
Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles

 ,
, 
Abstract
:1. Introduction
1.1. Single-Gene Markers, the Predominant Approach to Precision Oncology, Are Scarce
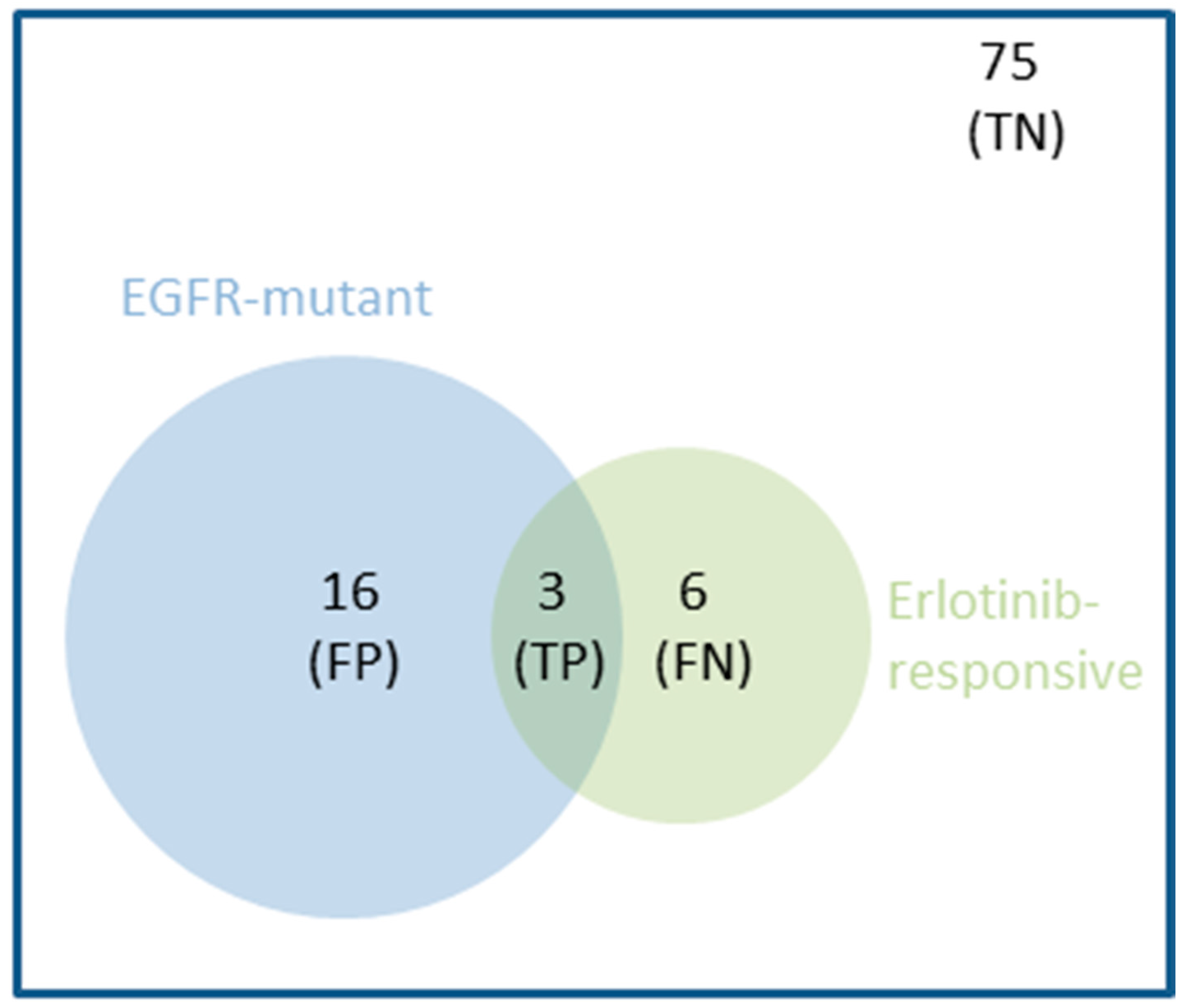
1.2. Even FDA-Approved Single-Gene Markers Can Be Weakly Predictive: An Example
1.3. Multi-Gene Models Built with Machine Learning Complement Single-Gene Markers
1.4. Learning from Preclinical Data Represents an Alternative When Clinical Data Is Unavailable
1.5. A Comparison of Multi-Gene vs. Single-Gene Models across In Vivo Preclinical Models Is Lacking
2. Materials and Methods
2.1. NIBR-PDXE Data
2.2. Processing Treatment Response Data for Modelling
2.3. Processing Molecular Profiling Data for Modelling
2.4. Processed Data Sets for Modelling
2.5. Measuring the Predictive Performance of a Classifier
2.6. Multi-Gene Classifiers with Built-In Feature Selection (FS)
2.7. Multi-Gene Classifiers with Optimal Model Complexity (OMC)
2.8. Random Model Based on the Prior Probability of Each Case
2.9. Single-Gene Markers
3. Results
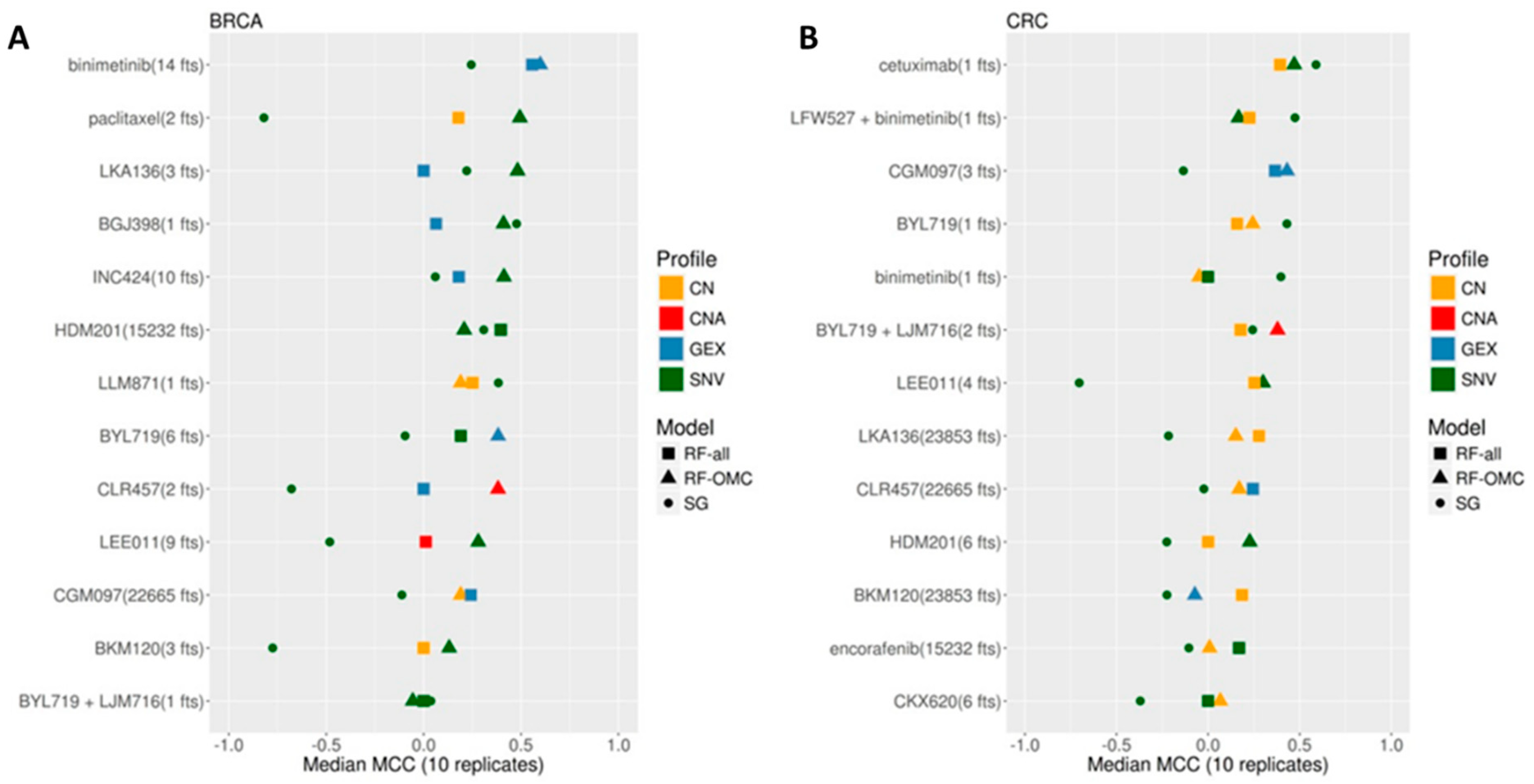
3.1. Establishing the Best Multi-Gene Predictor for Each Treatment and Cancer Type Pair
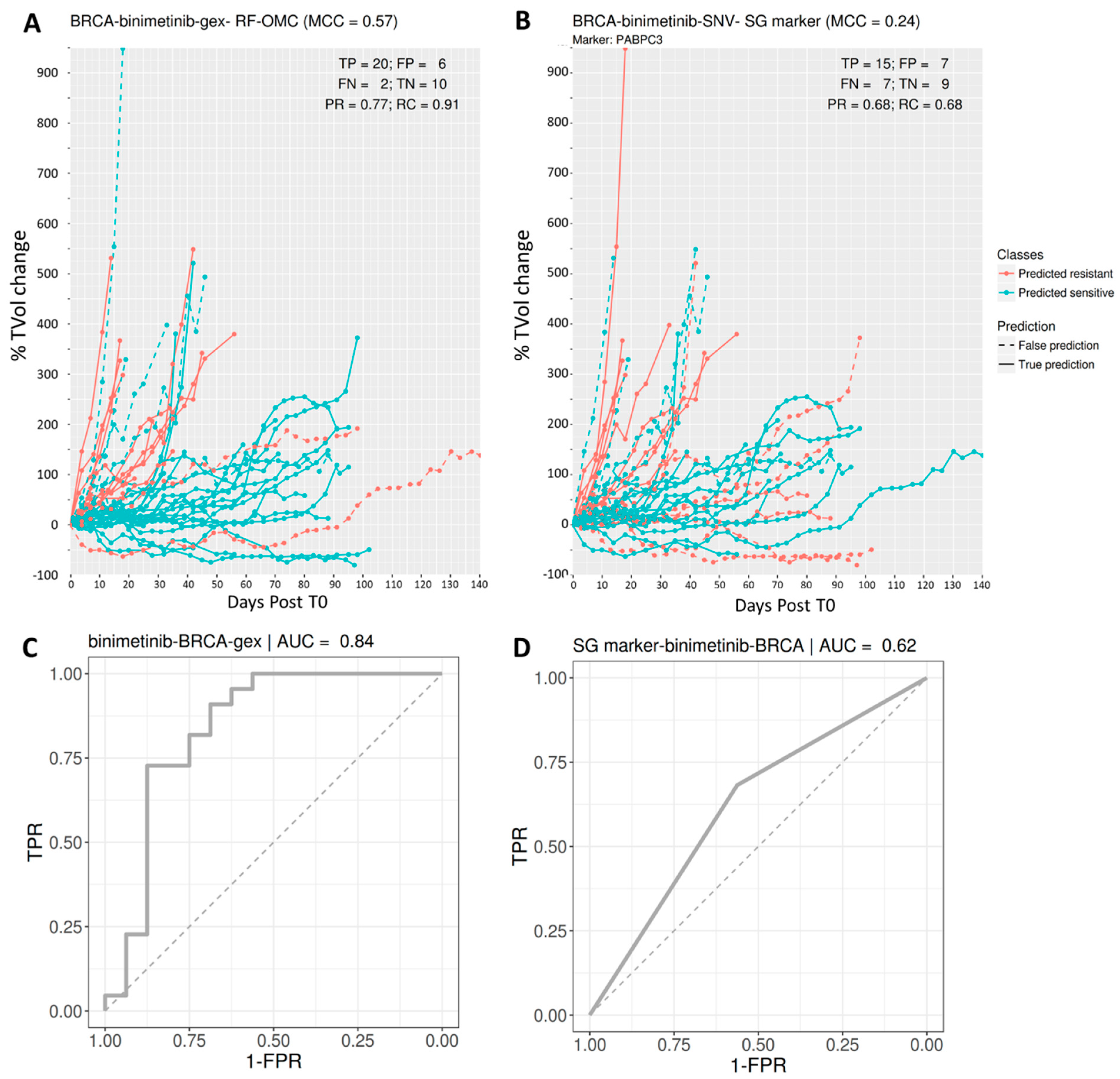
3.2. Predicting BRCA PDX Response to Binimetinib
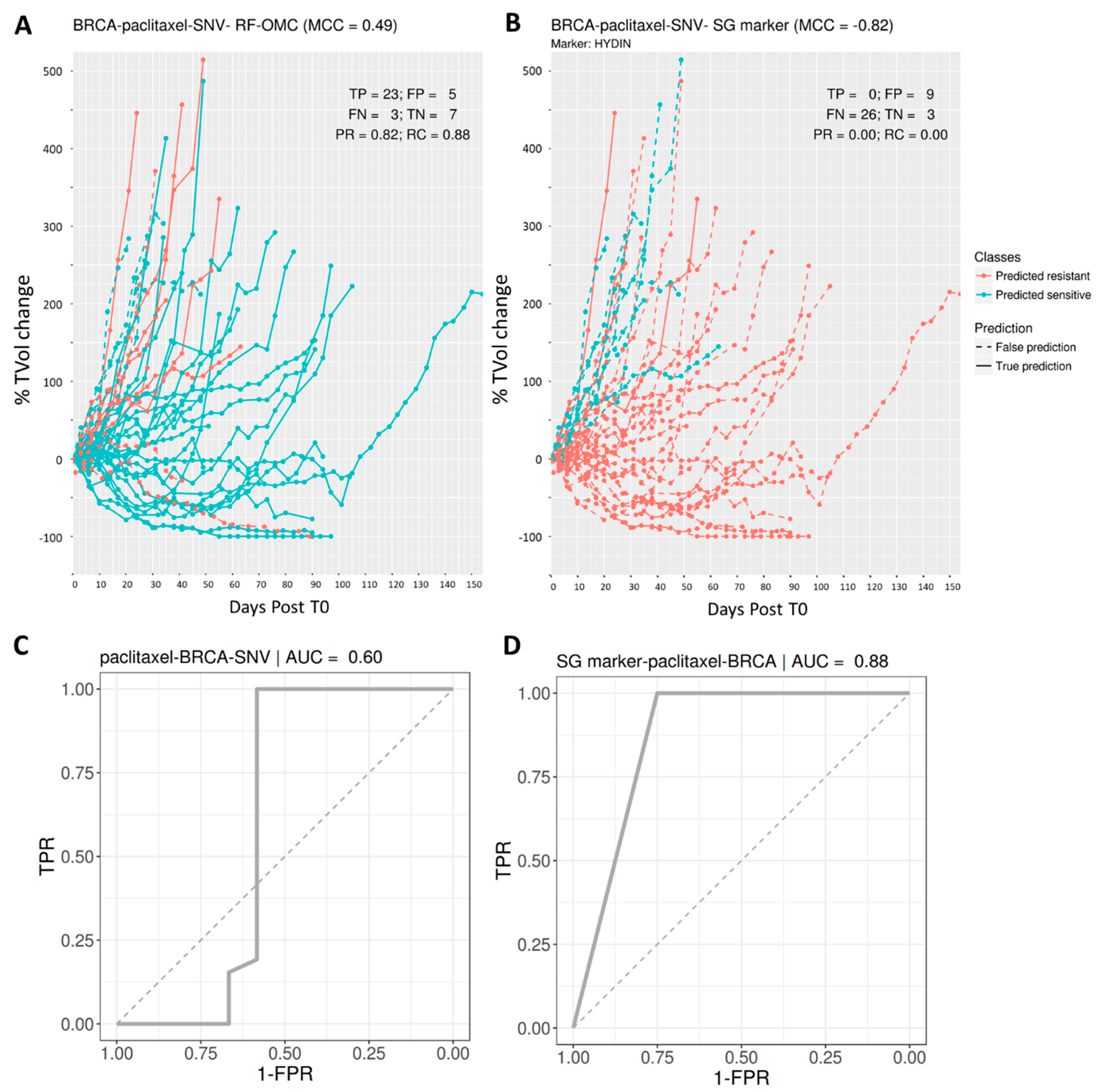
3.3. Predicting BRCA PDX Response to Paclitaxel
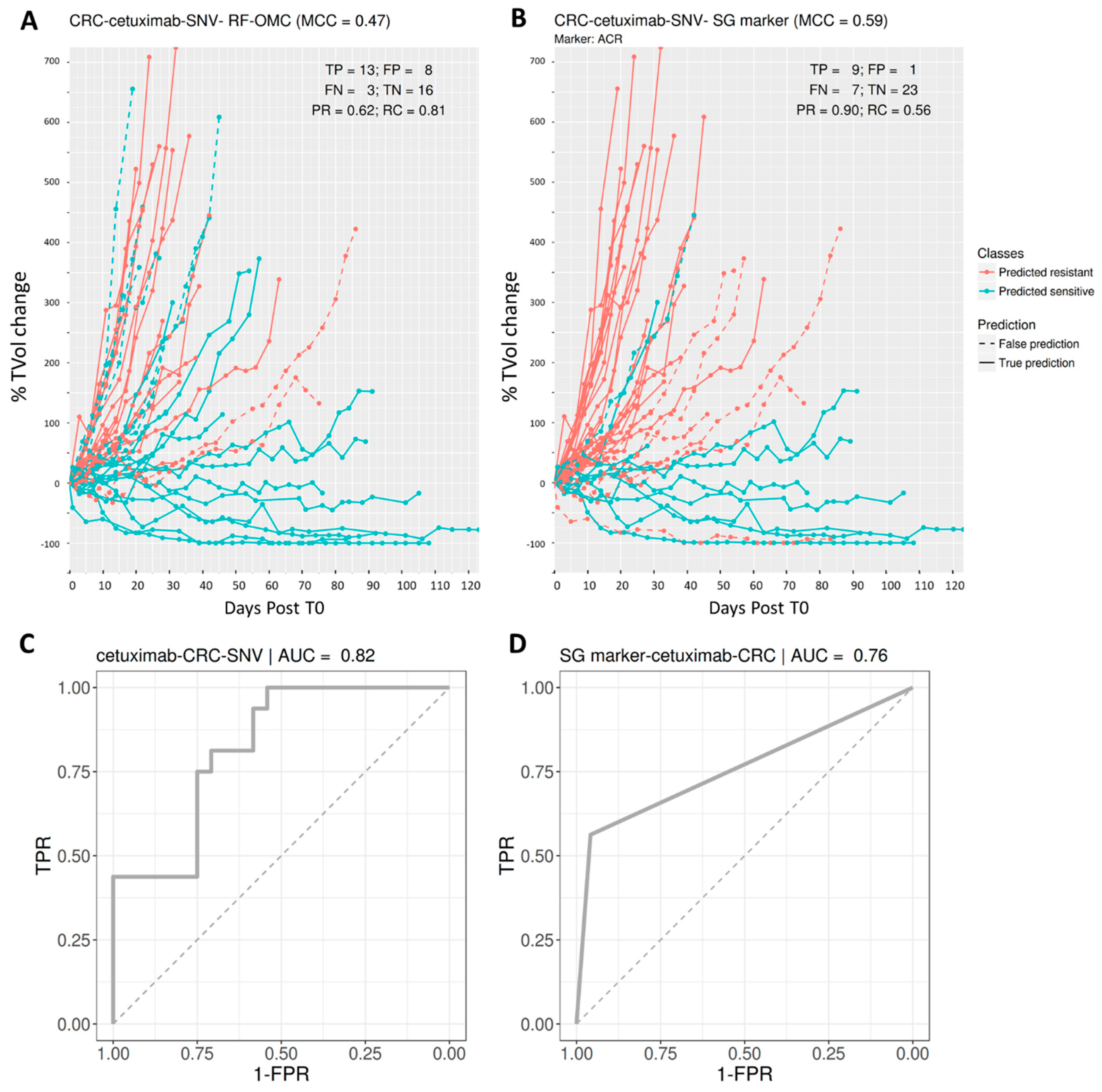
3.4. Predicting CRC PDX Response to Cetuximab
3.5. Multi-Gene Predictors Generally Offer Substantially Higher Recall Than Single-Gene Markers
3.6. Merging All the Profiles in a Single Set of Features Can Result in a Much Worse Prediction
3.7. OMC-Selected Features Are Much More Predictive Than Randomly Selected Features
3.8. RF-OMC Can Be Implemented with Alternative Ways of Ranking Features
4. Discussion
4.1. Advantages of Taking a Machine Learning Approach to This Problem
4.2. Limitations and Recommendations to Mitigate Them
4.3. Future Application to Clinical Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spear, B.B.; Heath-Chiozzi, M.; Huff, J. Clinical application of pharmacogenetics. Trends Mol. Med. 2001, 7, 201–204. [Google Scholar] [CrossRef]
- Rodríguez-Antona, C.; Taron, M. Pharmacogenomic biomarkers for personalized cancer treatment. J. Intern. Med. 2015, 277, 201–217. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.-J.; et al. Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients With Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Tsao, M.-S.; Sakurada, A.; Cutz, J.-C.; Zhu, C.-Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in Lung Cancer—Molecular and Clinical Predictors of Outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef]
- Ulivi, P.; Delmonte, A.; Chiadini, E.; Calistri, D.; Papi, M.; Mariotti, M.; Verlicchi, A.; Ragazzini, A.; Capelli, L.; Gamboni, A.; et al. Gene mutation analysis in EGFR wild type NSCLC responsive to erlotinib: Are there features to guide patient selection? Int. J. Mol. Sci. 2014, 16, 747–757. [Google Scholar] [CrossRef]
- Eckhardt, S.G.; Lieu, C. Is Precision Medicine an Oxymoron? JAMA Oncol. 2019, 5, 142–143. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Loboda, A.; Lenkala, D.; Wang, F.; LaCroix, B.; Karovic, S.; Wang, J.; Nebozhyn, M.; Chisamore, M.; Hardwick, J.; et al. Predicting Response to Histone Deacetylase Inhibitors Using High-Throughput Genomics. J. Natl. Cancer Inst. 2015, 107, djv247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naulaerts, S.; Dang, C.C.; Ballester, P.J. Precision and recall oncology: Combining multiple gene mutations for improved identification of drug-sensitive tumours. Oncotarget 2017, 8, 97025–97040. [Google Scholar] [CrossRef] [Green Version]
- Ballester, P.J.; Carmona, J. Artificial intelligence for the next generation of precision oncology. NPJ Precis. Oncol. 2021, 5, 79. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium, T.A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Covell, D.G. Data Mining Approaches for Genomic Biomarker Development: Applications Using Drug Screening Data from the Cancer Genome Project and the Cancer Cell Line Encyclopedia. PLoS ONE 2015, 10, e0127433. [Google Scholar] [CrossRef] [Green Version]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Vincent, K.M.; Findlay, S.D.; Postovit, L.M. Assessing breast cancer cell lines as tumour models by comparison of mRNA expression profiles. Breast Cancer Res. 2015, 17, 114. [Google Scholar] [CrossRef] [Green Version]
- Piyawajanusorn, C.; Nguyen, L.C.; Ghislat, G.; Ballester, P.J. A gentle introduction to understanding preclinical data for cancer pharmaco-omic modeling. Brief. Bioinform. 2021, bbab312. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Krepler, C.; Sproesser, K.; Brafford, P.; Beqiri, M.; Garman, B.; Xiao, M.; Shannan, B.; Watters, A.; Perego, M.; Zhang, G.; et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep. 2017, 21, 1953–1967. [Google Scholar] [CrossRef]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef]
- Li, J.; Ye, C.; Mansmann, U.R. Comparing Patient-Derived Xenograft and Computational Response Prediction for Targeted Therapy in Patients of Early-Stage Large Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274.e22. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Lemos, R.; Powis, G. The promise of patient-derived xenografts: The best laid plans of mice and men. Clin. Cancer Res. 2012, 18, 5160–5162. [Google Scholar] [CrossRef] [Green Version]
- Struss, W.J.; Black, P.C. Using PDX for Biomarker Development. In Patient-Derived Xenograft Models of Human Cancer; Springer: Cham, Switzerland, 2017; pp. 127–140. [Google Scholar]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in Bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-C.; Chen, H.-I.H.; Gorthi, A.; Mostavi, M.; Zheng, S.; Huang, Y.; Chen, Y. Deep learning of pharmacogenomics resources: Moving towards precision oncology. Brief. Bioinform. 2020, 21, 2066–2083. [Google Scholar] [CrossRef]
- Deyati, A.; Younesi, E.; Hofmann-Apitius, M.; Novac, N. Challenges and opportunities for oncology biomarker discovery. Drug Discov. Today 2013, 18, 614–624. [Google Scholar] [CrossRef]
- Ding, M.Q.; Chen, L.; Cooper, G.F.; Young, J.D.; Lu, X. Precision Oncology beyond Targeted Therapy: Combining Omics Data with Machine Learning Matches the Majority of Cancer Cells to Effective Therapeutics. Mol. Cancer Res. 2018, 16, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Qin, Y.; Zhang, N.; Wang, J.; Wang, H.; Zheng, X. DISIS: Prediction of Drug Response through an Iterative Sure Independence Screening. PLoS ONE 2015, 10, e0120408. [Google Scholar] [CrossRef]
- Berlow, N.; Haider, S.; Wan, Q.; Geltzeiler, M.; Davis, L.E.; Keller, C.; Pal, R. An Integrated Approach to Anti-Cancer Drug Sensitivity Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 995–1008. [Google Scholar] [CrossRef]
- Ammad-ud-din, M.; Khan, S.A.; Wennerberg, K.; Aittokallio, T. Systematic identification of feature combinations for predicting drug response with Bayesian multi-view multi-task linear regression. Bioinformatics 2017, 33, i359–i368. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, W.; Chen, Y.; Ma, Q.; Wei, J.; Liu, Q. Identifying anti-cancer drug response related genes using an integrative analysis of transcriptomic and genomic variations with cell line-based drug perturbations. Oncotarget 2016, 7, 9404–9419. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Ciriano, I.; van Westen, G.J.P.; Bouvier, G.; Nilges, M.; Overington, J.P.; Bender, A.; Malliavin, T.E. Improved large-scale prediction of growth inhibition patterns using the NCI60 cancer cell line panel. Bioinformatics 2016, 32, 85–95. [Google Scholar] [CrossRef]
- Nguyen, L.; Dang, C.C.; Ballester, P.J. Systematic assessment of multi-gene predictors of pan-cancer cell line sensitivity to drugs exploiting gene expression data. F1000Research 2016, 5, 2927. [Google Scholar] [CrossRef] [Green Version]
- Polano, M.; Chierici, M.; Bo, M.D.; Gentilini, D.; Di Cintio, F.; Baboci, L.; Gibbs, D.L.; Furlanello, C.; Toffoli, G. A pan-cancer approach to predict responsiveness to immune checkpoint inhibitors by machine learning. Cancers 2019, 11, 1562. [Google Scholar] [CrossRef] [Green Version]
- Naulaerts, S.; Menden, M.P.; Ballester, P.J. Concise polygenic models for cancer-specific identification of drug-sensitive tumors from their multi-omics profiles. Biomolecules 2020, 10, 963. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.C.; Peón, A.; Ballester, P.J. Unearthing new genomic markers of drug response by improved measurement of discriminative power. BMC Med. Genom. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gönen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-Ud-Din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In IJCAI’95, Proceedings of the 14th International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 1995; Volume 2, pp. 1137–1143. [Google Scholar] [CrossRef]
- Cawley, G.C.; Talbot, N.L.C. On Over-fitting in Model Selection and Subsequent Selection Bias in Performance Evaluation. J. Mach. Learn. Res. 2010, 11, 2079–2107. [Google Scholar]
- Varma, S.; Simon, R. Bias in error estimation when using cross-validation for model selection. BMC Bioinform. 2006, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Van Rijsbergen, C.J.; Van, C.J. Information Retrieval; Butterworths-Heinemann: Oxford, UK, 1979; ISBN 0408709294. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Probst, P.; Boulesteix, A.-L. To Tune or Not to Tune the Number of Trees in Random Forest. J. Mach. Learn. Res. 2018, 18, 6673–6690. [Google Scholar]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B.O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Probst, P.; Wright, M.N.; Boulesteix, A. Hyperparameters and tuning strategies for random forest. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2019, 9, e1301. [Google Scholar] [CrossRef] [Green Version]
- Chedzoy, O.B. Phi-Coefficient. In Encyclopedia of Statistical Sciences; John Wiley and Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Zhang, L.; Chen, X.; Guan, N.-N.; Liu, H.; Li, J.-Q. A Hybrid Interpolation Weighted Collaborative Filtering Method for Anti-cancer Drug Response Prediction. Front. Pharmacol. 2018, 9, 1017. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhao, Y.; Zhang, L.; Chen, X. Anti-cancer Drug Response Prediction Using Neighbor-Based Collaborative Filtering with Global Effect Removal. Mol. Ther. Nucleic Acids 2018, 13, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Azuaje, F. Computational models for predicting drug responses in cancer research. Brief. Bioinform. 2016, 18, 820–829. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Points of Significance: Model selection and overfitting. Nat. Methods 2016, 13, 703–704. [Google Scholar] [CrossRef]
- Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Beck, A.H.; Aerts, H.J.W.L.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2013, 504, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Hira, Z.M.; Gillies, D.F. A Review of Feature Selection and Feature Extraction Methods Applied on Microarray Data. Adv. Bioinform. 2015, 2015, 198363. [Google Scholar] [CrossRef]
- Felip, E.; Martinez, P. Can sensitivity to cytotoxic chemotherapy be predicted by biomarkers? Ann. Oncol. 2012, 23 (Suppl. 1), 189–192. [Google Scholar] [CrossRef]
- Kim, S.; Sundaresan, V.; Zhou, L.; Kahveci, T. Integrating Domain Specific Knowledge and Network Analysis to Predict Drug Sensitivity of Cancer Cell Lines. PLoS ONE 2016, 11, e0162173. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Gu, H.; Wang, Y.; Wang, J.; Qin, P. Autoencoder Based Feature Selection Method for Classification of Anticancer Drug Response. Front. Genet. 2019, 10, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parca, L.; Pepe, G.; Pietrosanto, M.; Galvan, G.; Galli, L.; Palmeri, A.; Sciandrone, M.; Ferrè, F.; Ausiello, G.; Helmer-Citterich, M. Modeling cancer drug response through drug-specific informative genes. Sci. Rep. 2019, 9, 15222. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Belkacemi, L.; Cheung, M.S.; Bose, R.N. Correlation between Gene Variants, Signaling Pathways, and Efficacy of Chemotherapy Drugs against Colon Cancers. Cancer Inform. 2016, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E. Avoiding common pitfalls in machine learning omic data science. Nat. Mater. 2019, 18, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Haury, A.-C.; Gestraud, P.; Vert, J.-P. The Influence of Feature Selection Methods on Accuracy, Stability and Interpretability of Molecular Signatures. PLoS ONE 2011, 6, e28210. [Google Scholar] [CrossRef]
- Eklund, M.; Norinder, U.; Boyer, S.; Carlsson, L. Choosing Feature Selection and Learning Algorithms in QSAR. J. Chem. Inf. Model. 2014, 54, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Delgado, M.; Cernadas, E.; Barro, S.; Amorim, D. Do we Need Hundreds of Classifiers to Solve Real World Classification Problems? J. Mach. Learn. Res. 2014, 15, 3133–3181. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Treatment | RF-OMC | Single-Gene Marker | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Profile | MCC | PR | RC | F1 | MCC | PR | RC | F1 | ||
| BRCA | binimetinib | GEX | 0.57 | 0.77 | 0.91 | 0.83 | 0.24 | 0.68 | 0.68 | 0.68 |
| BRCA | paclitaxel | SNV | 0.49 | 0.82 | 0.88 | 0.85 | −0.82 | 0 | 0 | NA |
| CRC | cetuximab | SNV | 0.47 | 0.62 | 0.81 | 0.70 | 0.59 | 0.9 | 0.56 | 0.69 |
| Cancer Type | Treatment_Name | Profile | nPDXs | MCC | Precision | Recall | Specificity | F1 |
|---|---|---|---|---|---|---|---|---|
| BRCA | BGJ398 | SNV | 38 | NA | 0.37 | 1.00 | 0.00 | 0.54 |
| BRCA | BKM120 | SNV | 38 | NA | NA | 0.03 | 1.00 | NA |
| BRCA | BYL719 | GEX | 38 | 0.02 | 0.64 | 0.77 | 0.29 | 0.69 |
| BRCA | BYL719 + LJM716 | SNV | 38 | NA | NA | 0.02 | 1.00 | NA |
| BRCA | CGM097 | CN | 38 | −0.17 | 0.07 | 0.04 | 0.88 | NA |
| BRCA | CLR457 | CNA | 38 | NA | NA | 0.04 | 0.95 | NA |
| BRCA | HDM201 | SNV | 38 | NA | 0.37 | 1.00 | 0.00 | 0.54 |
| BRCA | INC424 | SNV | 37 | 0.03 | 0.38 | 0.93 | 0.11 | 0.54 |
| BRCA | LEE011 | SNV | 38 | NA | NA | 0.07 | 0.93 | NA |
| BRCA | LKA136 | SNV | 38 | NA | 0.32 | 1.00 | 0.04 | 0.48 |
| BRCA | LLM871 | CN | 38 | NA | NA | NA | NA | NA |
| BRCA | binimetinib | GEX | 38 | 0.08 | 0.60 | 0.75 | 0.31 | 0.67 |
| BRCA | paclitaxel | SNV | 38 | NA | NA | 0.00 | 1.00 | NA |
| CRC | BKM120 | GEX | 39 | 0.00 | 0.64 | 0.82 | 0.21 | 0.71 |
| CRC | BYL719 | CN | 41 | 0.21 | 0.70 | 0.79 | 0.40 | 0.73 |
| CRC | BYL719 + LJM716 | CNA | 39 | NA | NA | 0.02 | 0.92 | NA |
| CRC | CGM097 | GEX | 36 | −0.04 | 0.27 | 0.18 | 0.78 | NA |
| CRC | CLR457 | CN | 40 | 0.02 | 0.70 | 0.89 | 0.13 | 0.78 |
| CRC | HDM201 | SNV | 39 | NA | 0.32 | 1.00 | 0.06 | 0.48 |
| CRC | LEE011 | SNV | 41 | 0.13 | 0.88 | 0.08 | 0.97 | NA |
| CRC | LKA136 | CN | 39 | −0.13 | 0.25 | 0.18 | 0.68 | NA |
| CRC | binimetinib | CN | 41 | 0.02 | 0.67 | 0.67 | 0.32 | 0.67 |
| CRC | cetuximab | SNV | 40 | 0.19 | 0.42 | 1.00 | 0.15 | 0.59 |
| CRC | CKX620 | CN | 38 | −0.09 | 0.76 | 0.97 | 0.00 | 0.85 |
| CRC | encorafenib | CN | 40 | −0.12 | 0.11 | 0.08 | 0.88 | NA |
| CRC | LFW527 + binimetinib | SNV | 31 | 0.36 | 0.70 | 0.63 | 0.67 | 0.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, L.C.; Naulaerts, S.; Bruna, A.; Ghislat, G.; Ballester, P.J. Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles. Biomedicines 2021, 9, 1319. https://doi.org/10.3390/biomedicines9101319
Nguyen LC, Naulaerts S, Bruna A, Ghislat G, Ballester PJ. Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles. Biomedicines. 2021; 9(10):1319. https://doi.org/10.3390/biomedicines9101319
Chicago/Turabian StyleNguyen, Linh C., Stefan Naulaerts, Alejandra Bruna, Ghita Ghislat, and Pedro J. Ballester. 2021. "Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles" Biomedicines 9, no. 10: 1319. https://doi.org/10.3390/biomedicines9101319
APA StyleNguyen, L. C., Naulaerts, S., Bruna, A., Ghislat, G., & Ballester, P. J. (2021). Predicting Cancer Drug Response In Vivo by Learning an Optimal Feature Selection of Tumour Molecular Profiles. Biomedicines, 9(10), 1319. https://doi.org/10.3390/biomedicines9101319