
Astrocytes, a Promising Opportunity to Control the Progress of Parkinson’s Disease

 , and
, and
Abstract
:1. Introduction
2. The Vulnerability of Dopaminergic Cells and Parkinson’s Disease
3. Astrocytes Modulate the Vulnerability of DA-Cells in Parkinson’s Disease
4. Are Astrocytes Involved in the Clinical Expression of Parkinson’s Disease?
5. Controlling Evolution PD with Astrocyte-Based Therapies
5.1. Astrocytes Provide Energy Resources to DA-Cells
5.2. Astrocytes Prevent Oxidative Stress in DA-Cells
5.3. Astrocytes Prevent Excitotoxicity in DA-Cells
5.4. Astrocytes Prevent Neuroinflammation in PD
5.5. Astrocytes Provide Neurotrophic Factors Which Are Necessary for DA-Cell Survival
5.6. The Global Activation of the Neuroprotective Functions of Astrocytes
6. Final Comments
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Braak, E. Neuroanatomy of parkinson disease. Changes in the neuronal cytoskeleton of a few disease-susceptible types of neurons lead to progressive destruction of circumscribed areas in the limbic and motor systems. Nervenarzt 2000, 71, 459–469. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Invited article: Nervous system pathology in sporadic parkinson disease. Neurology 2008, 70, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Maruszak, A.; Gaweda-Walerych, K.; Soltyszewski, I.; Zekanowski, C. Mitochondrial DNA in pathogenesis of alzheimer’s and parkinson’s diseases. Acta Neurobiol. Exp. Wars 2006, 66, 153–176. [Google Scholar] [PubMed]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in parkinson’s disease and huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.C.; Ko, H.W.; Bok, E.; Park, E.S.; Huh, S.H.; Nam, J.H.; Jin, B.K. The role of neuroinflammation on the pathogenesis of parkinson’s disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Fu, Q.; Cheng, L.; Zhai, M.; Wu, W.; Huang, L.; Du, G. Inflammatory response in parkinson’s disease (review). Mol. Med. Rep. 2014, 10, 2223–2233. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Morales, I.; Rodriguez-Sabate, C.; Sanchez, A.; Castro, R.; Brito, J.M.; Sabate, M. The degeneration and replacement of dopamine cells in parkinson’s disease: The role of aging. Front. Neuroanat. 2014, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing tnf-alpha-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s disease-related proteins pink1 and parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Cruz, N.F. Astrocyte activation in working brain: Energy supplied by minor substrates. Neurochem. Int. 2006, 48, 586–595. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. Pgc-1alpha, a potential therapeutic target for early intervention in parkinson’s disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [Green Version]
- Kuter, K.; Olech, L.; Glowacka, U.; Paleczna, M. Astrocyte support is important for the compensatory potential of the nigrostriatal system neurons during early neurodegeneration. J. Neurochem. 2019, 148, 63–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, H.L.; Teismann, P. Glutathione--a review on its role and significance in parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, D.A.; Newberg, A.B. Response to “potential role of n-acetyl cysteine in the cysteine proteome in parkinson’s disease?”. Clin. Pharmacol. Ther. 2020, 107, 1056. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-acetyl cysteine is associated with dopaminergic improvement in parkinson’s disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-acetyl cysteine may support dopamine neurons in parkinson’s disease: Preliminary clinical and cell line data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J. Neurochem. 2004, 89, 1241–1251. [Google Scholar] [CrossRef]
- Rai, S.N.; Yadav, S.K.; Singh, D.; Singh, S.P. Ursolic acid attenuates oxidative stress in nigrostriatal tissue and improves neurobehavioral activity in mptp-induced parkinsonian mouse model. J. Chem. Neuroanat. 2016, 71, 41–49. [Google Scholar] [CrossRef]
- Jin, X.Y.; Chen, H.; Li, D.D.; Li, A.L.; Wang, W.Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent mek inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 955–972. [Google Scholar] [CrossRef] [Green Version]
- Jing, L.; He, M.T.; Chang, Y.; Mehta, S.L.; He, Q.P.; Zhang, J.Z.; Li, P.A. Coenzyme q10 protects astrocytes from ros-induced damage through inhibition of mitochondria-mediated cell death pathway. Int. J. Biol. Sci. 2015, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Karakaya, S.; Kipp, M.; Beyer, C. Oestrogen regulates the expression and function of dopamine transporters in astrocytes of the nigrostriatal system. J. Neuroendocrinol. 2007, 19, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase b and a in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Rodriguez-Sabate, C.; Rodriguez, M. Striatal astrocytes engulf dopaminergic debris in parkinson’s disease: A study in an animal model. PLoS ONE 2017, 12, e0185989. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J. Astrocytes: News about brain health and diseases. Biomedicines 2020, 8, 394. [Google Scholar] [CrossRef] [PubMed]
- Cragnolini, A.B.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural Regen. Res. 2020, 15, 838–842. [Google Scholar] [PubMed]
- Cunnane, S.C.; Courchesne-Loyer, A.; St-Pierre, V.; Vandenberghe, C.; Pierotti, T.; Fortier, M.; Croteau, E.; Castellano, C.A. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of alzheimer’s disease. Ann. N. Y. Acad. Sci. 2016, 1367, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Mamik, M.K.; Asahchop, E.L.; Chan, W.F.; Zhu, Y.; Branton, W.G.; McKenzie, B.A.; Cohen, E.A.; Power, C. Insulin treatment prevents neuroinflammation and neuronal injury with restored neurobehavioral function in models of hiv/aids neurodegeneration. J. Neurosci. 2016, 36, 10683–10695. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M. Inhibitors of mao-b and comt: Their effects on brain dopamine levels and uses in parkinson’s disease. J. Neural. Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Rodriguez, M. Self-induced accumulation of glutamate in striatal astrocytes and basal ganglia excitotoxicity. Glia 2012, 60, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Sabate, M.; Rodriguez-Sabate, C.; Morales, I. The role of non-synaptic extracellular glutamate. Brain Res. Bull. 2013, 93, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Yanos, C.; Rodriguez-Sabate, C.; Sanchez, A.; Rodriguez, M. Striatal glutamate degenerates thalamic neurons. J. Neuropathol. Exp. Neurol. 2013, 72, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular mechanisms of glutamate toxicity in parkinson’s disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Dileone, M.; Del Rey, N.L.; Hernandez, L.F.; Obeso, J.A. Compensatory mechanisms in parkinson’s disease: Circuits adaptations and role in disease modification. Exp. Neurol. 2017, 298, 148–161. [Google Scholar] [CrossRef]
- Ferrarese, C.; Tremolizzo, L.; Rigoldi, M.; Sala, G.; Begni, B.; Brighina, L.; Ricci, G.; Albizzati, M.G.; Piolti, R.; Crosti, F.; et al. Decreased platelet glutamate uptake and genetic risk factors in patients with parkinson’s disease. Neurol. Sci. 2001, 22, 65–66. [Google Scholar] [CrossRef]
- Chung, E.K.; Chen, L.W.; Chan, Y.S.; Yung, K.K. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 2008, 511, 421–437. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters glt-1 and glast in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Rappold, P.M.; Tieu, K. Astrocytes and therapeutics for parkinson’s disease. Neurotherapeutics 2010, 7, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Fearnley, J.M.; Lees, A.J. Ageing and parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Rodriguez-Sabate, C.; Rodriguez, M. The astrocytic response to the dopaminergic denervation of the striatum. J. Neurochem. 2016, 139, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Nakamura, Y.; Li, W.; Hamanaka, G.; Arai, K.; Lo, E.H.; Hayakawa, K. Effects of o-glcnacylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow Metab. 2021, 41, 1523–1535. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human ipscs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Puertas-Avendano, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal anti-inflammatory drugs use and risk of parkinson disease: A dose-response meta-analysis. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef]
- Bortolanza, M.; Nascimento, G.C.; Socias, S.B.; Ploper, D.; Chehin, R.N.; Raisman-Vozari, R.; Del-Bel, E. Tetracycline repurposing in neurodegeneration: Focus on parkinson’s disease. J. Neural. Transm. 2018, 125, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Santa-Cecilia, F.V.; Socias, B.; Ouidja, M.O.; Sepulveda-Diaz, J.E.; Acuna, L.; Silva, R.L.; Michel, P.P.; Del-Bel, E.; Cunha, T.M.; Raisman-Vozari, R. Doxycycline suppresses microglial activation by inhibiting the p38 mapk and nf-kb signaling pathways. Neurotox Res. 2016, 29, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lizarraga, F.; Socias, S.B.; Avila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.; Binolfi, A.; Sepulveda-Diaz, J.E.; Del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of alpha-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, 41755. [Google Scholar] [CrossRef] [Green Version]
- Keijmel, S.P.; Delsing, C.E.; Bleijenberg, G.; van der Meer, J.W.M.; Donders, R.T.; Leclercq, M.; Kampschreur, L.M.; van den Berg, M.; Sprong, T.; Nabuurs-Franssen, M.H.; et al. Effectiveness of long-term doxycycline treatment and cognitive-behavioral therapy on fatigue severity in patients with q fever fatigue syndrome (qure study): A randomized controlled trial. Clin. Infect. Dis. 2017, 64, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, H.; Parishani, M.; Akbartabar Touri, M.; Ghavamzadeh, M.; Jafari Barmak, M.; Zarezade, V.; Delaviz, H.; Sadeghi, H. Pramipexole reduces inflammation in the experimental animal models of inflammation. Immunopharmacol. Immunotoxicol. 2017, 39, 80–86. [Google Scholar] [CrossRef]
- Shao, W.; Zhang, S.Z.; Tang, M.; Zhang, X.H.; Zhou, Z.; Yin, Y.Q.; Zhou, Q.B.; Huang, Y.Y.; Liu, Y.J.; Wawrousek, E.; et al. Suppression of neuroinflammation by astrocytic dopamine d2 receptors via alphab-crystallin. Nature 2013, 494, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Du, R.H.; Zhou, Y.; Xia, M.L.; Lu, M.; Ding, J.H.; Hu, G. Alpha-synuclein disrupts the anti-inflammatory role of drd2 via interfering beta-arrestin2-tab1 interaction in astrocytes. J. Neuroinflamm. 2018, 15, 258. [Google Scholar] [CrossRef] [Green Version]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-tumor necrosis factor therapy and incidence of parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of a1 astrocyte conversion by microglia is neuroprotective in models of parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Poyhonen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of neurotrophic factors in glial cells in the central nervous system: Expression and properties in neurodegeneration and injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Lindholm, P.; Saarma, M. Novel cdnf/manf family of neurotrophic factors. Dev. Neurobiol. 2010, 70, 360–371. [Google Scholar] [CrossRef]
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended treatment with glial cell line-derived neurotrophic factor in parkinson’s disease. J. Parkinsons Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudt, M.D.; Di Sebastiano, A.R.; Xu, H.; Jog, M.; Schmid, S.; Foster, P.; Hebb, M.O. Advances in neurotrophic factor and cell-based therapies for parkinson’s disease: A mini-review. Gerontology 2016, 62, 371–380. [Google Scholar] [CrossRef]
- Hegarty, S.V.; Lee, D.J.; O’Keeffe, G.W.; Sullivan, A.M. Effects of intracerebral neurotrophic factor application on motor symptoms in parkinson’s disease: A systematic review and meta-analysis. Parkinsonism Relat. Disord. 2017, 38, 19–25. [Google Scholar] [CrossRef]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, J.K.; Gardaneh, M.; Iwasiow, R.; Lanthier, P.; Gangaraju, S.; Ribecco-Lutkiewicz, M.; Tremblay, R.; Kiuchi, K.; Sikorska, M. Astrocyte-secreted gdnf and glutathione antioxidant system protect neurons against 6ohda cytotoxicity. Neurobiol. Dis. 2009, 33, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Q.; Zhang, B.; He, X.M.; Li, D.D.; Shi, J.S.; Zhang, F. Naringenin targets on astroglial nrf2 to support dopaminergic neurons. Pharmacol. Res. 2019, 139, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.T.; Liu, B.B.; Li, J.; Luo, L.; Liu, Q.; Geng, D.; Tang, Y.; Xia, Y.; Wu, D. Bdnf signaling is necessary for the antidepressant-like effect of naringenin. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liang, Y.; Wang, D.; Yan, Z.; Yin, H.; Wu, D.; Su, Q. Naringenin induces laxative effects by upregulating the expression levels of c-kit and scf, as well as those of aquaporin 3 in mice with loperamide-induced constipation. Int. J. Mol. Med. 2018, 41, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Gaba, B.; Khan, T.; Haider, M.F.; Alam, T.; Baboota, S.; Parvez, S.; Ali, J. Vitamin e loaded naringenin nanoemulsion via intranasal delivery for the management of oxidative stress in a 6-ohda parkinson’s disease model. Biomed. Res. Int. 2019, 2019, 2382563. [Google Scholar] [CrossRef] [PubMed]
- Sugumar, M.; Sevanan, M.; Sekar, S. Neuroprotective effect of naringenin against mptp-induced oxidative stress. Int. J. Neurosci. 2019, 129, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Jana, A.; Roy, A.; Pahan, K. Cinnamon and its metabolite protect the nigrostriatum in a mouse model of parkinson’s disease via astrocytic gdnf. J. Neuroimmune Pharmacol. 2019, 14, 503–518. [Google Scholar] [CrossRef]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural circuit-specialized astrocytes: Transcriptomic, proteomic, morphological, and functional evidence. Neuron 2017, 95, 531–549.e539. [Google Scholar] [CrossRef]
- Anderson, M.A.; Ao, Y.; Sofroniew, M.V. Heterogeneity of reactive astrocytes. Neurosci. Lett. 2014, 565, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Kohler, S.; Winkler, U.; Hirrlinger, J. Heterogeneity of astrocytes in grey and white matter. Neurochem. Res. 2021, 46, 3–14. [Google Scholar] [CrossRef]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charron, G.; Doudnikoff, E.; Canron, M.H.; Li, Q.; Vega, C.; Marais, S.; Baufreton, J.; Vital, A.; Oliet, S.H.; Bezard, E. Astrocytosis in parkinsonism: Considering tripartite striatal synapses in physiopathology? Front. Aging Neurosci. 2014, 6, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakh, B.S.; Deneen, B. The emerging nature of astrocyte diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Amiry-Moghaddam, M.; Bergersen, L.H.; Bjaalie, J.G.; Eriksson, J.; Gundersen, V.; Leergaard, T.B.; Morth, J.P.; Storm-Mathisen, J.; Torp, R.; et al. The glia doctrine: Addressing the role of glial cells in healthy brain ageing. Mech. Ageing Dev. 2013, 134, 449–459. [Google Scholar] [CrossRef]
- Chinta, S.J.; Lieu, C.A.; Demaria, M.; Laberge, R.M.; Campisi, J.; Andersen, J.K. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to parkinson’s disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Cadenas, E. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell 2014, 13, 1059–1067. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte heterogeneity: Impact to brain aging and disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Barres, B.A. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 2010, 20, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Bajo-Graneras, R.; Moratalla, R.; Perea, G.; Araque, A. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 2015, 349, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakh, B.S. Astrocyte-neuron interactions in the striatum: Insights on identity, form, and function. Trends Neurosci. 2019, 42, 617–630. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzeriat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to cns damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirko, S.; Irmler, M.; Gascon, S.; Bek, S.; Schneider, S.; Dimou, L.; Obermann, J.; De Souza Paiva, D.; Poirier, F.; Beckers, J.; et al. Astrocyte reactivity after brain injury-: The role of galectins 1 and 3. Glia 2015, 63, 2340–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in huntington’s disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- de Majo, M.; Koontz, M.; Rowitch, D.; Ullian, E.M. An update on human astrocytes and their role in development and disease. Glia 2020, 68, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain. Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.N.; Zou, L.; Goldman, S.A. Human glial chimeric mice to define the role of glial pathology in human disease. Methods Mol. Biol. 2019, 1936, 311–331. [Google Scholar]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hamalainen, R.H.; Koistinaho, J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef] [Green Version]
- Kostuk, E.W.; Cai, J.; Iacovitti, L. Subregional differences in astrocytes underlie selective neurodegeneration or protection in parkinson’s disease models in culture. Glia 2019, 67, 1542–1557. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.W.; Cao, S.N.; Zang, C.X.; Wang, L.; Yang, H.Y.; Bao, X.Q.; Zhang, D. Heat shock protein 70 suppresses neuroinflammation induced by alpha-synuclein in astrocytes. Mol. Cell Neurosci. 2018, 86, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, X.; Liu, T.; Liu, H.; Tong, L.; Jia, S.; Wang, Y.F. Neurochemical regulation of the expression and function of glial fibrillary acidic protein in astrocytes. Glia 2020, 68, 878–897. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for hla-dr in the substantia nigra of parkinson’s and alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S.; DeLanney, L.E.; Irwin, I.; Di Monte, D.; Langston, J.W. Astrocytes and parkinson’s disease. Prog. Brain Res. 1992, 94, 429–436. [Google Scholar]
- Saal, K.A.; Galter, D.; Roeber, S.; Bahr, M.; Tonges, L.; Lingor, P. Altered expression of growth associated protein-43 and rho kinase in human patients with parkinson’s disease. Brain Pathol. 2017, 27, 13–25. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Standaert, D.G. Ten unsolved questions about neuroinflammation in parkinson’s disease. Mov. Disord. 2020, 36, 16–24. [Google Scholar] [CrossRef]
- Neal, M.; Richardson, J.R. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 432–443. [Google Scholar] [CrossRef]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein gpnmb is selectively elevated in the substantia nigra of parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef]
- Navarrete, F.; Garcia-Gutierrez, M.S.; Aracil-Fernandez, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid cb1 and cb2 receptors, and monoacylglycerol lipase gene expression alterations in the basal ganglia of patients with parkinson’s disease. Neurotherapeutics 2018, 15, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.J.; Yeh, C.Y.; Terzieva, S.; Olabarria, M.; Kulijewicz-Nawrot, M.; Verkhratsky, A. Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 2014, 35, 15–23. [Google Scholar] [CrossRef]
- Cerbai, F.; Lana, D.; Nosi, D.; Petkova-Kirova, P.; Zecchi, S.; Brothers, H.M.; Wenk, G.L.; Giovannini, M.G. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE 2012, 7, e45250. [Google Scholar]
- O’Callaghan, J.P.; Miller, D.B. The concentration of glial fibrillary acidic protein increases with age in the mouse and rat brain. Neurobiol. Aging 1991, 12, 171–174. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Rocha, E.M.; Bai, Q.; El Ayadi, A.; Hinkle, D.; Burton, E.A.; Timothy Greenamyre, J. Astrocyte-specific dj-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of parkinson’s disease. Neurobiol. Dis. 2018, 115, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.J.; Eun, J.H.; Kim, B.G.; Jou, I.; Park, S.M.; Joe, E.H. A parkinson’s disease gene, dj-1, repairs brain injury through sox9 stabilization and astrogliosis. Glia 2018, 66, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, S.M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E.H. Pink1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced akt and increased p38 mapk activation, and downregulation of egfr. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef]
- Barodia, S.K.; McMeekin, L.J.; Creed, R.B.; Quinones, E.K.; Cowell, R.M.; Goldberg, M.S. Pink1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Parkinsons Dis. 2019, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.; Tang, A.M.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. Apoe2: Protective mechanism and therapeutic implications for alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Bossers, K.; Meerhoff, G.; Balesar, R.; van Dongen, J.W.; Kruse, C.G.; Swaab, D.F.; Verhaagen, J. Analysis of gene expression in parkinson’s disease: Possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol. 2009, 19, 91–107. [Google Scholar] [CrossRef]
- Xing, L.; Wang, D.; Wang, L.; Lan, W.; Pan, S. Differential proteomics analysis of mononuclear cells in cerebrospinal fluid of parkinson’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 15462–15466. [Google Scholar] [PubMed]
- Sanchez Campos, S.; Alza, N.P.; Salvador, G.A. Lipid metabolism alterations in the neuronal response to a53t alpha-synuclein and fe-induced injury. Arch. Biochem. Biophys. 2018, 655, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Haschka, D.; Volani, C.; Stefani, A.; Tymoszuk, P.; Mitterling, T.; Holzknecht, E.; Heidbreder, A.; Coassin, S.; Sumbalova, Z.; Seifert, M.; et al. Association of mitochondrial iron deficiency and dysfunction with idiopathic restless legs syndrome. Mov. Disord. 2019, 34, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.H.; Choi, D.J.; An, J.; Eun, J.H.; Jou, I.; Park, S. Astrocytes, microglia, and parkinson’s disease. Exp. Neurobiol. 2018, 27, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The role of astrocyte dysfunction in parkinson’s disease pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Linnartz, B.; Mendritzki, S.; Sczepan, T.; Lubbert, M.; Stichel, C.C.; Lubbert, H. Genetic mouse models for parkinson’s disease display severe pathology in glial cell mitochondria. Hum. Mol. Genet. 2011, 20, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Cha, S.H.; Choi, Y.R.; Jou, I.; Joe, E.H.; Park, S.M. Dj-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. Dj-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef] [Green Version]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Gorner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the parkinson’s disease-associated gene dj-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef] [Green Version]
- Mullett, S.J.; Hinkle, D.A. Dj-1 deficiency in astrocytes selectively enhances mitochondrial complex i inhibitor-induced neurotoxicity. J. Neurochem. 2011, 117, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullett, S.J.; Di Maio, R.; Greenamyre, J.T.; Hinkle, D.A. Dj-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 2013, 49, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano, R.M.; Casarejos, M.J.; Menendez-Cuervo, J.; Rodriguez-Navarro, J.A.; Garcia de Yebenes, J.; Mena, M.A. Glial dysfunction in parkin null mice: Effects of aging. J. Neurosci. 2008, 28, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Solano, R.M.; Menendez, J.; Casarejos, M.J.; Rodriguez-Navarro, J.A.; Garcia de Yebenes, J.; Mena, M.A. Midbrain neuronal cultures from parkin mutant mice are resistant to nitric oxide-induced toxicity. Neuropharmacology 2006, 51, 327–340. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Galvan, C.; Hellias, B.; Dotti, C.; Jensen, P.H. Astrocytic but not neuronal increased expression and redistribution of parkin during unfolded protein stress. J. Neurochem. 2002, 83, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Gai, W.P.; Xu, Y.H.; Sachdev, P.; Guillemin, G.J.; Jiang, X.M.; Ballard, J.W.; Horan, M.P.; Fang, Z.M.; Chong, B.H.; et al. Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl. Neurodegener. 2013, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of parkinson’s disease-related a53t alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.; Choi, D.J.; Yang, H.; Woo, J.H.; Chang, M.Y.; Kim, J.Y.; Sun, W.; Park, S.M.; Jou, I.; Lee, S.H.; et al. Pink1 expression increases during brain development and stem cell differentiation, and affects the development of gfap-positive astrocytes. Mol. Brain 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitner, E.B.; Dekel, H.; Zigdon, H.; Shachar, T.; Farfel-Becker, T.; Eilam, R.; Karlsson, S.; Futerman, A.H. Altered expression and distribution of cathepsins in neuronopathic forms of gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 2010, 19, 3583–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link gaucher and parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluss, J.H.; Mazza, M.C.; Li, Y.; Manzoni, C.; Lewis, P.A.; Cookson, M.R.; Mamais, A. Preclinical modeling of chronic inhibition of the parkinson’s disease associated kinase lrrk2 reveals altered function of the endolysosomal system in vivo. Mol. Neurodegener. 2021, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Mamais, A.; Dihanich, S.; Abeti, R.; Soutar, M.P.M.; Plun-Favreau, H.; Giunti, P.; Tooze, S.A.; Bandopadhyay, R.; Lewis, P.A. Inhibition of lrrk2 kinase activity stimulates macroautophagy. Biochim. Biophys. Acta 2013, 1833, 2900–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic lrrk2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase atp13a2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [Green Version]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via nlrp3 inflammasome activation. CNS Neurosci. Ther. 2016, 22, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Strokin, M.; Seburn, K.L.; Cox, G.A.; Martens, K.A.; Reiser, G. Severe disturbance in the ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated pla2g6. Hum. Mol. Genet. 2012, 21, 2807–2814. [Google Scholar] [CrossRef]
- Strokin, M.; Sergeeva, M.; Reiser, G. Proinflammatory treatment of astrocytes with lipopolysaccharide results in augmented ca2+ signaling through increased expression of via phospholipase a2 (ipla2). Am. J. Physiol. Cell Physiol. 2011, 300, C542–C549. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. Alpha-synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef]
- Houser, M.C.; Tansey, M.G. The gut-brain axis: Is intestinal inflammation a silent driver of parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3. [Google Scholar] [CrossRef]
- Xie, X.; Luo, X.; Liu, N.; Li, X.; Lou, F.; Zheng, Y.; Ren, Y. Monocytes, microglia, and cd200-cd200r1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J. Neurochem. 2017, 141, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Efremova, L.; Chovancova, P.; Adam, M.; Gutbier, S.; Schildknecht, S.; Leist, M. Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Arch. Toxicol. 2017, 91, 231–246. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on parkinson’s disease. Front. Cell Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in parkinson’s disease: The role of alpha-synuclein-specific t cells. Front. Immunol. 2019, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chen, S.; Liu, J. The role of t cells in the pathogenesis of parkinson’s disease. Prog. Neurobiol. 2018, 169, 1–23. [Google Scholar] [CrossRef]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Cheslow, L.; Alvarez, J.I. Glial-endothelial crosstalk regulates blood-brain barrier function. Curr. Opin. Pharmacol. 2016, 26, 39–46. [Google Scholar] [CrossRef]
- Chapouly, C.; Tadesse Argaw, A.; Horng, S.; Castro, K.; Zhang, J.; Asp, L.; Loo, H.; Laitman, B.M.; Mariani, J.N.; Straus Farber, R.; et al. Astrocytic tymp and vegfa drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain 2015, 138, 1548–1567. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Olsen, A.L.; Feany, M.B. Glial alpha-synuclein promotes neurodegeneration characterized by a distinct transcriptional program in vivo. Glia 2019, 67, 1933–1957. [Google Scholar] [CrossRef]
- Bruck, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Kery, R.; Chen, A.P.F.; Kirschen, G.W. Genetic targeting of astrocytes to combat neurodegenerative disease. Neural Regen. Res. 2020, 15, 199–211. [Google Scholar]
- Han, X.; Chen, M.; Wang, F.; Windrem, M.; Wang, S.; Shanz, S.; Xu, Q.; Oberheim, N.A.; Bekar, L.; Betstadt, S.; et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem. Cell 2013, 12, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Isobe, K.; Cheng, Z.; Nishio, N.; Suganya, T.; Tanaka, Y.; Ito, S. Ipscs, aging and age-related diseases. N. Biotechnol. 2014, 31, 411–421. [Google Scholar] [CrossRef]
- Tomov, N. Glial cells in intracerebral transplantation for parkinson’s disease. Neural Regen. Res. 2020, 15, 1173–1178. [Google Scholar] [CrossRef]
- Wei, Z.D.; Shetty, A.K. Treating parkinson’s disease by astrocyte reprogramming: Progress and challenges. Sci. Adv. 2021, 7, eabg3198. [Google Scholar] [CrossRef]
- Latchman, D.S. Transcription factors: An overview. Int. J. Biochem. Cell Biol. 1997, 29, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Brennand, K.; Panopoulos, A.D.; Herrerias, A.; Gage, F.H.; Izpisua-Belmonte, J.C. High-efficient generation of induced pluripotent stem cells from human astrocytes. PLoS ONE 2010, 5, e15526. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Zang, T.; Zou, Y.; Fang, S.; Smith, D.K.; Bachoo, R.; Zhang, C.L. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 2013, 15, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Miao, Q.; Yuan, J.; Han, S.; Zhang, P.; Li, S.; Rao, Z.; Zhao, W.; Ye, Q.; Geng, J.; et al. Ascl1 converts dorsal midbrain astrocytes into functional neurons in vivo. J. Neurosci. 2015, 35, 9336–9355. [Google Scholar] [CrossRef] [Green Version]
- Zarei-Kheirabadi, M.; Hesaraki, M.; Shojaei, A.; Kiani, S.; Baharvand, H. Generation of neural stem cells from adult astrocytes by using a single reprogramming factor. J. Cell Physiol. 2019, 234, 18697–18706. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microrna biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Deo, M.; Yu, J.Y.; Chung, K.H.; Tippens, M.; Turner, D.L. Detection of mammalian microrna expression by in situ hybridization with rna oligonucleotides. Dev. Dyn. 2006, 235, 2538–2548. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Le Bras, A. Aso-mediated conversion of glial cells into neurons. Lab. Anim. 2021, 50, 169. [Google Scholar] [CrossRef]
- Hu, X.; Qin, S.; Huang, X.; Yuan, Y.; Tan, Z.; Gu, Y.; Cheng, X.; Wang, D.; Lian, X.F.; He, C.; et al. Region-restrict astrocytes exhibit heterogeneous susceptibility to neuronal reprogramming. Stem. Cell Rep. 2019, 12, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Maimon, R.; Chillon-Marinas, C.; Snethlage, C.E.; Singhal, S.M.; McAlonis-Downes, M.; Ling, K.; Rigo, F.; Bennett, C.F.; Da Cruz, S.; Hnasko, T.S.; et al. Therapeutically viable generation of neurons with antisense oligonucleotide suppression of ptb. Nat. Neurosci. 2021, 24, 1089–1099. [Google Scholar] [CrossRef]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C.; Munoz-Lopez, M.; Real, P.J.; Macia, A.; Sanchez, L.; Ligero, G.; Garcia-Parez, J.L.; Menendez, P. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem. Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.; Chan, Y.K.; Samulski, R.J. Adeno-associated virus (aav) versus immune response. Viruses 2019, 11, 102. [Google Scholar]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial complex i deficiency in parkinson’s disease. Adv. Neurol. 1993, 60, 288–291. [Google Scholar]
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial dysfunction in astrocytes: A role in parkinson’s disease? Front. Cell Dev. Biol. 2020, 8, 608026. [Google Scholar] [CrossRef]
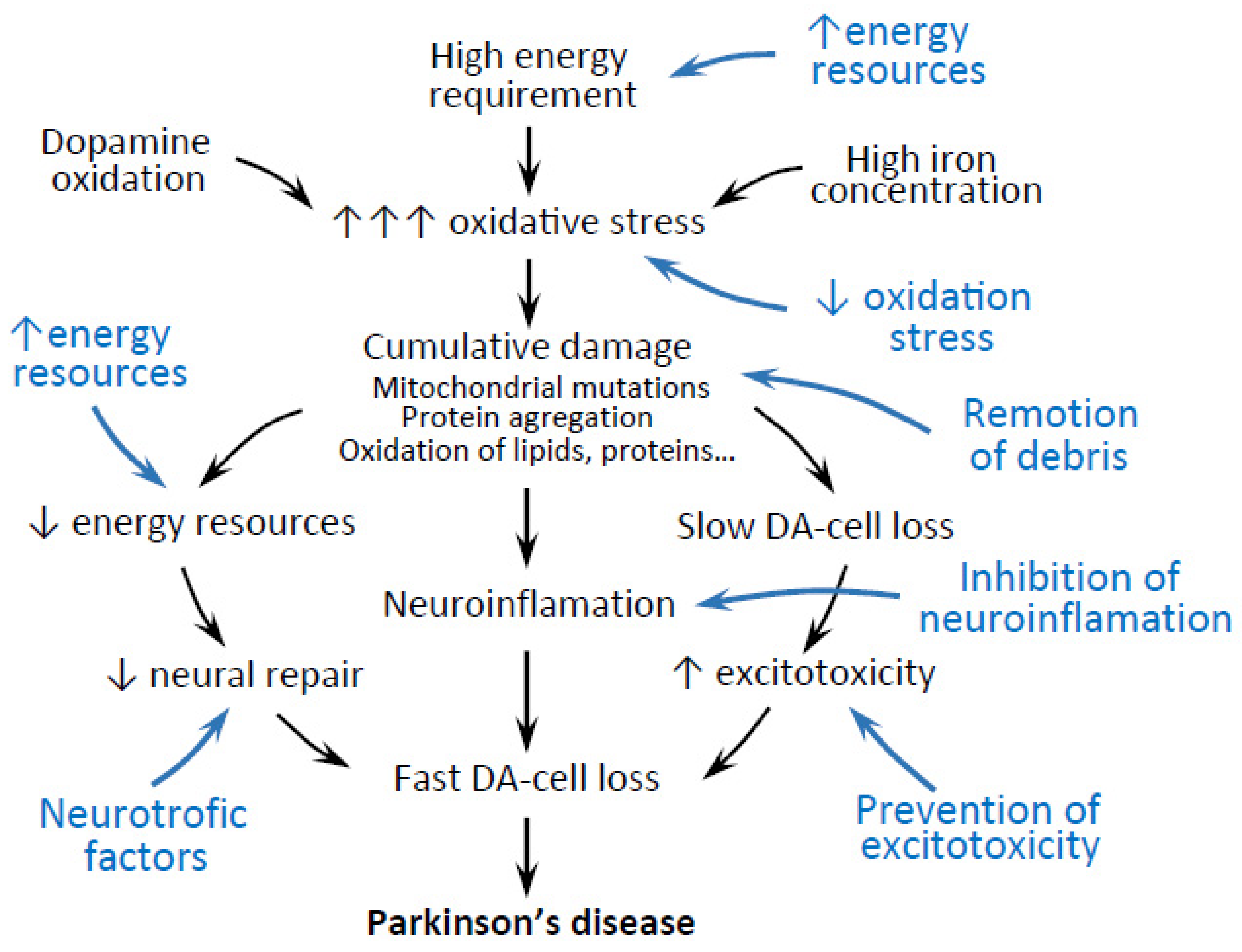
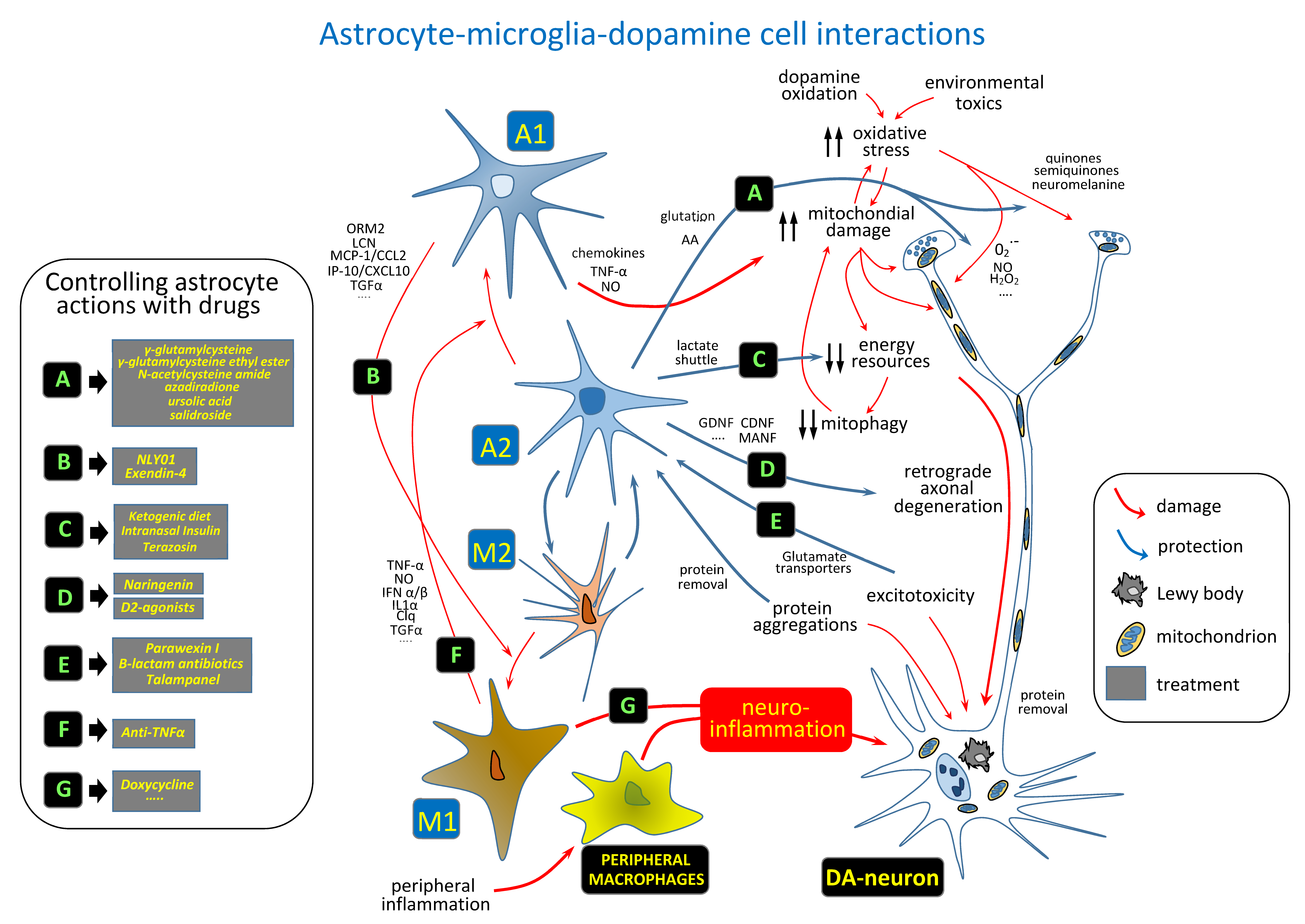

{kind=link}
{kind=link}
| DA-Cell Requirements | Astrocyte Support | |
|---|---|---|
| high energy requirements | energy resource | [21,22,23,24,25] |
| high oxidative stress | antioxidant activity | [26,27,28,29,30,31,32,33,34,35,36,37] |
| cumulative damage | transautophagy | [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57] |
| α-synuclein accumulation | α-synuclein remotion | [38] |
| mitochondrial damage | mitochondrial transfer | [58,59,60] |
| impaired mitophagy | transmitophagy | [61] |
| glutamatergic excitotoxicity | glutamate uptake | [44,45,46,47,48,49,50,51,52,53] |
| neuroinflammation | anti-inflammatory activity | [62,63,64,65,66,67,68,69,70,71,72,73,74] |
| need for trophic support | release of neurotrophic factors | [75,76,77,78,79,80,81,82,83,84,85,86,87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, A.; Morales, I.; Rodriguez-Sabate, C.; Sole-Sabater, M.; Rodriguez, M. Astrocytes, a Promising Opportunity to Control the Progress of Parkinson’s Disease. Biomedicines 2021, 9, 1341. https://doi.org/10.3390/biomedicines9101341
Sanchez A, Morales I, Rodriguez-Sabate C, Sole-Sabater M, Rodriguez M. Astrocytes, a Promising Opportunity to Control the Progress of Parkinson’s Disease. Biomedicines. 2021; 9(10):1341. https://doi.org/10.3390/biomedicines9101341
Chicago/Turabian StyleSanchez, Alberto, Ingrid Morales, Clara Rodriguez-Sabate, Miguel Sole-Sabater, and Manuel Rodriguez. 2021. "Astrocytes, a Promising Opportunity to Control the Progress of Parkinson’s Disease" Biomedicines 9, no. 10: 1341. https://doi.org/10.3390/biomedicines9101341
APA StyleSanchez, A., Morales, I., Rodriguez-Sabate, C., Sole-Sabater, M., & Rodriguez, M. (2021). Astrocytes, a Promising Opportunity to Control the Progress of Parkinson’s Disease. Biomedicines, 9(10), 1341. https://doi.org/10.3390/biomedicines9101341