Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact


Abstract
1. Introduction
2. NASH Pathogenesis in Brief
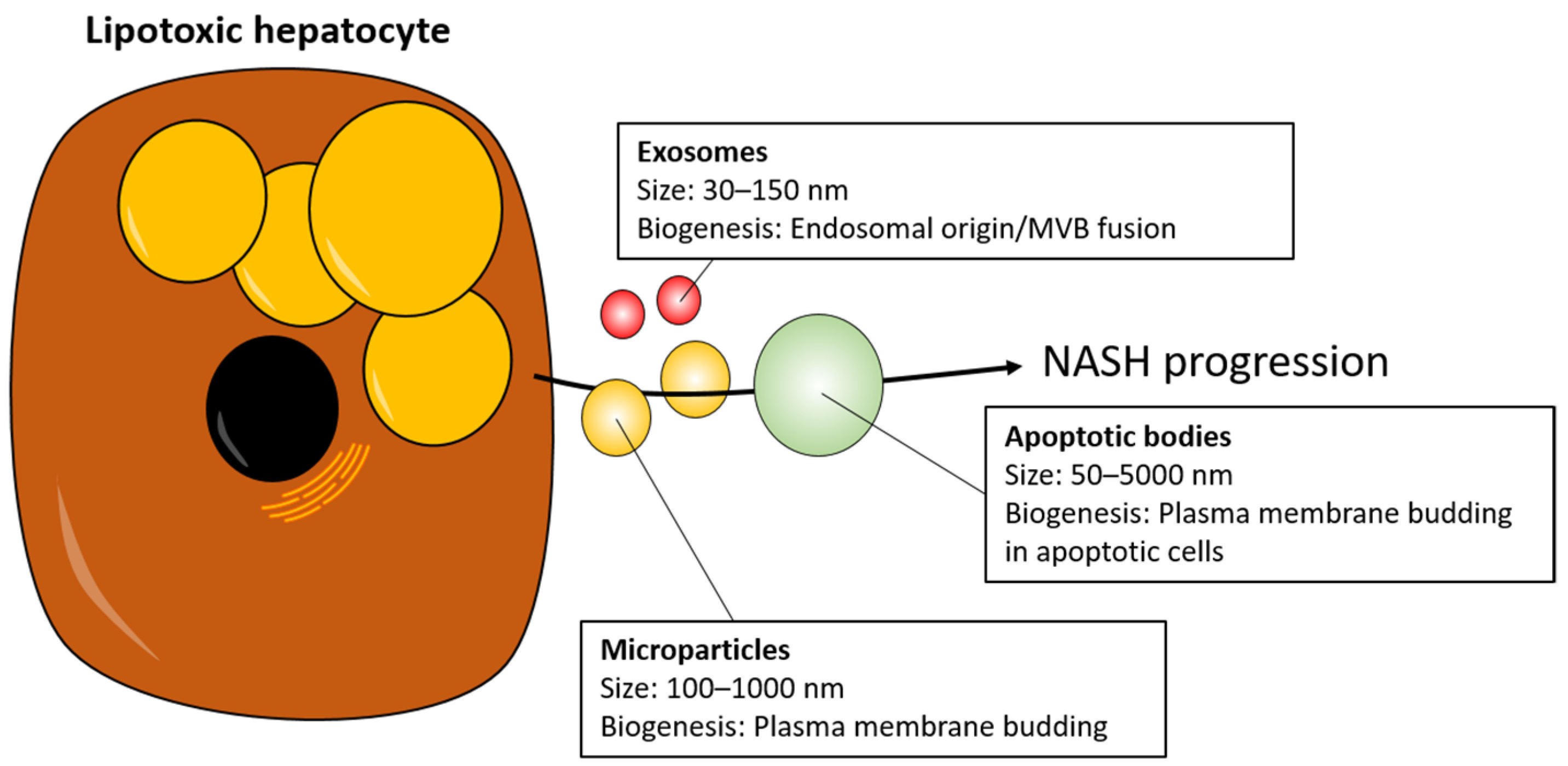
3. EVs as Mediators of NASH Progression
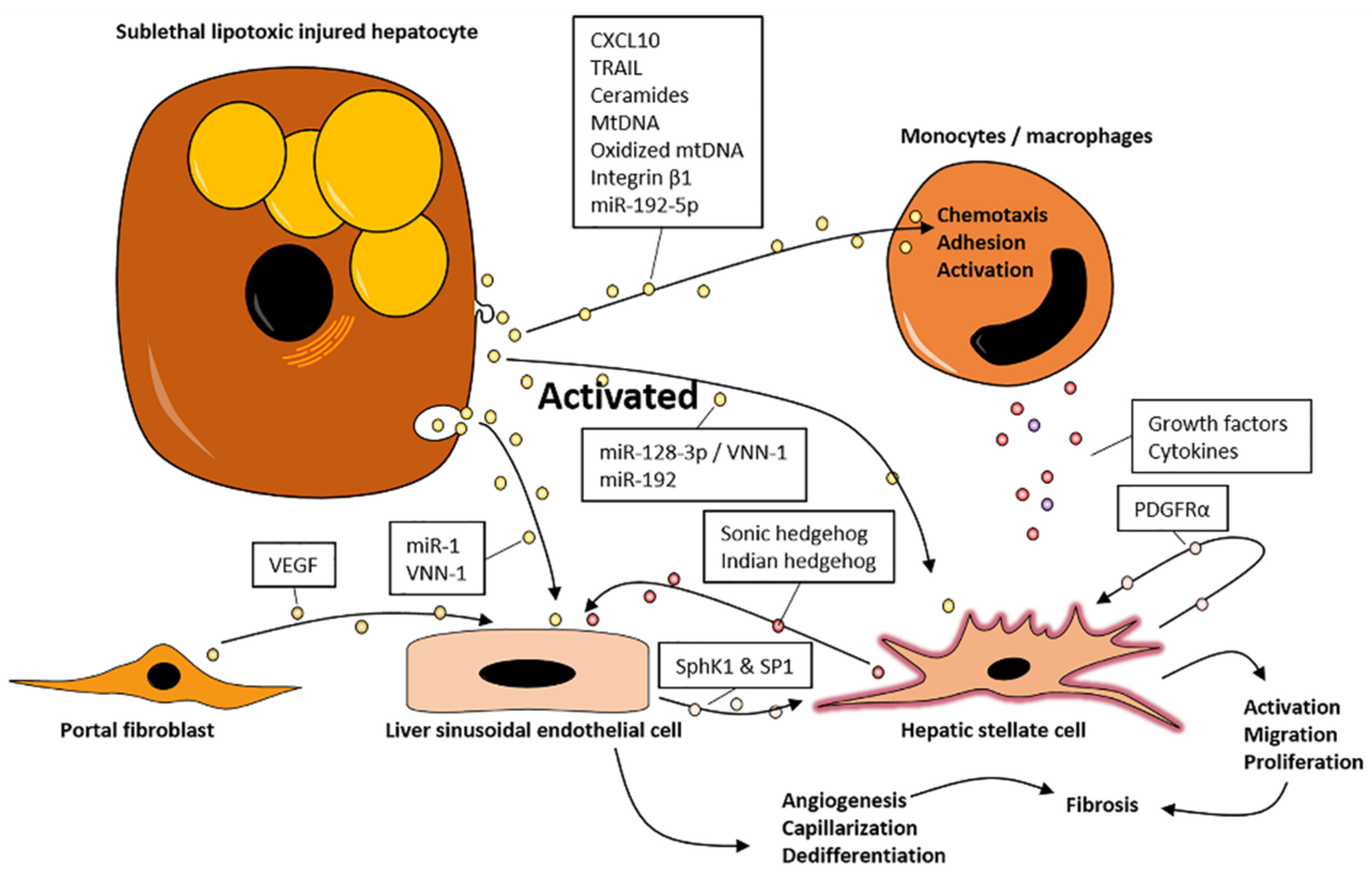
3.1. EVs Promote Inflammation
3.2. EVs Promote Fibrosis
4. EVs May Promote NASH via Organ Crosstalk
5. EVs as Biomarkers in Patients with NASH
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Dorairaj, V.; Sulaiman, S.A.; Abu, N.; Murad, N.A.A. Extracellular Vesicles in the Development of the Non-Alcoholic Fatty Liver Disease: An Update. Biomolecules 2020, 10, 1494. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Momen-Heravi, F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Huang-Doran, I.; Zhang, C.-Y.; Vidal-Puig, A. Extracellular Vesicles: Novel Mediators of Cell Communication in Metabolic Disease. Trends Endocrinol. Metab. 2017, 28, 3–18. [Google Scholar] [CrossRef]
- Hernández, A.; Arab, J.P.; Reyes, D.; Lapitz, A.; Moshage, H.; Banales, J.M.; Arrese, M. Extracellular Vesicles in NAFLD/ALD: From Pathobiology to Therapy. Cells 2020, 9, 817. [Google Scholar] [CrossRef]
- Narayanan, S.; Surette, F.A.; Hahn, Y.S. The Immune Landscape in Nonalcoholic Steatohepatitis. Immune Netw. 2016, 16, 147–158. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wong, V.W.; Peleg, N.; et al. Association between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625.e2. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Huber, Y.; Michel, M.; Nagel, M.; Galle, P.R.; Kostev, K.; Schattenberg, J.M. Impact of NAFLD on the Incidence of Cardiovascular Diseases in a Primary Care Population in Germany. Dig. Dis. Sci. 2020, 65, 2112–2119. [Google Scholar] [CrossRef]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Blüher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018, 555, 673–677. [Google Scholar] [CrossRef]
- Gehrke, N.; Schattenberg, J.M. Metabolic Inflammation-A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease? Gastroenterology 2020, 158, 1929–1947.e6. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef]
- Deng, Z.-B.; Liu, Y.; Liu, C.; Xiang, X.; Wang, J.; Cheng, Z.; Shah, S.V.; Zhang, S.; Zhang, L.; Zhuang, X.; et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009, 50, 1412–1420. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, J.; Man, K.; Chu, E.S.; Yau, T.O.; Sung, J.C.; Go, M.Y.; Deng, J.; Lu, L.; Wong, V.W.; et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mauer, A.S.; Hirsova, P.; Maiers, J.L.; Shah, V.H.; Malhi, H. Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G300–G313. [Google Scholar] [CrossRef]
- Dasgupta, D.; Nakao, Y.; Mauer, A.S.; Thompson, J.M.; Sehrawat, T.S.; Liao, C.-Y.; Krishnan, A.; Lucien, F.; Guo, Q.; Liu, M.; et al. IRE1A Stimulates Hepatocyte-derived Extracellular Vesicles That Promote Inflammation in Mice with Steatohepatitis. Gastroenterology 2020, 159, 1487–1503.e17. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef]
- Guo, Q.; Furuta, K.; Lucien, F.; Sanchez, L.H.G.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef]
- Cazanave, S.C.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Fingas, C.D.; Meng, X.W.; Finnberg, N.; El-Deiry, W.S.; Kaufmann, S.H.; Gores, G.J. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 2011, 286, 39336–39348. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pan, Q.; Cao, H.; Xin, F.; Zhao, Z.; Yang, R.; Zeng, J.; Zhou, H.; Fan, J.-G. Lipotoxic Hepatocyte-Derived Exosomal MicroRNA 192-5p Activates Macrophages Through Rictor/Akt/Forkhead Box Transcription Factor O1 Signaling in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 72, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Morello, E.; Bocca, C.; Foglia, B.; Benetti, E.; Novo, E.; Chiazza, F.; Rogazzo, M.; Fantozzi, R.; Povero, D.; et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0172575. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology 2020, 72, 412–429. [Google Scholar] [CrossRef]
- Arab, J.P.; Sehrawat, T.S.; Simonetto, D.A.; Verma, V.K.; Feng, D.; Tang, T.; Dreyer, K.; Yan, X.; Daley, W.L.; Sanyal, A.; et al. An Open-Label, Dose-Escalation Study to Assess the Safety and Efficacy of IL-22 Agonist F-652 in Patients With Alcohol-associated Hepatitis. Hepatology 2019, 72, 441–453. [Google Scholar] [CrossRef]
- McCommis, K.S.; Hodges, W.T.; Brunt, E.M.; Nalbantoglu, I.; McDonald, W.G.; Holley, C.L.; Fujiwara, H.; Schaffer, J.E.; Colca, J.R.; Finck, B.N. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017, 65, 1543–1556. [Google Scholar] [CrossRef]
- Skat-Rordam, J.; Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor gamma in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; Horcel, L.d.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663e4. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Kim, S.Y.; Ko, E.; Lee, J.-H.; Yi, H.-S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.; Lee, Y.; Park, S.; Jang, M.; Jo, E.; Kim, S.C.; Han, Y.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kaji, K.; Shirai, Y.; Yamazaki, M.; Uemura, M.; Yamao, J.; Fujimoto, M.; et al. Crosstalk between angiogenesis, cytokeratin-18, and insulin resistance in the progression of non-alcoholic steatohepatitis. World J. Gastroenterol. 2009, 15, 5193–5199. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.-E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Eguchi, A.; Niesman, I.R.; Andronikou, N.; Jeu, X.D.M.D.; Mulya, A.; Berk, M.; Lazic, M.; Thapaliya, S.; Parola, M.; et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci. Signal. 2013, 6, ra88. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Yang, L.; Liu, R.; Jung, Y.; Omenetti, A.; Syn, W.; Choi, S.S.; Cheong, Y.; Fearing, C.M.; Agboola, K.M.; et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 2009, 136, 320–330.e2. [Google Scholar] [CrossRef]
- Wang, R.; Ding, Q.; Yaqoob, U.; De Assuncao, T.M.; Verma, V.K.; Hirsova, P.; Cao, S.; Mukhopadhyay, D.; Huebert, R.C.; Shah, V.H. Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-dependent Migration. J. Biol. Chem. 2015, 290, 30684–30696. [Google Scholar] [CrossRef]
- Nojima, H.; Freeman, C.M.; Schuster, R.M.; Japtok, L.; Kleuser, B.; Edwards, M.J.; Gulbins, E.; Lentsch, A.B. Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate. J. Hepatol. 2016, 64, 60–68. [Google Scholar] [CrossRef]
- Lemoinne, S.; Cadoret, A.; Rautou, P.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef]
- Bedossa, P. Histological Assessment of NAFLD. Dig. Dis. Sci. 2016, 61, 1348–1355. [Google Scholar] [CrossRef]
- Li, X.; Chen, R.; Kemper, S.; Brigstock, D.R. Dynamic Changes in Function and Proteomic Composition of Extracellular Vesicles from Hepatic Stellate Cells during Cellular Activation. Cells 2020, 9, 290. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef]
- Chen, L.; Charrier, A.; Zhou, Y.; Chen, R.; Yu, B.; Agarwal, K.; Tsukamoto, H.; Lee, L.J.; Paulaitis, M.E.; Brigstock, D.R. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 2014, 59, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Velazquez, V.M.; Brigstock, D.R. Fibrogenic Signaling Is Suppressed in Hepatic Stellate Cells through Targeting of Connective Tissue Growth Factor (CCN2) by Cellular or Exosomal MicroRNA-199a-5p. Am. J. Pathol. 2016, 186, 2921–2933. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Kostallari, E.; Hirsova, P.; Prasnicka, A.; Verma, V.K.; Yaqoob, U.; Wongjarupong, N.; Roberts, L.R.; Shah, V.H. Hepatic stellate cell-derived platelet-derived growth factor receptor-alpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology 2018, 68, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wei, B.; De Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.H.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J. Hepatol. 2020, 73, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, M.-F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.-W.; Shen, D.; Zhang, J.-Z.; Zhou, N.; He, J.; et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Kranendonk, M.E.; Visseren, F.L.; Van Herwaarden, J.A.; Hoen, E.N.N.; De Jager, W.; Wauben, M.H.; Kalkhoven, E. Effect of extracellular vesicles of human adipose tissue on insulin signaling in liver and muscle cells. Obesity 2014, 22, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Chen, Q.; Wang, W.; Ling, Y.; Yan, Y.; Xia, P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 2020, 72, 156–166. [Google Scholar] [CrossRef]
- Spengler, E.K.; Loomba, R. Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin. Proc. 2015, 90, 1233–1246. [Google Scholar] [CrossRef]
- Mayo, R.; Crespo, J.; Martínez-Arranz, I.; Banales, J.M.; Arias, M.; Mincholé, I.; De La Fuente, R.A.; Jimenez-Agüero, R.; Alonso, C.; De Luis, D.A.; et al. Metabolomic-based noninvasive serum test to diagnose nonalcoholic steatohepatitis: Results from discovery and validation cohorts. Hepatol. Commun. 2018, 2, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xu, D.; Liu, Y.; Guo, X.; Li, W.; Guo, C.; Zhang, H.; Gao, Y.; Mao, Y.; Zhao, J. Combined Serum Biomarkers in Non-Invasive Diagnosis of Non-Alcoholic Steatohepatitis. PLoS ONE 2015, 10, e0131664. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Yamashita, H.; Ren, W.; Subramanian, M.G.; Myers, R.P.; Eguchi, A.; Simonetto, D.A.; Goodman, Z.D.; Harrison, S.A.; Sanyal, A.J.; et al. Characterization and Proteome of Circulating Extracellular Vesicles as Potential Biomarkers for NASH. Hepatol. Commun. 2020, 4, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Kornek, M.; Lynch, M.; Mehta, S.H.; Lai, M.; Exley, M.; Afdhal, N.H.; Schuppan, D. Circulating microparticles as disease-specific biomarkers of severity of inflammation in patients with hepatitis C or nonalcoholic steatohepatitis. Gastroenterology 2012, 143, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Scorletti, E.; Clough, G.F.; Englyst, N.A.; Byrne, C.D. Leukocyte extracellular vesicle concentration is inversely associated with liver fibrosis severity in NAFLD. J. Leukoc. Biol. 2018, 104, 631–639. [Google Scholar] [CrossRef]
- Pirola, C.J.; Fernandez Gianotti, T.; Castano, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.M.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S.C. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 8001–8002. [Google Scholar] [CrossRef]
- Murakami, Y.; Toyoda, H.; Tanahashi, T.; Tanaka, J.; Kumada, T.; Yoshioka, Y.; Kosaka, N.; Ochiya, T.; Taguchi, Y.-H. Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS ONE 2012, 7, e48366. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Rautou, P.; Bresson, J.; Sainte-Marie, Y.; Vion, A.; Paradis, V.; Renard, J.; Devue, C.; Heymes, C.; Letteron, P.; Elkrief, L.; et al. Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology 2012, 143, 166–176.e6. [Google Scholar] [CrossRef]
- Tamimi, T.I.A.-R.; Elgouhari, H.M.; Alkhouri, N.; Yerian, L.M.; Berk, M.P.; Lopez, R.; Schauer, P.R.; Zein, N.N.; Feldstein, A.E. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. J. Hepatol. 2011, 54, 1224–1229. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study Design & NAFLD Diagnosis | Cellular Source | EV Cargo |
|---|---|---|
| Cirrhotic NASH (n = 25, F4), pre-cirrhotic NASH (n = 25, F3) and healthy control (n = 25). Biopsy [63] | Total circulating and hepatocyte (ASGPR1- or SLC27A5-positive) | Proteomic signature of circulating EVs differentiates advanced NASH (F3 + F4) from healthy controls (AUROC = 0.77) and precirrhotic from cirrhotic NASH (AUROC = 0.80) |
| NAFLD (n = 67) vs. HCV patients (n = 42) or healthy controls (n = 44). Biopsy [64] | iNKT (Vα24/Vα11 positiv) or macrophages/monocytes (CD14+) | Number of iKT EVs to differentiate NAFLD from controls (AUROC= 0.92) and HCV (AUROC = 0.97) Number of CD14+ EVs differentiate NAFLD from controls (AUROC = 0.83) and HCV (AUROC > 0.99) |
| Advanced NAFLD, fibrosis 3 and 4 (n = 9) vs. early NAFLD, fibrosis 0–2 (n = 17). Biopsy [65] | Leukocytes (CD14+ or CD16+) Endothelial cells (either CD105+ CD31+ CD41/CD42−, CD105+ CD31− CD41/CD42−, or CD105− CD31+ CD41/CD42−) | ↓ Number of leucocyte and endothelial cell EVs in advanced NAFLD |
| NASH with mild (F1–2) fibrosis (n = 17) vs. steatosis (n = 8). Biopsy [29] | Not examined | ↑ Integrin β1 in NASH |
| NASH F0–1 fibrosis (n = 16) vs. bland steatosis (n = 16) or obese controls (n = 11). Biopsy for some [23] | Not examined | ↑ C16:0 ceramides and S1P in bland steatosis and NASH. Nominally increased in NASH vs. bland steatosis |
| Obese/high ALT (n = 9) vs. obese/normal ALT (n = 19) or lean/normal ALT (n = 19). Elevated ALT [28] | Hepatocyte (ARG1 positive, CD41 negative) | ↑ mtDNA in obese with high ALT |
| NASH (n = 47), steatosis (n = 30) and health controls (n = 19). Biopsy [66] | Not examined | ↑ miRNA-122, -192 and -375 in NASH vs. steatosis or healthy controls miRNA-122 could to a degree identify NASH (AUROC = 0.71) and fibrosis (AUROC = 0.61) |
| Advanced NAFLD (n = 3) vs. early NALFD (n = 3). Biopsy [39] † | Not examined | ↑ miRNA-122 and -192 in advanced NAFLD |
| NASH (n = 31) vs. healthy controls (n = 37). Biopsy [32] | Hepatocyte (ASPPR1 and CYP2E1 positive) | ↑ miR-192-5p in NASH |
| NASH (n = 12), hepatitis B (n = 4) and controls (n = 24). Biopsy [67] | Not examined | miRNA panel (miR-1225-5p, -1275, -368, -762, 320c, -451, -1974, -630, -1207-5p, -720, -1246, and -486-5p) distinguish NASH from HBV and controls with accuracies of 87.5% and 88.9%, respectively |
| NAFLD/NASH (n = 34) vs. healthy controls (n = 19). Biopsy [68] | Not examined | ↑ miRNA-16, -34a, and -122 in NAFLD/NASH miRNA-16 (AUROC = 0.96) and miRNA-122 (AUROC = 0.93) differentiates NAFLD from healthy controls |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ipsen, D.H.; Tveden-Nyborg, P. Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact. Biomedicines 2021, 9, 93. https://doi.org/10.3390/biomedicines9010093
Ipsen DH, Tveden-Nyborg P. Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact. Biomedicines. 2021; 9(1):93. https://doi.org/10.3390/biomedicines9010093
Chicago/Turabian StyleIpsen, David Højland, and Pernille Tveden-Nyborg. 2021. "Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact" Biomedicines 9, no. 1: 93. https://doi.org/10.3390/biomedicines9010093
APA StyleIpsen, D. H., & Tveden-Nyborg, P. (2021). Extracellular Vesicles as Drivers of Non-Alcoholic Fatty Liver Disease: Small Particles with Big Impact. Biomedicines, 9(1), 93. https://doi.org/10.3390/biomedicines9010093