
Genetics Is of the Essence to Face NAFLD



Abstract
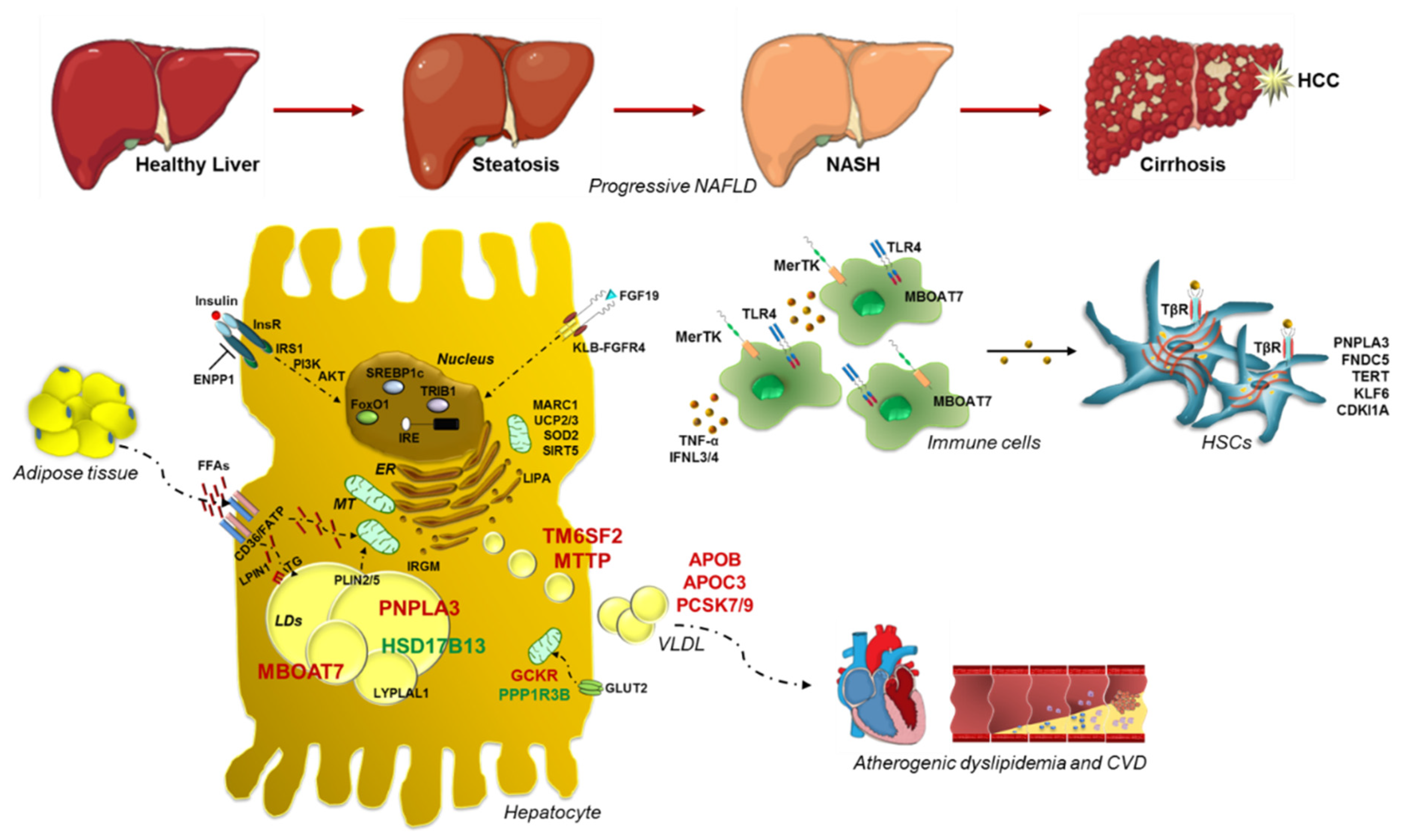
1. Introduction
2. Historical Overture to Discover the Link between Genetics and NAFLD
2.1. PNPLA3: Gambling on the Winning Horse
2.2. TM6SF2 Loss-of-Function in NAFLD
2.3. MBOAT7: A Common Modifier of Liver Damage
2.4. GCKR: The Jointing of Glucose Handling and Fatty Liver
2.5. Protective Inheritable Determinants: The HSD17B13 and PPP1R3B Variations
3. Genetic Signature of Glucose and Lipid Metabolism in NAFLD
4. Genetics of Lipid Droplets
5. Advanced Liver Injuries and Genetic Variants
6. Mitochondrial Dysfunctions: The Tipping Point in the Switching from Simple Steatosis to Steatohepatitis
7. Polygenic Risk Scores (PRSs): From Bench to Bedside and Back
8. Novel Insights into the Modelling of NAFLD: From Genetic Studies to Cellular Models
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACDRP | amish complex disease research program |
| ALD | alcoholic liver disease |
| ALT | alanine aminotransferase |
| APOB | apolipoprotein B |
| APOC3 | apolipoprotein C3 |
| ATGL | adipose triglyceride lipase |
| AUROC | area under receiver operating characteristics |
| BMI | body mass index |
| CCl4 | carbon tetrachloride CD36: cluster of differentiation 36 |
| CDKI1A | cyclin dependent kinase inhibitor 1A |
| CGI-58 | comparative gene identification-58 |
| CPN1 | carboxypeptidase n subunit 1 |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| CVD | cardiovascular disease |
| DAGs | diacylglycerols |
| DNL | de novo lipogenesis |
| ENPP1 | ectonucleotide pyrophosphatase/phosphodiesterase1 |
| ER | endothelial reticulum |
| EWAS | exome wide association studies |
| FATP5 | fatty acid transport proteins |
| FFAs | free fatty acids |
| FGF | fibroblast growth factor |
| FGFR4 | fibroblast growth factor receptor 4 |
| FNDC5 | fibronectin type III domain-containing protein 5 |
| FOXO1 | forkhead box protein O1 |
| GCKR | glucokinase regulator |
| GGT | γ-glutamyl transferase |
| GPR55G | protein-coupled receptor 55 |
| GWAS | genome-wide association study |
| HCC | hepatocellular carcinoma |
| HFD | high-fat diet |
| HNRNPUL1 | heterogeneous nuclear ribonucleoprotein U like 1 |
| HSCs | hepatic stellate cells |
| HSD17B13 | hydroxysteroid 17-β dehydrogenase 13 |
| IFNL3/4 | interferon λ3/λ4 |
| IL28 | interleukin 28 |
| InsR | insulin receptor |
| iPSCs | pluripotent stem cells |
| IR | insulin resistance |
| IRAS | Insulin Resistance Atherosclerosis Study |
| IRS1 | insulin receptor substrate |
| KLB | β-Klotho |
| KLF6 | krueppel-like factor 6 |
| KI | knock-in |
| KO | knock-out |
| LAL | lysosomal acid lipase |
| LD | lipid droplet |
| LDL | low-density lipoprotein |
| LIPA | lysosomal acid lipase |
| LPIAT1 | lyso-phosphatidylinositol (Lyso-PI) acyl-transferase1 |
| LPIN1 | Lipin1 |
| LPL | lipoprotein lipase |
| LYPLAL1 | lysophospholipase-like 1 |
| MARC1 | amidoxime-reducing component 1 |
| MBOAT7 | membrane bound o-acyltransferase domain-containing 7 |
| MCD | methionine choline deficient |
| MERTK | macrophage c-mer tyrosine kinase |
| MRI-PDFF | magnetic resonance imaging proton-density fat fraction |
| MTTP | microsomal triglyceride transfer protein |
| NAD | nicotinamide adenine dinucleotide |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NOMAS | Northern Manhattan Study |
| OR | odd ratio |
| PCs | phosphatidylcholines |
| PCSK7 | proprotein convertase subtilisin/kexin type 7 |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| PI | phosphatidylinositol |
| PI3K | Phosphoinositide 3-kinase |
| PLIN2 | perilipin 2 |
| PLIN5 | perilipin 5 |
| PNPLA2 | patatin-like phospholipase domain-containing 2 |
| PNPLA3 | patatin-like phospholipase domain-containing 3 |
| PPP1R3B | protein phosphatase 1 regulatory subunit 3B |
| PROTAC | proteolysis-targeting chimera |
| PRSs | polygenic risk scores |
| PS | phosphatidylserine |
| PUFAs | polyunsaturated fatty acids |
| ROC | receiver operating characteristics |
| ROS | reactive oxygen species |
| SIRTs | sirtuins |
| SNP | single nucleotide polymorphism |
| SOD2 | superoxide dismutase 2 |
| SREBP1c | sterol regulatory element-binding protein 1 |
| T2D | type 2 diabetes |
| TERT | telomerase reverse transcriptase |
| TG | triglyceride |
| TGF-β | transforming growth factor β |
| TLR4 | toll like receptor 4 |
| TM6SF2 | transmembrane 6 superfamily member 2 |
| TMC4 | transmembrane channel like 4 |
| TNF-α | tumor necrosis factor α |
| TRIB | tribbles homolog1 |
| UCP | uncoupling protein |
| VLDL | very-low density lipoproteins |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Henry, L.; Bush, H.; Mishra, A. Clinical and Economic Burden of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Wong, R.J.; Hu, M.; Perumpail, R.B.; Yoo, E.R.; Puri, P.; Younossi, Z.M.; Harrison, S.A.; Ahmed, A. Liver Transplantation for Nonalcoholic Steatohepatitis in the US: Temporal Trends and Outcomes. Dig. Dis. Sci. 2017, 62, 2915–2922. [Google Scholar] [CrossRef]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Paolini, E.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Nonalcoholic fatty liver disease or metabolic dysfunction-associated fatty liver disease diagnoses and cardiovascular diseases: From epidemiology to drug approaches. Eur. J. Clin. Investig. 2021, 51, e13519. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L. Genetics of nonalcoholic fatty liver disease. Metab. Clin. Exp. 2016, 65, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Erconi, V.; Valenti, L.; Gatti, S.; Fracanzani, A.L.; Dongiovanni, P. mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients 2019, 11, 2597. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Stender, S.; Kozlitina, J. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Dongiovanni, P. Alcohol or Gut Microbiota: Who Is the Guilty? Int. J. Mol. Sci. 2019, 20, 4568. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Dongiovanni, P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11, 2642. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L. A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Anstee, Q.M.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. BioMed Res. Int. 2015, 2015, 460190. [Google Scholar] [CrossRef]
- Struben, V.M.; Hespenheide, E.E.; Caldwell, S.H. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am. J. Med. 2000, 108, 9–13. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, L.E.; Scherzinger, A.L.; Stamm, E.R.; Hanley, A.J.; Norris, J.M.; Chen, Y.D.; Bryer-Ash, M.; Haffner, S.M.; Rotter, J.I. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity (Silver Spring Md.) 2009, 17, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef]
- Makkonen, J.; Pietilainen, K.H.; Rissanen, A.; Kaprio, J.; Yki-Jarvinen, H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: A study in monozygotic and dizygotic twins. J. Hepatol. 2009, 50, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef]
- Cui, J.; Chen, C.H.; Lo, M.T.; Schork, N.; Bettencourt, R.; Gonzalez, M.P.; Bhatt, A.; Hooker, J.; Shaffer, K.; Nelson, K.E.; et al. Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology 2016, 64, 1547–1558. [Google Scholar] [CrossRef]
- Tarnoki, A.D.; Tarnoki, D.L.; Bata, P.; Littvay, L.; Osztovits, J.; Jermendy, G.; Karlinger, K.; Lannert, A.; Preda, I.; Kiss, R.G.; et al. Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: A twin study. Liver Int. Off. J. Int. Assoc. Study Liver 2012, 32, 1287–1293. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Kanwal, F.; El-Serag, H.B.; Thrift, A.P. Acculturation and Nonalcoholic Fatty Liver Disease Risk Among Hispanics of Mexican Origin: Findings From the National Health and Nutrition Examination Survey. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 310–312. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Fleischman, M.W.; Budoff, M.; Zeb, I.; Li, D.; Foster, T. NAFLD prevalence differs among hispanic subgroups: The Multi-Ethnic Study of Atherosclerosis. World J. Gastroenterol. 2014, 20, 4987–4993. [Google Scholar] [CrossRef]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Stender, S.; Pietrelli, A.; Mancina, R.M.; Cespiati, A.; Petta, S.; Pelusi, S.; Pingitore, P.; Badiali, S.; Maggioni, M.; et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med. 2018, 283, 356–370. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L. Evaluation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Bantel, H.; Rau, M.; Schattenberg, J.M.; Grunhage, F.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Weber, S.N.; et al. Could inherited predisposition drive non-obese fatty liver disease? Results from German tertiary referral centers. J. Hum. Genet. 2018, 63, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid Res. 2009, 50, 2111–2116. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Q.; Wu, K.; Fan, D. I148M variant of PNPLA3 confer increased risk for nonalcoholic fatty liver disease not only in European population, but also in Chinese population. Hepatology 2011, 54, 2275. [Google Scholar] [CrossRef]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [PubMed]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Kursawe, R.; D’Adamo, E.; Dykas, D.J.; Zhang, C.K.; Bale, A.E.; Calí, A.M.; Narayan, D.; Shaw, M.M.; Pierpont, B.; et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology 2010, 52, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, L.J.; Patel, P.R.; Mann, J.P. PNPLA3: A Determinant of Response to Low-Fructose Diet in Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 154, 1207–1208. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Petta, S.; Lombardi, R.; Pisano, G.; Russello, M.; Consonni, D.; Di Marco, V.; Cammà, C.; Mensi, L.; Dongiovanni, P.; et al. Liver and Cardiovascular Damage in Patients With Lean Nonalcoholic Fatty Liver Disease, and Association With Visceral Obesity. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 1604–1611.e1601. [Google Scholar] [CrossRef]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef]
- Min, H.K.; Sookoian, S.; Pirola, C.J.; Cheng, J.; Mirshahi, F.; Sanyal, A.J. Metabolic profiling reveals that PNPLA3 induces widespread effects on metabolism beyond triacylglycerol remodeling in Huh-7 hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G66–G76. [Google Scholar] [CrossRef]
- Banini, B.A.; D, P.K.; Cazanave, S.; Seneshaw, M.; Mirshahi, F.; Santhekadur, P.K.; Wang, L.; Guan, H.P.; Oseini, A.; Alonso, C.; et al. Identification of a metabolic, transcriptomic and molecular signature of PNPLA3-mediated acceleration of steatohepatitis. Hepatology 2020. [Google Scholar] [CrossRef]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Mottillo, E.P.; Mladenovic-Lucas, L.; Zhou, L.; Granneman, J.G. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat. Metab. 2019, 1, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Rajagopal, V.; Smith, C.; Cohick, E. Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells 2020, 9, 2247. [Google Scholar] [CrossRef]
- Negoita, F.; Blomdahl, J.; Wasserstrom, S.; Winberg, M.E.; Osmark, P.; Larsson, S.; Stenkula, K.G.; Ekstedt, M.; Kechagias, S.; Holm, C.; et al. PNPLA3 variant M148 causes resistance to starvation-mediated lipid droplet autophagy in human hepatocytes. J. Cell. Biochem. 2019, 120, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4, e127902. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Merkel, D.; Kovarova, M.; Hoene, M. Dissociation of Fatty Liver and Insulin Resistance in I148M PNPLA3 Carriers: Differences in Diacylglycerol (DAG) FA18:1 Lipid Species as a Possible Explanation. Nutrients 2018, 10, 1314. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef]
- Qadri, S.; Lallukka-Bruck, S.; Luukkonen, P.K.; Zhou, Y.; Gastaldelli, A. The PNPLA3-I148M variant increases polyunsaturated triglycerides in human adipose tissue. Liver Int. 2020, 40, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Qadri, S.; Lehtimäki, T.E.; Juuti, A.; Sammalkorpi, H.; Penttilä, A.K.; Hakkarainen, A.; Orho-Melander, M.; Arola, J.; Yki-Järvinen, H. The PNPLA3-I148M variant confers an antiatherogenic lipid profile in insulin-resistant patients. J. Clin. Endocrinol. Metab. 2020, 106, e300–e315. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Valenti, L.; Motta, B.M.; Soardo, G.; Iavarone, M.; Donati, B.; Sangiovanni, A.; Carnelutti, A.; Dongiovanni, P.; Rametta, R.; Bertelli, C.; et al. PNPLA3 I148M polymorphism, clinical presentation, and survival in patients with hepatocellular carcinoma. PLoS ONE 2013, 8, e75982. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Trepo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between the PNPLA3 (rs738409 C > G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177. [Google Scholar] [CrossRef]
- Carpino, G.; Pastori, D. PNPLA3 variant and portal/periportal histological pattern in patients with biopsy-proven non-alcoholic fatty liver disease: A possible role for oxidative stress. Sci. Rep. 2017, 7, 15756. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Tardelli, M.; Herac, M.; Claudel, T.; Trauner, M. Metabolic regulation of hepatic PNPLA3 expression and severity of liver fibrosis in patients with NASH. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 1098–1110. [Google Scholar] [CrossRef]
- Grimaudo, S.; Pipitone, R.M.; Pennisi, G.; Celsa, C.; Camma, C.; Di Marco, V.; Barcellona, M.R.; Boemi, R.; Enea, M.; Giannetti, A.; et al. Association Between PNPLA3 rs738409 C > G Variant and Liver-Related Outcomes in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 935–944.e933. [Google Scholar] [CrossRef]
- Valenti, L.; Rumi, M.; Galmozzi, E.; Aghemo, A.; Del Menico, B.; De Nicola, S.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Rametta, R.; et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011, 53, 791–799. [Google Scholar] [CrossRef]
- Guyot, E.; Sutton, A.; Rufat, P.; Laguillier, C.; Mansouri, A.; Moreau, R.; Ganne-Carrie, N.; Beaugrand, M.; Charnaux, N.; Trinchet, J.C.; et al. PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J. Hepatol. 2013, 58, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, Y.S.; Kirstein, M.M. The PNPLA3 rs738409 GG genotype is associated with poorer prognosis in 239 patients with autoimmune hepatitis. Aliment. Pharmacol. Ther. 2020, 51, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; Gilyard, S.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J. Biol. Chem. 2016, 291, 10659–10676. [Google Scholar] [CrossRef]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lu, H.; Guo, Y.; Zhu, T.; Garcia-Barrio, M.T.; Jiang, Z.; Willer, C.J.; Zhang, J.; Chen, Y.E. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016, 150, 1208–1218. [Google Scholar] [CrossRef]
- Ruhanen, H.; Nidhina Haridas, P.A.; Eskelinen, E.-L.; Eriksson, O.; Olkkonen, V.M.; Käkelä, R. Depletion of TM6SF2 disturbs membrane lipid composition and dynamics in HuH7 hepatoma cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. TM6SF2 may drive postprandial lipoprotein cholesterol toxicity away from the vessel walls to the liver in NAFLD. J. Hepatol. 2016, 64, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef]
- Li, T.T.; Li, T.H.; Peng, J.; He, B.; Liu, L.S.; Wei, D.H.; Jiang, Z.S.; Zheng, X.L.; Tang, Z.H. TM6SF2: A novel target for plasma lipid regulation. Atherosclerosis 2018, 268, 170–176. [Google Scholar] [CrossRef]
- Ehrhardt, N.; Doche, M.E.; Chen, S.; Mao, H.Z.; Walsh, M.T.; Bedoya, C.; Guindi, M.; Xiong, W.; Ignatius Irudayam, J.; Iqbal, J.; et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum. Mol. Genet. 2017, 26, 2719–2731. [Google Scholar] [CrossRef]
- O’Hare, E.A.; Yang, R.; Yerges-Armstrong, L.M.; Sreenivasan, U.; McFarland, R.; Leitch, C.C.; Wilson, M.H.; Narina, S.; Gorden, A.; Ryan, K.A.; et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology 2017, 65, 1526–1542. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef]
- Mancina, R.M.; Sentinelli, F.; Incani, M.; Bertoccini, L.; Russo, C.; Romeo, S.; Baroni, M.G. Transmembrane-6 superfamily member 2 (TM6SF2) E167K variant increases susceptibility to hepatic steatosis in obese children. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2016, 48, 100–101. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Grandone, A.; Cozzolino, D.; Marzuillo, P.; Cirillo, G.; Di Sessa, A.; Ruggiero, L.; Di Palma, M.R.; Perrone, L.; Miraglia del Giudice, E. TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL-cholesterol and increased liver injury in obese children. Pediatric Obes. 2016, 11, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Que, S.; Zhou, L.; Zheng, S.; Romeo, S.; Mardinoglu, A. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: A meta-analysis. Sci. Rep. 2017, 7, 9273. [Google Scholar] [CrossRef]
- Pirola, C.J.; Sookoian, S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015, 62, 1742–1756. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease-related risk of cardiovascular disease and other cardiac complications. Diabetes Obes. Metab. 2021. [Google Scholar] [CrossRef]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N.; et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Eslam, M.; Mangia, A.; Berg, T.; Chan, H.L.; Irving, W.L.; Dore, G.J.; Abate, M.L.; Bugianesi, E.; Adams, L.A.; Najim, M.A.; et al. Diverse impacts of the rs58542926 E167K variant in TM6SF2 on viral and metabolic liver disease phenotypes. Hepatology 2016, 64, 34–46. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Mallardi, P.; Fernández Gianotti, T.; Burgueño, A.L.; San Martino, J.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Raksayot, M.; Chuaypen, N.; Khlaiphuengsin, A.; Pinjaroen, N.; Treeprasertsuk, S.; Poovorawan, Y.; Tanaka, Y.; Tangkijvanich, P. Independent and additive effects of PNPLA3 and TM6SF2 polymorphisms on the development of non-B, non-C hepatocellular carcinoma. J. Gastroenterol. 2019, 54, 427–436. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, P.; De, L.; Li, B.; Su, S. The roles of transmembrane 6 superfamily member 2 rs58542926 polymorphism in chronic liver disease: A meta-analysis of 24,147 subjects. Mol. Genet. Genom. Med. 2019, 7, e824. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Buch, S.; Nischalke, H.D.; Weiss, K.H.; Gotthardt, D.; Fischer, J.; Rosendahl, J.; Marot, A.; Elamly, M.; Casper, M.; et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am. J. Gastroenterol. 2018, 113, 1475–1483. [Google Scholar] [CrossRef]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2016, 48, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Trepo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouze, E.; Imbeaud, S.; Gustot, T.; Deviere, J.; Debette, S.; et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 2019, 144, 533–544. [Google Scholar] [CrossRef]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.; Wahlers, A.; Begli, N.H.; Leopold, Y.; Kloters-Plachky, P.; Mehrabi, A.; Mohr, I.; Sander, J.; Rupp, C.; Gotthardt, D.N.; et al. The MBOAT7 rs641738 variant is associated with an improved outcome in primary sclerosing cholangitis. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 646–652. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef]
- Zarini, S.; Hankin, J.A.; Murphy, R.C.; Gijon, M.A. Lysophospholipid acyltransferases and eicosanoid biosynthesis in zebrafish myeloid cells. Prostaglandins Other Lipid Mediat. 2014, 113–115, 52–61. [Google Scholar] [CrossRef][Green Version]
- Gijon, M.A.; Riekhof, W.R.; Zarini, S.; Murphy, R.C.; Voelker, D.R. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J. Biol. Chem. 2008, 283, 30235–30245. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Read, R.W.; Hansen, G.M.; Payne, B.J.; Small, D.; Sands, A.T.; Zambrowicz, B.P. Congenital hydrocephalus in genetically engineered mice. Vet. Pathol. 2012, 49, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Inoue, T.; Sasaki, J.; Kubo, T.; Matsuda, S.; Nakasaki, Y.; Hattori, M.; Tanaka, F.; Udagawa, O.; Kono, N.; et al. LPIAT1 regulates arachidonic acid content in phosphatidylinositol and is required for cortical lamination in mice. Mol. Biol. Cell 2012, 23, 4689–4700. [Google Scholar] [CrossRef] [PubMed]
- Ruis-Gonzalez, M.D.; Canete, M.D.; Gomez-Chaparro, J.L.; Abril, N.; Canete, R.; Lopez-Barea, J. Alterations of protein expression in serum of infants with intrauterine growth restriction and different gestational ages. J. Proteom. 2015, 119, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Johansen, A.; Rosti, R.O.; Musaev, D.; Sticca, E.; Harripaul, R.; Zaki, M.; Caglayan, A.O.; Azam, M.; Sultan, T.; Froukh, T.; et al. Mutations in MBOAT7, Encoding Lysophosphatidylinositol Acyltransferase I, Lead to Intellectual Disability Accompanied by Epilepsy and Autistic Features. Am. J. Hum. Genet. 2016, 99, 912–916. [Google Scholar] [CrossRef]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The genetics of alcohol dependence and alcohol-related liver disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trepo, E. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e1216. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265. [Google Scholar] [CrossRef]
- Viitasalo, A.; Eloranta, A.M.; Atalay, M.; Romeo, S.; Pihlajamaki, J.; Lakka, T.A. Association of MBOAT7 gene variant with plasma ALT levels in children: The PANIC study. Pediatric Res. 2016, 80, 651–655. [Google Scholar] [CrossRef]
- Di Sessa, A.; Umano, G.R.; Cirillo, G.; Del Prete, A.; Iacomino, R.; Marzuillo, P.; Del Giudice, E.M. The Membrane-bound O-Acyltransferase7 rs641738 Variant in Pediatric Nonalcoholic Fatty Liver Disease. J. Pediatric Gastroenterol. Nutr. 2018, 67, 69–74. [Google Scholar] [CrossRef]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Thabet, K.; Chan, H.L.Y.; Petta, S.; Mangia, A.; Berg, T.; Boonstra, A.; Brouwer, W.P.; Abate, M.L.; Wong, V.W.; Nazmy, M.; et al. The membrane-bound O-acyltransferase domain-containing 7 variant rs641738 increases inflammation and fibrosis in chronic hepatitis B. Hepatology 2017, 65, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Thabet, K.; Asimakopoulos, A.; Shojaei, M.; Romero-Gomez, M.; Mangia, A.; Irving, W.L.; Berg, T.; Dore, G.J.; Gronbaek, H.; Sheridan, D.; et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat. Commun. 2016, 7, 12757. [Google Scholar] [CrossRef]
- Pelusi, S.; Baselli, G. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 3682. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Huang, C.X.; Li, G.Y.; Chen, K.H.; Han, L.; Tang, L.; Luo, H.Q.; Bao, M.H. Meta-analysis of the association between MBOAT7 rs641738, TM6SF2 rs58542926 and nonalcoholic fatty liver disease susceptibility. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 533–541. [Google Scholar] [CrossRef]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; Gazzi, C.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Lack of evidence supporting a role of TMC4-rs641738 missense variant-MBOAT7- intergenic downstream variant-in the Susceptibility to Nonalcoholic Fatty Liver Disease. Sci. Rep. 2018, 8, 5097. [Google Scholar] [CrossRef]
- Basyte-Bacevice, V.; Skieceviciene, J.; Valantiene, I.; Sumskiene, J.; Petrenkiene, V.; Kondrackiene, J.; Petrauskas, D.; Lammert, F.; Kupcinskas, J. TM6SF2 and MBOAT7 Gene Variants in Liver Fibrosis and Cirrhosis. Int. J. Mol. Sci. 2019, 20, 1277. [Google Scholar] [CrossRef]
- Koo, B.K.; Joo, S.K.; Kim, D. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2018, 33, 1277–1285. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically-characterised cohort. J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- Teo, K.; Abeysekera, K.W.M.; Adams, L.; Aigner, E.; Anstee, Q.M.; Banales, J.M.; Banerjee, R.; Basu, P.; Berg, T.; Bhatnagar, P.; et al. rs641738C > T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol. 2021, 74, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Fracanzani, A.L.; Dongiovanni, P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. EBioMedicine 2020, 57, 102866. [Google Scholar] [CrossRef] [PubMed]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Umano, G.R.; Caprio, S.; Di Sessa, A.; Chalasani, N.; Dykas, D.J.; Pierpont, B.; Bale, A.E.; Santoro, N. The rs626283 Variant in the MBOAT7 Gene is Associated with Insulin Resistance and Fatty Liver in Caucasian Obese Youth. Am. J. Gastroenterol. 2018, 113, 376–383. [Google Scholar] [CrossRef]
- Xia, M.; Chandrasekaran, P.; Rong, S.; Fu, X.; Mitsche, M.A. Hepatic Deletion of Mboat7 (Lpiat1) Causes Activation of SREBP-1c and Fatty Liver. J. Lipid Res. 2021, 62, 100031. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef]
- Fondevila, M.F.; Fernandez, U.; Gonzalez-Rellan, M.J.; Da Silva Lima, N.; Buque, X.; Gonzalez-Rodriguez, A.; Alonso, C.; Iruarrizaga-Lejarreta, M.; Delgado, T.C.; Varela-Rey, M.; et al. The L-α-lysophosphatidylinositol/GPR55 system induces the development of non-alcoholic steatosis and steatohepatitis. Hepatology 2021, 73, 606–624. [Google Scholar] [CrossRef]
- Thangapandi, V.R.; Knittelfelder, O. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J.; Valenti, L. Genetic pathways in nonalcoholic fatty liver disease: Insights from systems biology. Hepatology 2020, 72, 330–346. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic Insights for Drug Development in NAFLD. Trends Pharmacol. Sci. 2019, 40, 506–516. [Google Scholar] [CrossRef]
- Petta, S.; Miele, L.; Bugianesi, E.; Cammà, C.; Rosso, C.; Boccia, S.; Cabibi, D.; Di Marco, V.; Grimaudo, S.; Grieco, A.; et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e87523. [Google Scholar] [CrossRef]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef]
- Raimondo, A.; Rees, M.G.; Gloyn, A.L. Glucokinase regulatory protein: Complexity at the crossroads of triglyceride and glucose metabolism. Curr. Opin. Lipidol. 2015, 26, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Caprio, S.; Pierpont, B.; Van Name, M.; Savoye, M.; Parks, E.J. Hepatic De Novo Lipogenesis in Obese Youth Is Modulated by a Common Variant in the GCKR Gene. J. Clin. Endocrinol. Metab. 2015, 100, E1125–E1132. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Zain, S.M.; Mohamed, R.; Rampal, S.; Chin, K.F.; Basu, R.C.; Cheah, P.L.; Mahadeva, S.; Mohamed, Z. Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: An interaction study with adiponutrin gene. J. Gastroenterol. 2014, 49, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Shima, T.; Mizuno, M.; Mitsumoto, Y.; Umemura, A.; Kanbara, Y.; Tanaka, S.; Sumida, Y.; Yasui, K.; Takahashi, M.; et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 2018, 13, e0185490. [Google Scholar] [CrossRef] [PubMed]
- Kozian, D.H.; Barthel, A.; Cousin, E.; Brunnhöfer, R.; Anderka, O.; März, W.; Böhm, B.; Winkelmann, B.; Bornstein, S.R.; Schmoll, D. Glucokinase-activating GCKR polymorphisms increase plasma levels of triglycerides and free fatty acids, but do not elevate cardiovascular risk in the Ludwigshafen Risk and Cardiovascular Health Study. Horm. Metab. Res. 2010, 42, 502–506. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Horiguchi, Y.; Araki, M.; Motojima, K. 17beta-Hydroxysteroid dehydrogenase type 13 is a liver-specific lipid droplet-associated protein. Biochem. Biophys. Res. Commun. 2008, 370, 235–238. [Google Scholar] [CrossRef]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef]
- Kozlitina, J.; Stender, S.; Hobbs, H.H.; Cohen, J.C. HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N. Engl. J. Med. 2018, 379, 1876–1877. [Google Scholar] [CrossRef]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res. 2019, 60, 176–185. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Tukiainen, T.; Juuti, A.; Sammalkorpi, H.; Haridas, P.A.N.; Niemelä, O.; Arola, J.; Orho-Melander, M.; Hakkarainen, A.; Kovanen, P.T.; et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight 2020, 5, e132158. [Google Scholar] [CrossRef]
- Gellert-Kristensen, H.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase-Lowering Effect of a HSD17B13 Variant. Hepatology 2020, 71, 56–66. [Google Scholar] [CrossRef]
- Yang, J.; Trepo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouze, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Lu, C.; Fang, S.; Weng, Q.; Lv, X.; Meng, M.; Zhu, J.; Zheng, L.; Hu, Y.; Gao, Y.; Wu, X.; et al. Integrated analysis reveals critical glycolytic regulators in hepatocellular carcinoma. Cell Commun. Signal. CCS 2020, 18, 97. [Google Scholar] [CrossRef]
- Mann, J.P.; Pietzner, M.; Wittemans, L.B.; Rolfe, E.L.; Kerrison, N.D.; Imamura, F.; Forouhi, N.G.; Fauman, E.; Allison, M.E.; Griffin, J.L.; et al. Insights into genetic variants associated with NASH-fibrosis from metabolite profiling. Hum. Mol. Genet. 2020, 29, 3451–3463. [Google Scholar] [CrossRef]
- Stender, S.; Smagris, E.; Lauridsen, B.K.; Kofoed, K.F.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Pennacchio, L.A.; Dickel, D.E.; Cohen, J.C.; Hobbs, H.H. Relationship between genetic variation at PPP1R3B and levels of liver glycogen and triglyceride. Hepatology 2018, 67, 2182–2195. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Mannisto, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.B.; Shewale, S.V.; Sequeira, R.N.; Millar, J.S.; Hand, N.J. Hepatic protein phosphatase 1 regulatory subunit 3B (Ppp1r3b) promotes hepatic glycogen synthesis and thereby regulates fasting energy homeostasis. J. Biol. Chem. 2017, 292, 10444–10454. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Rametta, R.; Meroni, M.; Valenti, L. The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development—A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Pelusi, S.; Petta, S.; Rosso, C.; Borroni, V.; Fracanzani, A.L.; Dongiovanni, P.; Craxi, A.; Bugianesi, E.; Fargion, S.; Valenti, L. Renin-Angiotensin System Inhibitors, Type 2 Diabetes and Fibrosis Progression: An Observational Study in Patients with Nonalcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0163069. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef]
- Kitamoto, A.; Kitamoto, T.; Nakamura, T.; Ogawa, Y.; Yoneda, M.; Hyogo, H.; Ochi, H.; Mizusawa, S.; Ueno, T.; Nakao, K.; et al. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocr. J. 2014, 61, 683–689. [Google Scholar] [CrossRef]
- Cefalù, A.B.; Pirruccello, J.P.; Noto, D.; Gabriel, S.; Valenti, V.; Gupta, N.; Spina, R.; Tarugi, P.; Kathiresan, S.; Averna, M.R. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2021–2025. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Moulin, P.; Roy, P.; Samson-Bouma, M.E.; Collardeau-Frachon, S.; Chebel-Dumont, S.; Peretti, N.; Dumortier, J.; Zoulim, F.; Fontanges, T.; et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J. Hepatol. 2014, 61, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Hariri, A.; Nelson-Williams, C.; Foo, J.N.; Zhang, X.M.; Dziura, J.; Lifton, R.P.; Shulman, G.I. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 362, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 2011, 53, 467–474. [Google Scholar] [CrossRef]
- Valenti, L.; Nobili, V.; Al-Serri, A.; Rametta, R.; Leathart, J.B.; Zappa, M.A.; Dongiovanni, P.; Fracanzani, A.L.; Alterio, A.; Roviaro, G.; et al. The APOC3 T-455C and C-482T promoter region polymorphisms are not associated with the severity of liver damage independently of PNPLA3 I148M genotype in patients with nonalcoholic fatty liver. J. Hepatol. 2011, 55, 1409–1414. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Baselli, G.; Mancina, R.M.; Ruscica, M.; Longo, M.; Rametta, R.; Cespiati, A.; Pelusi, S.; Ferri, N.; et al. PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J. Lipid Res. 2019, 60, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Huang, J.; Qi, Q.; Li, Y.; Bray, G.A.; Rood, J.; Sacks, F.M.; Qi, L. PCSK7 genotype modifies effect of a weight-loss diet on 2-year changes of insulin resistance: The POUNDS LOST trial. Diabetes Care 2015, 38, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, Y.; Duval, S.; Sachan, V.; Essalmani, R.; Susan-Resiga, D.; Roubtsova, A.; Hamelin, J.; Gerhardy, S.; Kirchhofer, D.; Tagliabracci, V.S.; et al. Proprotein convertase 7 (PCSK7) reduces apoA-V levels. FEBS J. 2020, 287, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Ferri, N.; Macchi, C.; Meroni, M.; Lanti, C.; Ricci, C.; Maggioni, M.; Fracanzani, A.L.; Badiali, S.; Fargion, S.; et al. Liver fat accumulation is associated with circulating PCSK9. Ann. Med. 2016, 48, 384–391. [Google Scholar] [CrossRef]
- Trinder, M.; Francis, G.A.; Brunham, L.R. Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2020, 5, 390–399. [Google Scholar] [CrossRef]
- Costet, P.; Cariou, B.; Lambert, G.; Lalanne, F.; Lardeux, B.; Jarnoux, A.L.; Grefhorst, A.; Staels, B.; Krempf, M. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J. Biol. Chem. 2006, 281, 6211–6218. [Google Scholar] [CrossRef]
- Kotowski, I.K.; Pertsemlidis, A.; Luke, A.; Cooper, R.S.; Vega, G.L.; Cohen, J.C.; Hobbs, H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006, 78, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Grimaudo, S.; Bartesaghi, S.; Rametta, R.; Marra, F.; Mancina, R.M.; Pihlajamäki, J.; Kakol-Palm, D.; Andréasson, A.C.; Don-giovanni, P.; Fracanzani, A.L.; et al. PCSK9 rs11591147 R46L loss-of-function variant protects against liver damage in individuals with NAFLD. Liver Int. 2021, 41, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gomaraschi, M.; Fracanzani, A.L.; Dongiovanni, P.; Pavanello, C.; Giorgio, E.; Da Dalt, L.; Norata, G.D.; Calabresi, L.; Consonni, D.; Lombardi, R.; et al. Lipid accumulation impairs lysosomal acid lipase activity in hepatocytes: Evidence in NAFLD patients and cell cultures. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2019, 1864, 158523. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Ćorić, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef]
- Lyons, H.; Vouyoukas, E.; Higgins, M.; Maciejko, J.J. Clinical and Histologic Liver Improvement in Siblings With Lysosomal Acid Lipase Deficiency After Enzyme Replacement. J. Pediatric Gastroenterol. Nutr. 2020, 70, 635–639. [Google Scholar] [CrossRef]
- Malinová, V.; Balwani, M.; Sharma, R.; Arnoux, J.B.; Kane, J.; Whitley, C.B.; Marulkar, S.; Abel, F. Sebelipase alfa for lysosomal acid lipase deficiency: 5-year treatment experience from a phase 2 open-label extension study. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 2203–2214. [Google Scholar] [CrossRef]
- Auinger, A.; Valenti, L.; Pfeuffer, M.; Helwig, U.; Herrmann, J.; Fracanzani, A.L.; Dongiovanni, P.; Fargion, S.; Schrezenmeir, J.; Rubin, D. A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis. Horm. Metab. Res. 2010, 42, 854–859. [Google Scholar] [CrossRef]
- Valenti, L.; Motta, B.M.; Alisi, A.; Sartorelli, R.; Buonaiuto, G.; Dongiovanni, P.; Rametta, R.; Pelusi, S.; Fargion, S.; Nobili, V. LPIN1 rs13412852 polymorphism in pediatric nonalcoholic fatty liver disease. J. Pediatric Gastroenterol. Nutr. 2012, 54, 588–593. [Google Scholar] [CrossRef]
- Faulkner, C.S.; White, C.M.; Shah, V.H.; Jophlin, L.L. A single nucleotide polymorphism of PLIN2 is associated with nonalcoholic steatohepatitis and causes phenotypic changes in hepatocyte lipid droplets: A pilot study. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2020, 1865, 158637. [Google Scholar] [CrossRef]
- Magné, J.; Aminoff, A.; Perman Sundelin, J.; Mannila, M.N.; Gustafsson, P.; Hultenby, K.; Wernerson, A.; Bauer, G.; Listenberger, L.; Neville, M.J.; et al. The minor allele of the missense polymorphism Ser251Pro in perilipin 2 (PLIN2) disrupts an α-helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3090–3099. [Google Scholar] [CrossRef]
- Drevinge, C.; Dalen, K.T.; Mannila, M.N.; Täng, M.S.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Mardani, I.; Haugen, F.; Fogelstrand, P.; et al. Perilipin 5 is protective in the ischemic heart. Int. J. Cardiol. 2016, 219, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, P.F.; Lin, H.F.; Liu, K.; Chang, M.H.; Ni, Y.H. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J. Hepatol. 2016, 65, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Schwerbel, K.; Kamitz, A.; Krahmer, N.; Hallahan, N.; Jähnert, M.; Gottmann, P.; Lebek, S.; Schallschmidt, T.; Arends, D.; Schumacher, F.; et al. Immunity-related GTPase induces lipophagy to prevent excess hepatic lipid accumulation. J. Hepatol. 2020, 73, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Eslam, M.; McLeod, D.; Kelaeng, K.S.; Mangia, A.; Berg, T.; Thabet, K.; Irving, W.L.; Dore, G.J.; Sheridan, D.; Grønbæk, H.; et al. IFN-λ3, not IFN-λ4, likely mediates IFNL3-IFNL4 haplotype-dependent hepatic inflammation and fibrosis. Nat. Genet. 2017, 49, 795–800. [Google Scholar] [CrossRef]
- Petta, S.; Grimaudo, S.; Cammà, C.; Cabibi, D.; Di Marco, V.; Licata, G.; Pipitone, R.M.; Craxì, A. IL28B and PNPLA3 polymorphisms affect histological liver damage in patients with non-alcoholic fatty liver disease. J. Hepatol. 2012, 56, 1356–1362. [Google Scholar] [CrossRef]
- Eslam, M.; Hashem, A.M.; Leung, R.; Romero-Gomez, M.; Berg, T.; Dore, G.J.; Chan, H.L.; Irving, W.L.; Sheridan, D.; Abate, M.L.; et al. Interferon-λ rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nat. Commun. 2015, 6, 6422. [Google Scholar] [CrossRef]
- Eslam, M.; Hashem, A.M.; Romero-Gomez, M.; Berg, T.; Dore, G.J.; Mangia, A.; Chan, H.L.Y.; Irving, W.L.; Sheridan, D.; Abate, M.L.; et al. FibroGENE: A gene-based model for staging liver fibrosis. J. Hepatol. 2016, 64, 390–398. [Google Scholar] [CrossRef]
- Petta, S.; Valenti, L. Interferon lambda 4 rs368234815 TT > δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. J. Hepatol. 2017, 66, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Valenti, L.; Svegliati-Baroni, G.; Ruscica, M.; Pipitone, R.M.; Dongiovanni, P.; Rychlicki, C.; Ferri, N.; Cammà, C.; Fracanzani, A.L.; et al. Fibronectin Type III Domain-Containing Protein 5 rs3480 A > G Polymorphism, Irisin, and Liver Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2017, 102, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Metwally, M.; Bayoumi, A.; Romero-Gomez, M.; Thabet, K.; John, M.; Adams, L.A.; Huo, X.; Aller, R.; García-Monzón, C.; Teresa Arias-Loste, M.; et al. A polymorphism in the Irisin-encoding gene (FNDC5) associates with hepatic steatosis by differential miRNA binding to the 3’UTR. J. Hepatol. 2019, 70, 494–500. [Google Scholar] [CrossRef]
- Gorden, A.; Yang, R.; Yerges-Armstrong, L.M.; Ryan, K.A.; Speliotes, E.; Borecki, I.B.; Harris, T.B.; Chu, X.; Wood, G.C.; Still, C.D.; et al. Genetic variation at NCAN locus is associated with inflammation and fibrosis in non-alcoholic fatty liver disease in morbid obesity. Hum. Hered. 2013, 75, 34–43. [Google Scholar] [CrossRef]
- Cai, W.; Weng, D.H.; Yan, P.; Lin, Y.T.; Dong, Z.H.; Mailamuguli; Yao, H. Genetic polymorphisms associated with nonalcoholic fatty liver disease in Uyghur population: A case-control study and meta-analysis. Lipids Health Dis. 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Cicione, C.; Degirolamo, C.; Moschetta, A. Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology 2012, 56, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al. beta-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 2020, 72, 411–419. [Google Scholar] [CrossRef]
- Markan, K.R.; Naber, M.C.; Small, S.M.; Peltekian, L.; Kessler, R.L.; Potthoff, M.J. FGF21 resistance is not mediated by downregulation of beta-klotho expression in white adipose tissue. Mol. Metab. 2017, 6, 602–610. [Google Scholar] [CrossRef]
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela-Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Martinez-Chantar, M.L.; Maratos-Flier, E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 2010, 139, 456–463. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Gallego-Escuredo, J.M.; Catalán, V.; Rodríguez, A.; Domingo, P.; Moncada, R.; Valentí, V.; Salvador, J.; Giralt, M.; Villarroya, F.; et al. FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin. Nutr. (Edinb. Scotl. ) 2017, 36, 861–868. [Google Scholar] [CrossRef]
- Miele, L.; Beale, G.; Patman, G.; Nobili, V.; Leathart, J.; Grieco, A.; Abate, M.; Friedman, S.L.; Narla, G.; Bugianesi, E.; et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology 2008, 135, 282–291.e281. [Google Scholar] [CrossRef] [PubMed]
- Rametta, R.; Meroni, M. From Environment to Genome and Back: A Lesson from HFE Mutations. Int. J. Mol. Sci. 2020, 21, 3505. [Google Scholar] [CrossRef]
- Petta, S.; Valenti, L.; Marra, F.; Grimaudo, S.; Tripodo, C.; Bugianesi, E.; Cammà, C.; Cappon, A.; Di Marco, V.; Di Maira, G.; et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Rüeger, S.; Bochud, P.Y.; Dufour, J.F.; Müllhaupt, B.; Semela, D.; Heim, M.H.; Moradpour, D.; Cerny, A.; Malinverni, R.; Booth, D.R.; et al. Impact of common risk factors of fibrosis progression in chronic hepatitis C. Gut 2015, 64, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e407. [Google Scholar] [CrossRef] [PubMed]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Aravinthan, A.; Mells, G.; Allison, M.; Leathart, J.; Kotronen, A.; Yki-Jarvinen, H.; Daly, A.K.; Day, C.P.; Anstee, Q.M.; Alexander, G. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle (Georget. Tex.) 2014, 13, 1489–1494. [Google Scholar] [CrossRef]
- Calado, R.T.; Regal, J.A.; Kleiner, D.E.; Schrump, D.S.; Peterson, N.R.; Pons, V.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS ONE 2009, 4, e7926. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Paolini, E.; Alisi, A.; Miele, L.; De Caro, E.R.; Pisano, G.; Maggioni, M.; Soardo, G.; Valenti, L.V.; et al. The rs599839 A > G Variant Disentangles Cardiovascular Risk and Hepatocellular Carcinoma in NAFLD Patients. Cancers (Basel) 2021, 13, 1783. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Petta, S.; Longo, M.; Alisi, A.; Soardo, G.; Valenti, L.; Miele, L.; Grimaudo, S.; Pennisi, G.; et al. Neurotensin up-regulation is associated with advanced fibrosis and hepatocellular carcinoma in patients with MAFLD. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2020, 1865, 158765. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786. [Google Scholar] [CrossRef]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al. The UCP2 -866 G > A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1574–1580. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Izaola, O.; González Sagrado, M.; Conde, R.; Alvarez, T.; Pacheco, D.; Velasco, M.C. Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576. [Google Scholar]
- Zhong, L.; Mostoslavsky, R. Fine tuning our cellular factories: Sirtuins in mitochondrial biology. Cell Metab. 2011, 13, 621–626. [Google Scholar] [CrossRef]
- Dong, C.; Della-Morte, D.; Wang, L.; Cabral, D.; Beecham, A.; McClendon, M.S.; Luca, C.C.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS ONE 2011, 6, e27157. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.E. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef] [PubMed]
- Innes, H.; Buch, S.; Hutchinson, S.; Guha, I.N.; Morling, J.R.; Barnes, E.; Irving, W.; Forrest, E.; Pedergnana, V.; Goldberg, D.; et al. Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159, 1276–1289.e1277. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Juuti, A.; Sammalkorpi, H.; Penttilä, A.K.; Orešič, M.; Hyötyläinen, T.; Arola, J.; Orho-Melander, M.; Yki-Järvinen, H. MARC1 variant rs2642438 increases hepatic phosphatidylcholines and decreases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2020, 73, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.V.; Schneider, K.M.; Conlon, D.M.; Park, J.; Vujkovic, M.; Zandvakili, I.; Ko, Y.A.; Trautwein, C.; Center, R.; Carr, R.M.; et al. A genome-first approach to mortality and metabolic phenotypes in MTARC1 p.Ala165Thr (rs2642438) heterozygotes and homozygotes. Med (New York N.Y.) 2021, 2, 851–863.e853. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Pacifico, L.; Chiesa, C.; Perla, F.M.; Ceci, F.; Angeloni, A.; D’Erasmo, L.; Di Martino, M.; Arca, M. Genetic and metabolic predictors of hepatic fat content in a cohort of Italian children with obesity. Pediatric Res. 2019, 85, 671–677. [Google Scholar] [CrossRef]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood predictors of adult fatty liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef]
- Koo, B.K.; Joo, S.K.; Kim, D.; Lee, S.; Bae, J.M.; Park, J.H.; Kim, J.H.; Chang, M.S.; Kim, W. Development and Validation of a Scoring System, Based on Genetic and Clinical Factors, to Determine Risk of Steatohepatitis in Asian Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 2592–2599.e2510. [Google Scholar] [CrossRef]
- Larrieta-Carrasco, E.; Flores, Y.N.; Macías-Kauffer, L.R.; Ramírez-Palacios, P.; Quiterio, M.; Ramírez-Salazar, E.G.; León-Mimila, P.; Rivera-Paredez, B.; Cabrera-Álvarez, G.; Canizales-Quinteros, S.; et al. Genetic variants in COL13A1, ADIPOQ and SAMM50, in addition to the PNPLA3 gene, confer susceptibility to elevated transaminase levels in an admixed Mexican population. Exp. Mol. Pathol. 2018, 104, 50–58. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Wang, K.; Wang, Z.; Sun, X.; Zhong, L.; Deng, G.; Song, G.; Sun, B.; Peng, Z.; et al. Additive Effects of the Risk Alleles of PNPLA3 and TM6SF2 on Non-alcoholic Fatty Liver Disease (NAFLD) in a Chinese Population. Front. Genet. 2016, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, Y.; Zhang, S.; Wang, X.; Shen, J.; Zhang, S. Interaction of TM6SF2 E167K and PNPLA3 I148M variants in NAFLD in northeast China. Ann. Hepatol. 2019, 18, 456–460. [Google Scholar] [CrossRef]
- Chen, L.; Du, S.; Lu, L.; Lin, Z.; Jin, W.; Hu, D.; Jiang, X.; Xin, Y.; Xuan, S. The additive effects of the TM6SF2 E167K and PNPLA3 I148M polymorphisms on lipid metabolism. Oncotarget 2017, 8, 74209–74216. [Google Scholar] [CrossRef] [PubMed]
- Gellert-Kristensen, H.; Richardson, T.G.; Davey Smith, G.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 2020, 72, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Kim, N.; Shin, H.Y. Modeling Non-Alcoholic Fatty Liver Disease (NAFLD) Using “Good-Fit” Genome-Editing Tools. Cells 2020, 9, 2572. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): New perspectives for a fairy-tale ending? Metab. Clin. Exp. 2021, 117, 154708. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Erconi, V.; Carli, F.; Macchi, C.; Fortunato, F.; Ronchi, D.; Sabatini, S.; Paolini, E.; De Caro, E.R.; et al. TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A. Modelling PNPLA3-Associated Non-Alcoholic Fatty Liver Disease Using Human Induced Pluripotent Stem Cells. Hepatology 2021. [Google Scholar] [CrossRef]
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10, bio054189. [Google Scholar] [CrossRef] [PubMed]
- Sinton, M.C.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Wernig-Zorc, S.; Thomson, J.P.; Carter, R.N.; Lyall, M.J.; Walker, P.D.; Thakker, A.; Meehan, R.R.; et al. A human pluripotent stem cell model for the analysis of metabolic dysfunction in hepatic steatosis. iScience 2021, 24, 101931. [Google Scholar] [CrossRef] [PubMed]
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Dessein, A. Clinical utility of polygenic risk scores for predicting NAFLD disorders. J. Hepatol. 2021, 74, 769–770. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Variant | Gene | Global MAF | Function | Effect | Impact | Phenotype |
|---|---|---|---|---|---|---|
| rs738409 C > G | PNPLA3 | 0.26 (G) | Lipid remodeling | p.I148M | Loss-of-function | ↑ NAFLD, NASH, fibrosis, HCC |
| rs58542926 C > T | TM6SF2 | 0.07 (T) | VLDL secretion | p.E167K | Loss-of-function | ↑ NAFLD, NASH, fibrosis |
| rs641738 C > T | TMC4/ MBOAT7 | 0.37 (T) | Lipid remodeling | p.G17E | Loss-of-function | ↑ NAFLD, NASH, fibrosis, HCC |
| rs1260326 C > T | GCKR | 0.29 (T) | Regulation of DNL | p.P446L | Loss-of-function | ↑ NAFLD, NASH, fibrosis |
| rs72613567 T > TA | HSD17B13 | 0.18 (TA) | Lipid remodeling | Truncated protein | Loss-of-function | ↓ NASH, fibrosis, HCC |
| rs4841132 G > A | PPP1R3B | 0.09 (A) | Glycogen synthesis | Non-coding | Gain-of-function | ↓ NAFLD, fibrosis, HCC |
| rs1801278 C > T | IRS1 | 0.05 (T) | Insulin signaling | p.G972R | Loss-of-function | ↑ Fibrosis |
| rs1044498 A > C | ENPP1 | 0.34 (C) | Insulin signaling | p.K121Q | Gain-of-function | ↑ Fibrosis |
| rs2954021 G > A | TRIB1 | 0.45 (A) | Regulation of DNL | Non-coding | Gain-of-function | ↑ NAFLD |
| rs12137855 C > T | LYPLAL1 | 0.16 (T) | Lipid metabolism | Intronic | Loss-of-function | ↑ NAFLD |
| Several | APOB | NA | VLDL secretion | Protein change | Loss-of-function | ↑ NAFLD NASH, fibrosis, HCC |
| Several | MTTP | NA | VLDL secretion | Protein change | Loss-of-function | ↑NAFLD |
| rs236918 G > C | PCSK7 | 0.26 (C) | Membrane transferrin receptor shedding and regulation of circulating lipids | Intronic | Gain-of-function | ↑ NASH, fibrosis |
| Several | PCSK9 | NA | LDL uptake | Protein change | Loss-of-function | No evidence of association with steatosis |
| Several | LIPA | NA | Lipid remodeling | Protein change | LAL deficiency | ↑ NAFLD, NASH, fibrosis |
| rs56225452 G > A | FATP5 | 0.16 (A) | FFAs uptake | Non-coding | Gain-of-function | ↑ NASH, fibrosis |
| rs13412852 C > T | LPIN1 | 0.21 (T) | Lipid metabolism | Intronic | Not Defined | ↓ NASH, fibrosis |
| rs35568725 A > G | PLIN2 | 0.02 (G) | Lipid remodeling | p.S251P | Loss-of-function | ↑ NAFLD, NASH, IR, atherosclerosis |
| rs884164 A > G | PLIN5 | 0.19 (G) | Lipid remodeling | Non-coding | Loss-of-function | ↑ oxidative stress |
| rs17618244 G > A | KLB | 0.15 (A) | FGF19/FGFR4 pathway | p.R728Q | Loss-of-function | ↓ NASH, fibrosis |
| rs4374383 G > A | MERTK | 0.45 (A) | Innate immunity | Intronic | Loss-of-function | ↓ Fibrosis |
| rs3750861 G > A | KLF6 | 0.07 (A) | HSCs activation | Splice variant IVS1-27G | Loss-of-function | ↓ Fibrosis |
| Several | TERT | NA | Telomere maintenance | Protein change | Loss-of-function | ↑ Fibrosis, HCC |
| rs12979860 C > T | IL28B | 0.36 (T) | Innate immunity | Alternative IFNL3/4 transcription | Loss-of-function | ↓ NASH, Fibrosis |
| rs3480 A > G | FNDC5 | 0.42 (G) | HSCs activation | Non-coding | Loss-of-function | ↓ Fibrosis |
| rs4880 C > T | SOD2 | 0.33 (T) | Mitochondrial antioxidant | p.A16V | Loss-of-function | ↑ Fibrosis |
| rs695366 G > A | UCP2 | 0.26 (A) | Mitochondrial lipid metabolism Oxphos | −866 promoter variant | Gain-of-function | ↓ NASH, fibrosis |
| rs2642438 G > A | MARC1 | 0.19 (A) | Mitochondrial detoxification | p.A165T | Loss-of-function | ↓ NAFLD, NASH, fibrosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meroni, M.; Longo, M.; Tria, G.; Dongiovanni, P. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 1359. https://doi.org/10.3390/biomedicines9101359
Meroni M, Longo M, Tria G, Dongiovanni P. Genetics Is of the Essence to Face NAFLD. Biomedicines. 2021; 9(10):1359. https://doi.org/10.3390/biomedicines9101359
Chicago/Turabian StyleMeroni, Marica, Miriam Longo, Giada Tria, and Paola Dongiovanni. 2021. "Genetics Is of the Essence to Face NAFLD" Biomedicines 9, no. 10: 1359. https://doi.org/10.3390/biomedicines9101359
APA StyleMeroni, M., Longo, M., Tria, G., & Dongiovanni, P. (2021). Genetics Is of the Essence to Face NAFLD. Biomedicines, 9(10), 1359. https://doi.org/10.3390/biomedicines9101359