguchi, R.; Shinsato, Y.; et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int. J. Exp. Pathol. 2019, 100, 144–152. [Google Scholar] [CrossRef]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef]
- Goetzke, R.; Sechi, A.; de Laporte, L.; Neuss, S.; Wagner, W. Why the impact of mechanical stimuli on stem cells remains a challenge. Cell Mol. Life Sci. 2018, 75, 3297–3312. [Google Scholar] [CrossRef]
- Groll, J.; Boland, T.; Blunk, T.; Burdick, J.A.; Cho, D.W.; Dalton, P.D.; Derby, B.; Forgacs, G.; Li, Q.; Mironov, V.A.; et al. Biofabrication: Reappraising the definition of an evolving field. Biofabrication 2016, 8, 013001. [Google Scholar] [CrossRef]
- Lee, K.; Silva, E.A.; Mooney, D.J. Growth factor delivery-based tissue engineering: General approaches and a review of recent developments. J. R. Soc. Interface 2011, 8, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Di Nardo, P.; Minieri, M.; Ahluwalia, A. Engineering the stem cell niche and the differentiative micro-and macroenvironment: Technologies and tools for applying biochemical, physical and structural stimuli and their effects on stem cells. In Stem Cell Engineering; Springer: Berlin, Germany, 2011; pp. 41–59. [Google Scholar]
- Sharon, N.; Chawla, R.; Mueller, J.; Vanderhooft, J.; Whitehorn, L.J.; Rosenthal, B.; Gürtler, M.; Estanboulieh, R.R.; Shvartsman, D.; Gifford, D.K.; et al. A peninsular structure coordinates asynchronous differentiation with morphogenesis to generate pancreatic islets. Cell 2019, 176, 790–804.e713. [Google Scholar] [CrossRef] [PubMed]
- Caddeo, S.; Boffito, M.; Sartori, S. Tissue engineering approaches in the design of healthy and pathological in vitro tissue models. Front. Bioeng. Biotechnol. 2017, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease modeling in stem cell-derived 3D organoid systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Ko, U.H.; Oh, Y.; Lim, A.; Sohn, J.-W.; Shin, J.H.; Kim, H.; Han, Y.-M. Islet-like organoids derived from human pluripotent stem cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci. Rep. 2016, 6, 35145. [Google Scholar] [CrossRef]
- Gao, B.; Jing, C.; Ng, K.; Pingguan-Murphy, B.; Yang, Q. Fabrication of three-dimensional islet models by the geometry-controlled hanging-drop method. Acta Mech. Sin. 2019, 35, 329–337. [Google Scholar] [CrossRef]
- Guo-Parke, H.; McCluskey, J.T.; Kelly, C.; Hamid, M.; McClenaghan, N.H.; Flatt, P.R. Configuration of electrofusion-derived human insulin-secreting cell line as pseudoislets enhances functionality and therapeutic utility. J. Endocrinol. 2012, 214, 257–265. [Google Scholar] [CrossRef]
- Greggio, C.; de Franceschi, F.; Figueiredo-Larsen, M.; Gobaa, S.; Ranga, A.; Semb, H.; Lutolf, M.; Grapin-Botton, A. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development 2013, 140, 4452–4462. [Google Scholar] [CrossRef]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.; van de Wetering, M.; Sojoodi, M.; Li, V.S.; Schuijers, J.; Gracanin, A. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef]
- Candiello, J.; Grandhi, T.S.P.; Goh, S.K.; Vaidya, V.; Lemmon-Kishi, M.; Eliato, K.R.; Ros, R.; Kumta, P.N.; Rege, K.; Banerjee, I. 3D heterogeneous islet organoid generation from human embryonic stem cells using a novel engineered hydrogel platform. Biomaterials 2018, 177, 27–39. [Google Scholar] [CrossRef]
- Youngblood, R.L.; Sampson, J.P.; Lebioda, K.R.; Shea, L.D. Microporous scaffolds support assembly and differentiation of pancreatic progenitors into β-cell clusters. Acta Biomater. 2019, 96, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Van Hoof, D.; Mendelsohn, A.D.; Seerke, R.; Desai, T.A.; German, M.S. Differentiation of human embryonic stem cells into pancreatic endoderm in patterned size-controlled clusters. Stem Cell Res. 2011, 6, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sekine, K.; Kin, T.; Takebe, T.; Taniguchi, H. Self-condensation culture enables vascularization of tissue fragments for efficient therapeutic transplantation. Cell Rep. 2018, 23, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Takebe, T.; Taniguchi, H. Methods for generating vascularized islet-like organoids via self-condensation. Curr. Protoc. Stem Cell Biol. 2018, 45, e49. [Google Scholar] [CrossRef]
- Ibrahim, M.; Richardson, M.K. Beyond organoids: In vitro vasculogenesis and angiogenesis using cells from mammals and zebrafish. Reprod. Toxicol. 2017. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, M.; Zhou, T.; Tan, L.; Chong, C.N.; Zhang, T.; Dong, X.; Xiang, J.Z.; Yu, A.S.; Yue, L.; et al. An isogenic human esc platform for functional evaluation of genome-wide-association-study-identified diabetes genes and drug discovery. Cell Stem Cell 2016, 19, 326–340. [Google Scholar] [CrossRef]
- Maxwell, K.G.; Augsornworawat, P.; Velazco-Cruz, L.; Kim, M.H.; Asada, R.; Hogrebe, N.J.; Morikawa, S.; Urano, F.; Millman, J.R. Gene-edited human stem cell-derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Z.; Song, E.; Xu, T. Islet organoid as a promising model for diabetes. Protein Cell 2021, 1–19. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786. [Google Scholar] [CrossRef]
- Petersen, O.W.; Rønnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Stendahl, J.C.; Kaufman, D.B.; Stupp, S.I. Extracellular matrix in pancreatic islets: Relevance to scaffold design and transplantation. Cell Transpl. 2009, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, S.; Ma, M.; Kim, S.M.; Guye, P.; Pancoast, J.R.; Anderson, D.G.; Weiss, R.; Lee, R.T.; Hammond, P.T. Microbead-based biomimetic synthetic neighbors enhance survival and function of rat pancreatic β-cells. Sci. Rep. 2013, 3, 2863. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-I.; Wang, Y. Cell responses to surface and architecture of tissue engineering scaffolds. In Regenerative Medicine and Tissue Engineering-Cells and Biomaterials; InTech: Shanghai, China, 2011. [Google Scholar]
- Daly, A.C.; Freeman, F.E.; Gonzalez-Fernandez, T.; Critchley, S.E.; Nulty, J.; Kelly, D.J. 3D bioprinting for cartilage and osteochondral tissue engineering. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef]
- Khorsandi, L.; Khodadadi, A.; Nejad-Dehbashi, F.; Saremy, S. Three-dimensional differentiation of adipose-derived mesenchymal stem cells into insulin-producing cells. Cell Tissue Res. 2015, 361, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Bogdani, M.; Korpos, E.; Simeonovic, C.J.; Parish, C.R.; Sorokin, L.; Wight, T.N. Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr. Diabetes Rep. 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.; Schlörer, E.; Greiner, L. EUS elastography of the pancreas: Feasibility and pattern description of the normal pancreas, chronic pancreatitis, and focal pancreatic lesions. Gastrointest. Endosc. 2007, 65, 971–978. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773. [Google Scholar] [CrossRef]
- Badylak, S.F.; Freytes, D.O.; Gilbert, T.W. Extracellular matrix as a biological scaffold material: Structure and function. Acta Biomater. 2009, 5, 1–13. [Google Scholar] [CrossRef]
- Goh, S.K.; Bertera, S.; Olsen, P.; Candiello, J.E.; Halfter, W.; Uechi, G.; Balasubramani, M.; Johnson, S.A.; Sicari, B.M.; Kollar, E.; et al. Perfusion-decellularized pancreas as a natural 3D scaffold for pancreatic tissue and whole organ engineering. Biomaterials 2013, 34, 6760–6772. [Google Scholar] [CrossRef]
- Jiang, K.; Chaimov, D.; Patel, S.N.; Liang, J.P.; Wiggins, S.C.; Samojlik, M.M.; Rubiano, A.; Simmons, C.S.; Stabler, C.L. 3-D physiomimetic extracellular matrix hydrogels provide a supportive microenvironment for rodent and human islet culture. Biomaterials 2019, 198, 37–48. [Google Scholar] [CrossRef]
- Ng, W.L.; Chua, C.K.; Shen, Y.-F. Print Me An Organ! Why We Are Not There Yet. Prog. Polym. Sci. 2019, 97, 101145. [Google Scholar] [CrossRef]
- Sullivan, D.C.; Mirmalek-Sani, S.-H.; Deegan, D.B.; Baptista, P.M.; Aboushwareb, T.; Atala, A.; Yoo, J.J. Decellularization methods of porcine kidneys for whole organ engineering using a high-throughput system. Biomaterials 2012, 33, 7756–7764. [Google Scholar] [CrossRef]
- Mao, G.-H.; Chen, G.-A.; Bai, H.-Y.; Song, T.-R.; Wang, Y.-X. The reversal of hyperglycaemia in diabetic mice using PLGA scaffolds seeded with islet-like cells derived from human embryonic stem cells. Biomaterials 2009, 30, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Gibly, R.F.; Zhang, X.; Graham, M.L.; Hering, B.J.; Kaufman, D.B.; Lowe, W.L., Jr.; Shea, L.D. Extrahepatic islet transplantation with microporous polymer scaffolds in syngeneic mouse and allogeneic porcine models. Biomaterials 2011, 32, 9677–9684. [Google Scholar] [CrossRef] [PubMed]
- Blomeier, H.; Zhang, X.; Rives, C.; Brissova, M.; Hughes, E.; Baker, M.; Powers, A.C.; Kaufman, D.B.; Shea, L.D.; Lowe Jr, W.L. Polymer scaffolds as synthetic microenvironments for extrahepatic islet transplantation. Transplantation 2006, 82, 452. [Google Scholar] [CrossRef]
- Chun, S.; Huang, Y.; Xie, W.; Hou, Y.; Huang, R.; Song, Y.; Liu, X.; Zheng, W.; Shi, Y.; Song, C. Adhesive growth of pancreatic islet cells on a polyglycolic acid fibrous scaffold. In Transplantation Proceedings; Elsevier: Houston, TX, USA, 2008; pp. 1658–1663. [Google Scholar]
- Coronel, M.M.; Stabler, C.L. Engineering a local microenvironment for pancreatic islet replacement. Curr. Opin. Biotechnol. 2013, 24, 900–908. [Google Scholar] [CrossRef]
- Pedraza, E.; Brady, A.C.; Fraker, C.A.; Molano, R.D.; Sukert, S.; Berman, D.M.; Kenyon, N.S.; Pileggi, A.; Ricordi, C.; Stabler, C.L. Macroporous three-dimensional PDMS scaffolds for extrahepatic islet transplantation. Cell Transplant. 2013, 22, 1123–1135. [Google Scholar] [CrossRef]
- Tokito, F.; Shinohara, M.; Maruyama, M.; Inamura, K.; Nishikawa, M.; Sakai, Y. High density culture of pancreatic islet-like 3D tissue organized in oxygen-permeable porous scaffolds with external oxygen supply. J. Biosci. Bioeng. 2020. [Google Scholar] [CrossRef]
- Weber, L.M.; Anseth, K.S. Hydrogel encapsulation environments functionalized with extracellular matrix interactions increase islet insulin secretion. Matrix Biol. 2008, 27, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Salvay, D.M.; Rives, C.B.; Zhang, X.; Chen, F.; Kaufman, D.B.; Lowe Jr, W.L.; Shea, L.D. Extracellular matrix protein-coated scaffolds promote the reversal of diabetes after extrahepatic islet transplantation. Transplantation 2008, 85, 1456. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.J.; Antipenko, S.V.; Anderson, J.M.; Jaimes, K.F.; Viera, L.; Stephen, B.R.; Bryant, S.M.; Yancey, B.D.; Hughes, K.J.; Cui, W.; et al. Enhanced rat islet function and survival in vitro using a biomimetic self-assembled nanomatrix gel. Tissue Eng. Part A 2011, 17, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, G.; Luca, A.D.; de Koning, E.; Engelse, M.; van Blitterswijk, C.A.; Karperien, M.; van Apeldoorn, A.A.; Moroni, L. Hybrid polycaprolactone/alginate scaffolds functionalized with VEGF to promote de novo vessel formation for the transplantation of islets of langerhans. Adv. Healthc. Mater. 2016, 5, 1606–1616. [Google Scholar] [CrossRef]
- Chow, L.W.; Wang, L.J.; Kaufman, D.B.; Stupp, S.I. Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets. Biomaterials 2010, 31, 6154–6161. [Google Scholar] [CrossRef] [PubMed]
- Wex, C.; Fröhlich, M.; Brandstädter, K.; Bruns, C.; Stoll, A. Experimental analysis of the mechanical behavior of the viscoelastic porcine pancreas and preliminary case study on the human pancreas. J. Mech. Behav. Biomed. Mater. 2015, 41, 199–207. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef]
- Diekjürgen, D.; Grainger, D.W. Polysaccharide matrices used in 3D in vitro cell culture systems. Biomaterials 2017, 141, 96–115. [Google Scholar] [CrossRef]
- Phelps, E.A.; Headen, D.M.; Taylor, W.R.; Thulé, P.M.; García, A.J. Vasculogenic bio-synthetic hydrogel for enhancement of pancreatic islet engraftment and function in type 1 diabetes. Biomaterials 2013, 34, 4602–4611. [Google Scholar] [CrossRef]
- Borg, D.J.; Welzel, P.B.; Grimmer, M.; Friedrichs, J.; Weigelt, M.; Wilhelm, C.; Prewitz, M.; Stißel, A.; Hommel, A.; Kurth, T.; et al. Macroporous biohybrid cryogels for co-housing pancreatic islets with mesenchymal stromal cells. Acta Biomater. 2016, 44, 178–187. [Google Scholar] [CrossRef]
- Wang, W.; Jin, S.; Ye, K. Development of islet organoids from H9 human embryonic stem cells in biomimetic 3D scaffolds. Stem Cells Dev. 2017, 26, 394–404. [Google Scholar] [CrossRef]
- Song, S.; Roy, S. Progress and challenges in macroencapsulation approaches for type 1 diabetes (T1D) treatment: Cells, biomaterials, and devices. Biotechnol. Bioeng. 2016, 113, 1381–1402. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the cancer microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Vijayavenkataraman, S.; Lu, W.; Fuh, J. 3D bioprinting of skin: A state-of-the-art review on modelling, materials, and processes. Biofabrication 2016, 8, 032001. [Google Scholar] [CrossRef]
- Cui, H.; Nowicki, M.; Fisher, J.P.; Zhang, L.G. 3D bioprinting for organ regeneration. Adv. Healthc. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Daoud, J.T.; Petropavlovskaia, M.S.; Patapas, J.M.; Degrandpré, C.E.; DiRaddo, R.W.; Rosenberg, L.; Tabrizian, M. Long-term in vitro human pancreatic islet culture using three-dimensional microfabricated scaffolds. Biomaterials 2011, 32, 1536–1542. [Google Scholar] [CrossRef]
- Marchioli, G.; van Gurp, L.; van Krieken, P.; Stamatialis, D.; Engelse, M.; van Blitterswijk, C.; Karperien, M.; de Koning, E.; Alblas, J.; Moroni, L. Fabrication of three-dimensional bioplotted hydrogel scaffolds for islets of Langerhans transplantation. Biofabrication 2015, 7, 025009. [Google Scholar] [CrossRef] [PubMed]
- Duin, S.; Schutz, K.; Ahlfeld, T.; Lehmann, S.; Lode, A.; Ludwig, B.; Gelinsky, M. 3D bioprinting of functional islets of langerhans in an alginate/methylcellulose hydrogel blend. Adv. Healthc. Mater. 2019, 8, e1801631. [Google Scholar] [CrossRef] [PubMed]
- Milojević, M.; Harih, G.; Vihar, B.; Vajda, J.; Gradišnik, L.; Zidarič, T.; Stana Kleinschek, K.; Maver, U.; Maver, T. Hybrid 3D printing of advanced hydrogel-based wound dressings with tailorable properties. Pharmaceutics 2021, 13, 564. [Google Scholar] [CrossRef]
- Milojević, M.; Gradišnik, L.; Stergar, J.; Skelin Klemen, M.; Stožer, A.; Vesenjak, M.; Dobnik Dubrovski, P.; Maver, T.; Mohan, T.; Stana Kleinschek, K.; et al. Development of multifunctional 3D printed bioscaffolds from polysaccharides and NiCu nanoparticles and their application. Appl. Surf. Sci. 2019, 488, 836–852. [Google Scholar] [CrossRef]
- Kim, J.; Shim, I.K.; Hwang, D.G.; Lee, Y.N.; Kim, M.; Kim, H.; Kim, S.-W.; Lee, S.; Kim, S.C.; Cho, D.-W.; et al. 3D cell printing of islet-laden pancreatic tissue-derived extracellular matrix bioink constructs for enhancing pancreatic functions. J. Mater. Chem. B 2019, 7, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Klak, M.; Kowalska, P.; Dobrzański, T.; Tymicki, G.; Cywoniuk, P.; Gomółka, M.; Kosowska, K.; Bryniarski, T.; Berman, A.; Dobrzyń, A.; et al. Bionic organs: Shear forces reduce pancreatic islet and mammalian cell viability during the process of 3D bioprinting. Micromachines 2021, 12, 304. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Vacanti, J. Advances in tissue engineering. J. Pediatric Surg. 2016, 51, 8–12. [Google Scholar] [CrossRef]
- Hinton, T.J.; Jallerat, Q.; Palchesko, R.N.; Park, J.H.; Grodzicki, M.S.; Shue, H.J.; Ramadan, M.H.; Hudson, A.R.; Feinberg, A.W. Three-dimensional printing of complex biological structures by freeform reversible embedding of suspended hydrogels. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef]
- Stumberger, G.; Vihar, B. Freeform perfusable microfluidics embedded in hydrogel matrices. Materials 2018, 11, 2529. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, Z.; Hu, N.; Zhou, Y.; Maggio, L.; Miri, A.K.; Fragasso, A.; Jin, X.; Khademhosseini, A.; Zhang, Y.S. Coaxial extrusion bioprinting of 3D microfibrous constructs with cell-favorable gelatin methacryloyl microenvironments. Biofabrication 2018, 10, 024102. [Google Scholar] [CrossRef]
- Mistry, P.; Aied, A.; Alexander, M.; Shakesheff, K.; Bennett, A.; Yang, J. Bioprinting using mechanically robust core-shell cell-laden hydrogel strands. Macromol. BioSci. 2017, 17. [Google Scholar] [CrossRef]
- Milojevic, M.; Vihar, B.; Banovic, L.; Misko, M.; Gradisnik, L.; Zidaric, T.; Maver, U. Core/shell printing scaffolds for tissue engineering of tubular structures. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Liu, X.; Carter, S.D.; Renes, M.J.; Kim, J.; Rojas-Canales, D.M.; Penko, D.; Angus, C.; Beirne, S.; Drogemuller, C.J.; Yue, Z.; et al. Development of a coaxial 3D printing platform for biofabrication of implantable islet-containing constructs. Adv. Healthc. Mater. 2019, 8, e1801181. [Google Scholar] [CrossRef]
- Mammoto, T.; Mammoto, A.; Ingber, D.E. Mechanobiology and developmental control. Annu. Rev. Cell Dev. Biol. 2013, 29, 27–61. [Google Scholar] [CrossRef]
- Daoud, J.; Rosenberg, L.; Tabrizian, M. Pancreatic islet culture and preservation strategies: Advances, challenges, and future outlook. Cell Transpl. 2010, 19, 1523–1535. [Google Scholar] [CrossRef]
- Li, Z.; Sun, H.; Zhang, J.; Zhang, H.; Meng, F.; Cui, Z. Development of in vitro 3D Tissueflex® islet model for diabetic drug efficacy testing. PLoS ONE 2013, 8, e72612. [Google Scholar] [CrossRef]
- Vunjak-Novakovic, G.; Searby, N.; de Luis, J.; Freed, L.E. Microgravity studies of cells and tissues. Ann. N. Y. Acad. Sci. 2002, 974, 504–517. [Google Scholar] [CrossRef]
- Rutzky, L.P.; Bilinski, S.; Kloc, M.; Phan, T.; Zhang, H.; Katz, S.M.; Stepkowski, S.M. Microgravity culture condition reduces immunogenicity and improves function of pancreatic islets1. Transplantation 2002, 74, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.A.; Platton, S.L.; Dennison, A.R.; James, R.F. Immunohistochemical evidence that culture in the high aspect rotating vessel can up-regulate hormone expression in growth dedifferentiated PHHI-derived islet cells. In vitro cellular & developmental biology. Animal 2007, 43, 210–214. [Google Scholar] [CrossRef]
- Hou, Y.; Song, C.; Xie, W.J.; Wei, Z.; Huang, R.P.; Liu, W.; Zhang, Z.L.; Shi, Y.B. Excellent effect of three-dimensional culture condition on pancreatic islets. Diabetes Res. Clin. Pract. 2009, 86, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.E.; Paget, M.B.; Downing, R. Preservation of glucose responsiveness in human islets maintained in a rotational cell culture system. Mol. Cell Endocrinol. 2005, 238, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.E.; Paget, M.B.; Bailey, C.J.; Downing, R. Sustained insulin secretory response in human islets co-cultured with pancreatic duct-derived epithelial cells within a rotational cell culture system. Diabetologia 2009, 52, 477–485. [Google Scholar] [CrossRef][Green Version]
- Ahadian, S.; Civitarese, R.; Bannerman, D.; Mohammadi, M.H.; Lu, R.; Wang, E.; Davenport-Huyer, L.; Lai, B.; Zhang, B.; Zhao, Y.; et al. Organ-on-a-chip platforms: A convergence of advanced materials, cells, and microscale technologies. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef]
- Low, L.; Tagle, D. Tissue chips–innovative tools for drug development and disease modeling. Lab Chip 2017, 17, 3026–3036. [Google Scholar] [CrossRef]
- Lee, S.H.; Hong, S.; Song, J.; Cho, B.; Han, E.J.; Kondapavulur, S.; Kim, D.; Lee, L.P. Microphysiological analysis platform of pancreatic islet β-cell spheroids. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Zhang, B.; Radisic, M. Synergistic engineering: Organoids meet organs-on-a-chip. Cell Stem Cell 2017, 21, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-G.; Lee, H.; Cho, D.-W. 3D printing of organs-on-chips. Bioengineering 2017, 4, 10. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Regnault, C.; Dheeman, D.S.; Hochstetter, A. Microfluidic devices for drug assays. High Throughput 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.S.; Wang, Y.; Harvat, T.A.; Oberholzer, J.; Eddington, D.T. Microfluidic device for multimodal characterization of pancreatic islets. Lab Chip 2009, 9, 97–106. [Google Scholar] [CrossRef]
- Sankar, K.S.; Green, B.J.; Crocker, A.R.; Verity, J.E.; Altamentova, S.M.; Rocheleau, J.V. Culturing pancreatic islets in microfluidic flow enhances morphology of the associated endothelial cells. PLoS ONE 2011, 6, e24904. [Google Scholar] [CrossRef]
- Lee, D.; Wang, Y.; Mendoza-Elias, J.E.; Adewola, A.F.; Harvat, T.A.; Kinzer, K.; Gutierrez, D.; Qi, M.; Eddington, D.T.; Oberholzer, J. Dual microfluidic perifusion networks for concurrent islet perifusion and optical imaging. Biomed. Microdevices 2012, 14, 7–16. [Google Scholar] [CrossRef]
- Shackman, J.G.; Dahlgren, G.M.; Peters, J.L.; Kennedy, R.T. Perfusion and chemical monitoring of living cells on a microfluidic chip. Lab Chip 2005, 5, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Dishinger, J.F.; Kennedy, R.T. Serial immunoassays in parallel on a microfluidic chip for monitoring hormone secretion from living cells. Anal. Chem. 2007, 79, 947–954. [Google Scholar] [CrossRef]
- Easley, C.J.; Rocheleau, J.V.; Head, W.S.; Piston, D.W. Quantitative measurement of zinc secretion from pancreatic islets with high temporal resolution using droplet-based microfluidics. Anal. Chem. 2009, 81, 9086–9095. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.-J.; Aspinwall, C.A.; Battiste, M.A.; Kennedy, R.T. Detection of secretion from single pancreatic β-cells using extracellular fluorogenic reactions and confocal fluorescence microscopy. Anal. Chem. 2000, 72, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lo, J.F.; Mendoza-Elias, J.E.; Adewola, A.F.; Harvat, T.A.; Kinzer, K.P.; Lee, D.; Qi, M.; Eddington, D.T.; Oberholzer, J. Application of microfluidic technology to pancreatic islet research: First decade of endeavor. Bioanalysis 2010, 2, 1729–1744. [Google Scholar] [CrossRef] [PubMed]
- Castiello, F.R.; Heileman, K.; Tabrizian, M. Microfluidic perfusion systems for secretion fingerprint analysis of pancreatic islets: Applications, challenges and opportunities. Lab Chip 2016, 16, 409–431. [Google Scholar] [CrossRef]
- Nguyen, D.T.; van Noort, D.; Jeong, I.K.; Park, S. Endocrine system on chip for a diabetes treatment model. Biofabrication 2017, 9, 015021. [Google Scholar] [CrossRef]
- Bauer, S.; Wennberg Huldt, C.; Kanebratt, K.P.; Durieux, I.; Gunne, D.; Andersson, S.; Ewart, L.; Haynes, W.G.; Maschmeyer, I.; Winter, A.; et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: Towards a novel human ex vivo type 2 diabetes model. Sci. Rep. 2017, 7, 14620. [Google Scholar] [CrossRef]
- Patel, S.N.; Ishahak, M.; Chaimov, D.; Velraj, A.; LaShoto, D.; Hagan, D.W.; Buchwald, P.; Phelps, E.A.; Agarwal, A.; Stabler, C.L. Organoid microphysiological system preserves pancreatic islet function within 3D matrix. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A.; Cohrs, C.M.; Tsata, V.; Chouinard, J.A.; Selck, C.; Stertmann, J.; Reichelt, S.; Rose, T.; Ehehalt, F.; Weitz, J.; et al. Using pancreas tissue slices for in situ studies of islet of Langerhans and acinar cell biology. Nat. Protoc. 2014, 9, 2809–2822. [Google Scholar] [CrossRef] [PubMed]
- Speier, S. Experimental Approaches for High-Resolution In vivo Imaging of Islet of Langerhans Biology. Curr. Diabetes Rep. 2011, 11, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Speier, S.; Rupnik, M. A novel approach to in situ characterization of pancreatic ß-cells. Pflügers Arch. Eur. J. Physiol. 2003, 446, 553–558. [Google Scholar] [CrossRef]
- Cohrs, C.M.; Chen, C.; Jahn, S.R.; Stertmann, J.; Chmelova, H.; Weitz, J.; Bähr, A.; Klymiuk, N.; Steffen, A.; Ludwig, B.; et al. Vessel network architecture of adult human islets promotes distinct cell-cell interactions in situ and is altered after transplantation. Endocrinology 2017, 158, 1373–1385. [Google Scholar] [CrossRef]
- Marciniak, A.; Selck, C.; Friedrich, B.; Speier, S. Mouse pancreas tissue slice culture facilitates long-term studies of exocrine and endocrine cell physiology in situ. PLoS ONE 2013, 8, e78706. [Google Scholar] [CrossRef]
- Weitz, J.R.; Makhmutova, M.; Almaça, J.; Stertmann, J.; Aamodt, K.; Brissova, M.; Speier, S.; Rodriguez-Diaz, R.; Caicedo, A. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 2017. [Google Scholar] [CrossRef]
- Meneghel-Rozzo, T.; Rozzo, A.; Poppi, L.; Rupnik, M. In vivo and in vitro development of mouse pancreatic ß-cells in organotypic slices. Cell Tissue Res. 2004, 316, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Dolenšek, J.; Stožer, A.; Skelin Klemen, M.; Miller, E.W.; Slak Rupnik, M. The relationship between membrane potential and calcium dynamics in glucose-stimulated beta cell syncytium in acute mouse pancreas tissue slices. PLoS ONE 2013, 8, e82374. [Google Scholar] [CrossRef]
- Dolenšek, J.; Špelič, D.; Klemen, M.; Žalik, B.; Gosak, M.; Rupnik, M.; Stožer, A. Membrane potential and calcium dynamics in beta cells from mouse pancreas tissue slices: Theory, experimentation, and analysis. Sensors 2015, 15, 27393. [Google Scholar] [CrossRef] [PubMed]
- Skelin Klemen, M.; Dolenšek, J.; Stožer, A.; Rupnik, M. Measuring exocytosis in endocrine tissue slices. In Exocytosis Methods; Thorn, P., Ed.; Neuromethods; Humana Press: New York, NY, USA, 2014; Volume 83, pp. 127–146. [Google Scholar]
- Skelin, M.; Rupnik, M. cAMP increases the sensitivity of exocytosis to Ca2+ primarily through protein kinase A in mouse pancreatic beta cells. Cell Calcium 2011, 49, 89–99. [Google Scholar] [CrossRef]
- Dolensek, J.; Skelin, M.; Rupnik, M.S. Calcium dependencies of regulated exocytosis in different endocrine cells. Physiol. Res. 2011, 60, S29–S38. [Google Scholar] [CrossRef] [PubMed]
- Stozer, A.; Dolensek, J.; Skelin, M.; Rupnik, M. Cell physiology in tissue slices: Studying beta cells in the islets of Langerhans = Celicna fiziologija v tkivnih rezinah: Preucevanje celic beta v Langerhansovih otockih. Acta Med. Biotech. 2013, 6, 20–32. [Google Scholar]
- Markovic, R.; Stozer, A.; Gosak, M.; Dolensek, J.; Marhl, M.; Rupnik, M.S. Progressive glucose stimulation of islet beta cells reveals a transition from segregated to integrated modular functional connectivity patterns. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Gosak, M.; Stožer, A.; Markovič, R.; Dolenšek, J.; Perc, M.; Rupnik, M.S.; Marhl, M. Critical and supercritical spatiotemporal calcium dynamics in beta cells. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Stožer, A.; Gosak, M.; Dolenšek, J.; Perc, M.; Marhl, M.; Rupnik, M.S.; Korošak, D. Functional connectivity in islets of Langerhans from mouse pancreas tissue slices. PLoS Comput. Comput. Biology 2013, 9, e1002923. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Rupnik, M.; Gaisano, H.Y. Unperturbed islet α-cell function examined in mouse pancreas tissue slices. J. Physiol. 2011, 589, 395–408. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Rupnik, M.S.; Karimian, N.; Herrera, P.L.; Gilon, P.; Feng, Z.-P.; Gaisano, H.Y. In situ electrophysiological examination of pancreatic α cells in the streptozotocin-induced diabetes model, revealing the cellular basis of glucagon hypersecretion. Diabetes 2013, 62, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C. Science and Principles of Biodegradable and Bioresorbable Medical Polymers: Materials and Properties; Woodhead Publishing: Duxford, UK, 2016. [Google Scholar]
- Haq, M.A.; Su, Y.; Wang, D. Mechanical properties of PNIPAM based hydrogels: A review. Mater. Sci. Eng. C 2017, 70, 842–855. [Google Scholar] [CrossRef]
- Arslan-Yildiz, A.; el Assal, R.; Chen, P.; Guven, S.; Inci, F.; Demirci, U. Towards artificial tissue models: Past, present, and future of 3D bioprinting. Biofabrication 2016, 8, 014103. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In vitro model | Description | Advantages | Disadvantages |
|---|---|---|---|
| Monolayer monocultures | Cells are grown on the flat surface of a plastic cell culture container and covered with culturing medium. | Ease of use, low cost, high throughput, high reproducibility, standardization. | Non-physiological structural support (stiff, nonpermeable, 2D substrate) and cellular alignment resulting in loss of phenotype, changes cell shape. Static environmental conditions. |
| Monolayer monocultures with ECM | Surface of the cell culture container is coated using ECM or ECM substitutes on which the cells are cultured. | Cost-efficient. Mimic some aspects of cell-ECM interaction, e.g., allows for the modulation of cell behaviour and signalling. Especially useful for epithelial cultures. | Loss of phenotype, lack of more complex 3D architecture, altered cell shape. Static environmental conditions. |
| Monolayer co-cultures | Monolayer cultures containing more than one cell type. | Ease of use, low cost, allows for the study of interactions between cell types, synergistic/antagonistic effects of medium consumption and metabolite production. | Difficult to analyse the contribution of each cell type to co-culture function. Depending on cell growth rate, one cell type can rapidly dominate the culturing container. Static environmental conditions. |
| 3D cultures (organoids) | Cells grown in a 3D environment. The 3D environment can be created by an ECM or by a nonadherent surface. | Cost efficient, ease of use. High cell density, mimics the 3D architecture of native tissues. Prolonged cell viability and functionality. Self-assembly. | Limited by nutrient/oxygen diffusion allowing for only small sample sizes. Difficult to functionally analyse entrapped cells. Limited control over shape/size. Static environmental conditions. |
| 3D cultures (scaffolds) | Cells grown on artificial supports (scaffolds) which mimic the native ECM | Improved nutrient/oxygen diffusion. Mimics the 3D architecture of native tissues. Prolonged cell viability and functionality. Adjustable physico-chemical properties and their gradients. | Complex design and manufacturing procedures. Difficult to functionally analyse entrapped cells. Limited range of biocompatible materials. Static environmental conditions. |
| 3D co-cultures (spheroids, scaffolds) | More than one cell type is grown in a 3D environment. | Reproduce important functions of the target tissue/organ. Prolonged cell viability and functionality. | Complex/costly. Difficult to functionally analyse entrapped cells. Static environmental conditions. |
| Bioreactors | Cells grown in reactors that mimic physiological conditions | Simulate “micro gravity”, mimic physicochemical gradients, constant perfusion. High throughput. | Homogenization of cell-medium suspension does not allow for implementation of chemical and structural gradients. Shear stress. Cells tend to aggregate which leads to central necrosis. Costly. |
| Organ-on-a-chip | A microfluidic cell culture device containing continuously perfused chambers inhabited by living cells. | Simulate tissue- and organ-level physiology. Sensor integration and real-time measurement of multiple analytes. | Complex, multistep fabrication methods. Costly with low throughput. |
| Tissue slices | Tissue slices are cultured in plates or on supports. | In vivo environment is preserved. | Requires hyperoxic conditions. Cell viability limited over short periods of time. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milojević, M.; Rožanc, J.; Vajda, J.; Činč Ćurić, L.; Paradiž, E.; Stožer, A.; Maver, U.; Vihar, B. In Vitro Disease Models of the Endocrine Pancreas. Biomedicines 2021, 9, 1415. https://doi.org/10.3390/biomedicines9101415
Milojević M, Rožanc J, Vajda J, Činč Ćurić L, Paradiž E, Stožer A, Maver U, Vihar B. In Vitro Disease Models of the Endocrine Pancreas. Biomedicines. 2021; 9(10):1415. https://doi.org/10.3390/biomedicines9101415
Chicago/Turabian StyleMilojević, Marko, Jan Rožanc, Jernej Vajda, Laura Činč Ćurić, Eva Paradiž, Andraž Stožer, Uroš Maver, and Boštjan Vihar. 2021. "In Vitro Disease Models of the Endocrine Pancreas" Biomedicines 9, no. 10: 1415. https://doi.org/10.3390/biomedicines9101415
APA StyleMilojević, M., Rožanc, J., Vajda, J., Činč Ćurić, L., Paradiž, E., Stožer, A., Maver, U., & Vihar, B. (2021). In Vitro Disease Models of the Endocrine Pancreas. Biomedicines, 9(10), 1415. https://doi.org/10.3390/biomedicines9101415