Mithramycin and Analogs for Overcoming Cisplatin Resistance in Ovarian Cancer

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
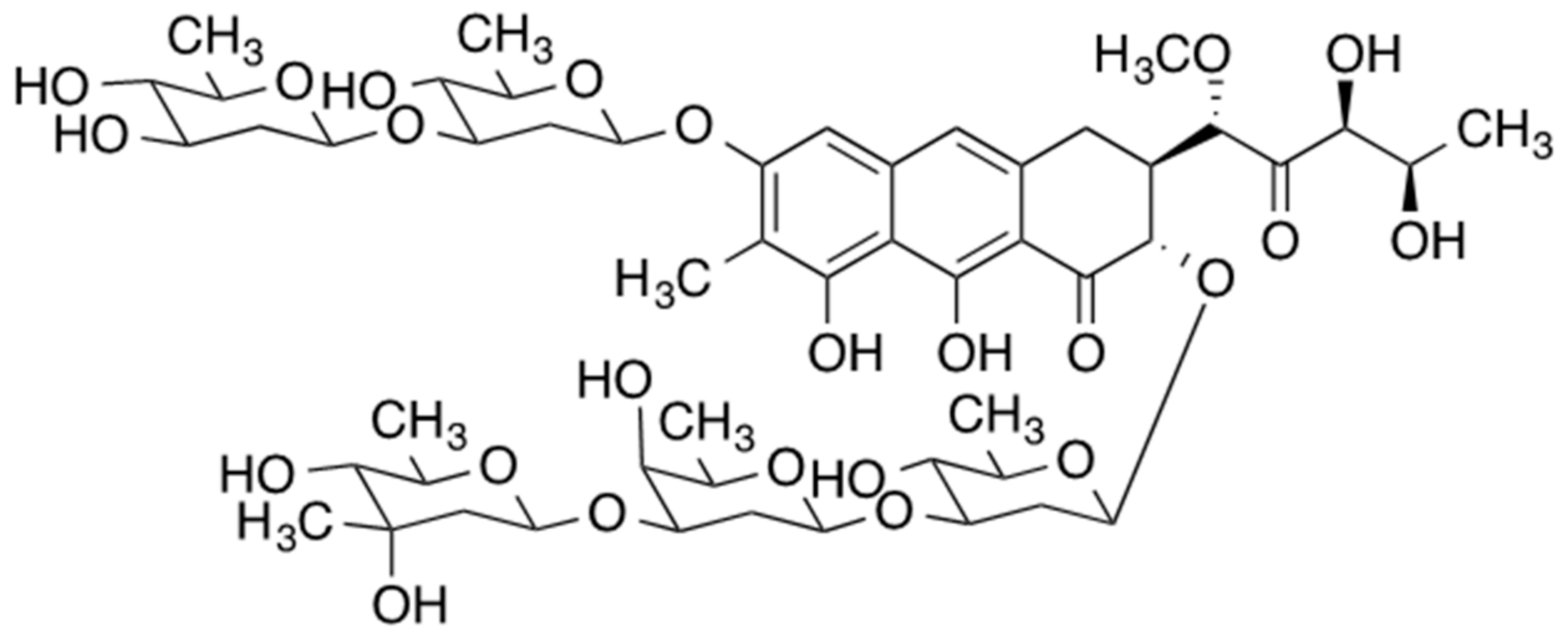
2. Mithramycin
3. Use of Mithramycin in Ovarian Cancer
4. Recent Clinical Development
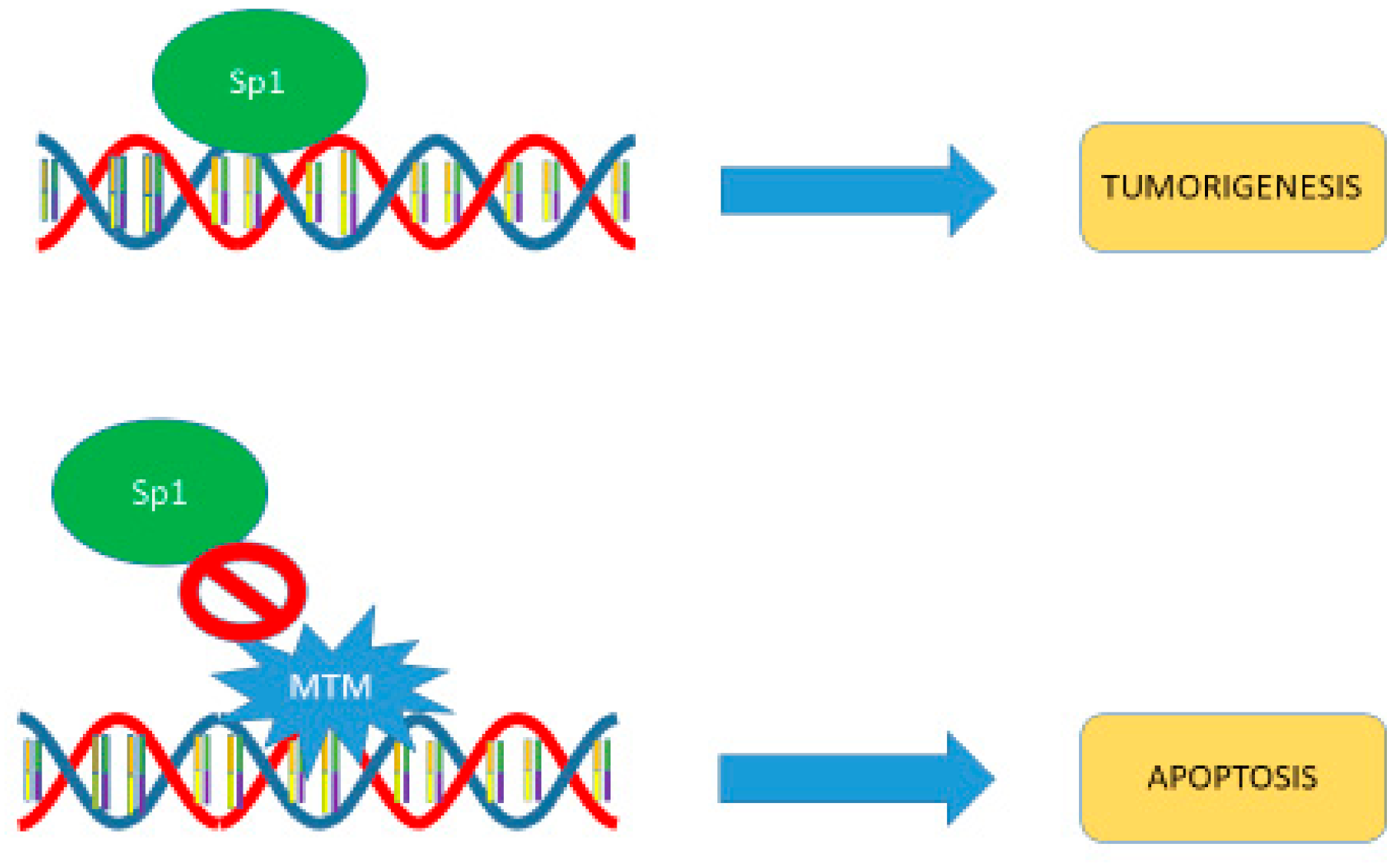
5. Mechanism of Action
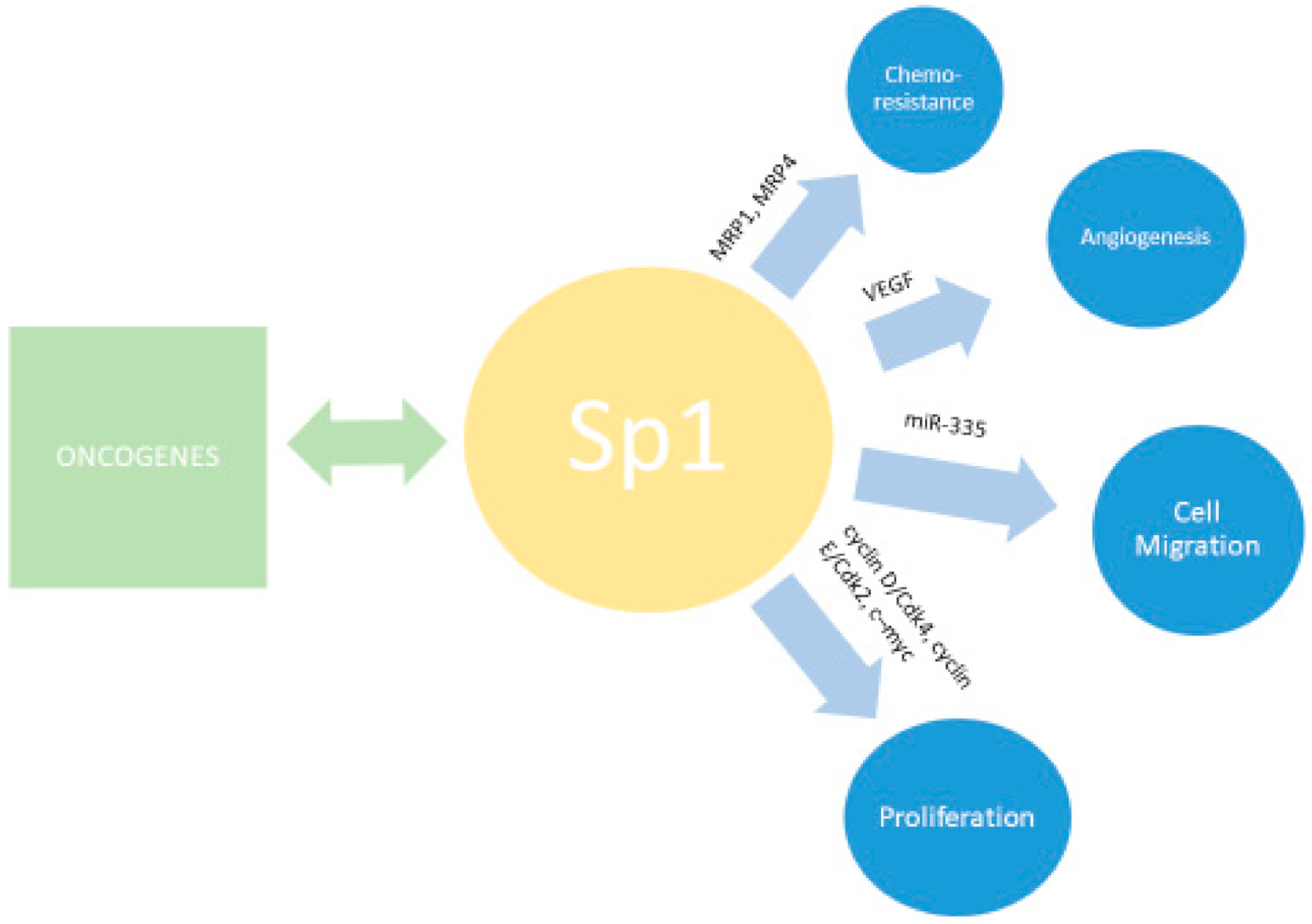
6. SP1
7. Development of Mithramycin Analogues
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer Statistics. Cancer Rehabil. 2018, 70, 7–30. [Google Scholar] [CrossRef]
- Colombo, N.; Sessa, C.; Du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Int. J. Gynecol. Cancer 2019, 29, 728–760. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Ovarian Cancer (Version 1.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed on 1 August 2020).
- Salani, R.; Khanna, N.; Frimer, M.; Bristow, R.E.; Chen, L.-M. An update on post-treatment surveillance and diagnosis of recurrence in women with gynecologic ma-lignancies: Society of Gynecologic Oncology (SGO) recommendations. Gynecol. Oncol. 2017, 146, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Fleming, G.F.; Seidman, J.D.; Yemelyanova, A.; Lengyel, E. Epithelial Ovarian Cancer. In Principles and practice of gynecologic oncology; Chi, D.S., Berchuck, A., Dizon, D.S., Yashar, C.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2017; Chapter 23; pp. 611–705. [Google Scholar]
- Hanker, L.C.; Liobl, S.; Burchardi, N.; Pfisterer, J.; Meier, W.; Pujade-Lauraine, E.; Ray-Conquard, I.; Sehouli, J.; Harter, P.; du Bois, A.; et al. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary tax-ane/platinum-based therapy. Ann. Oncol. 2012, 23, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Matulonis, U.A.; Secord, A.A.; Nemunaitis, J.; Roman, L.D.; Blagden, S.P.; Banerjee, S.; McGuire, W.P.; Ghamande, S.; Birrer, M.J.; et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Plati-num-resistant, Recurrent Ovarian Cancer. Clin. Cancer Res. 2020, 26, 1009–1016. [Google Scholar] [CrossRef]
- Sehouli, J.; Stengel, D.; Harter, P.; Kurzeder, C.; Belau, A.; Bogenrieder, T.; Markmann, S.; Mahner, S.; Mueller, L.; Lorenz, R.; et al. Topotecan Weekly Versus Conventional 5-Day Schedule in Patients With Platinum-Resistant Ovarian Cancer: A Randomized Multicenter Phase II Trial of the North-Eastern German Society of Gynecological Oncology Ovarian Cancer Study Group. J. Clin. Oncol. 2011, 29, 242–248. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Lopez-Martinez, D.; Liang, C.-C.; Cohn, M.A. Cellular response to DNA interstrand crosslinks: The Fanconi anemia pathway. Cell. Mol. Life Sci. 2016, 73, 3097–3114. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D. Mechanisms of Drug Resistance in High-Grade Serous Ovarian Cancer. Hematol. Clin. N. Am. 2018, 32, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Batty, D.P.; Wood, R.D. Damage recognition in nucleotide excision repair of DNA. Gene 2000, 241, 193–204. [Google Scholar] [CrossRef]
- Bhagwat, N.; Olsen, A.L.; Wang, A.T.; Hanada, K.; Stuckert, P.; Kanaar, R.; D’Andrea, A.; Niedernhofer, L.J.; McHugh, P.J. XPF-ERCC1 Participates in the Fanconi Anemia Pathway of Cross-Link Repair. Mol. Cell. Biol. 2009, 29, 6427–6437. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF–ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper Transporters and the Cellular Pharmacology of the Platinum-Containing Cancer Drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar] [CrossRef]
- Chen, H.H.W.; Chen, W.-C.; Liang, Z.-D.; Tsai, W.-B.; Long, Y.; Aiba, I.; Fu, S.; Broaddus, R.; Liu, J.; Feun, L.G.; et al. Targeting drug transport mechanisms for improving platinum-based cancer chemotherapy. Expert Opin. Ther. Targets 2015, 19, 1307–1317. [Google Scholar] [CrossRef]
- Lee, J.; Minasian, L.M.; Doong, H. New strategies in ovarian cancer treatment. Cancer 2019, 125, 4623–4629. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 230076, Mithramycin a. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mithramycin-a (accessed on 1 August 2020).
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS-FLI1 fusion transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.-R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an Inhibitor of the EWS-FLI1 Oncogenic Transcription Factor by High-Throughput Screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef]
- Wohlert, S.E.; Künzel, E.; Machinek, R.; Méndez, C.; Salas, J.A.; Rohr, J. The Structure of Mithramycin Reinvestigated. J. Nat. Prod. 1999, 62, 119–121. [Google Scholar] [CrossRef]
- Kofman, S.; Medrek, T.J.; Alexander, R.W. Mithramycin in the treatment of embryonal cancer. Cancer 1964, 17, 938–948. [Google Scholar] [CrossRef]
- Sewell, I.A.; Ellis, H. A trial of mithramycin in the treatment of advanced malignant disease. Br. J. Cancer 1966, 20, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Baum, M. A clinical trial of mithramycin in the treatment of advanced malignant disease. Br. J. Cancer 1968, 22, 176–183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, M.; Mathur, A.; Zhang, Y.; Xi, S.; Atay, S.; Hong, J.A.; Datrice, N.; Upham, T.; Kemp, C.D.; Ripley, R.T.; et al. Mithramycin Represses Basal and Cigarette Smoke–Induced Expression of ABCG2 and In-hibits Stem Cell Signaling in Lung and Esophageal Cancer Cells. Cancer Res. 2012, 72, 4178–4192. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. Plicamycin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548735/ (accessed on 19 February 2020).
- Sissung, T.M.; Huang, P.A.; Hauke, R.J.; McCrea, E.M.; Peer, C.J.; Barbier, R.H.; Strope, J.D.; Ley, A.M.; Zhang, M.; Hong, J.A.; et al. Severe Hepatotoxicity of Mithramycin Therapy Caused by Altered Expression of Hepatocellular Bile Transporters. Mol. Pharmacol. 2019, 96, 158–167. [Google Scholar] [CrossRef]
- Hou, C.; Weidenbach, S.; Cano, K.E.; Wang, Z.; Mitra, P.; Ivanov, D.N.; Rohr, J.; Tsodikov, O. Structures of mithramycin analogues bound to DNA and implications for targeting transcription factor FLI1. Nucleic Acids Res. 2016, 44, 8990–9004. [Google Scholar] [CrossRef]
- Weidenbach, S.; Hou, C.; Chen, J.-M.; Tsodikov, O.; Rohr, J. Dimerization and DNA recognition rules of mithramycin and its analogues. J. Inorg. Biochem. 2016, 156, 40–47. [Google Scholar] [CrossRef]
- Sastry, M.; Patel, D.J. Solution structure of the mithramycin dimer-DNA complex. Biochemistry 1993, 32, 6588–6604. [Google Scholar] [CrossRef]
- Demicheli, C.; Albertini, J.P.; Garnier-Suillerot, A. Interaction of mithramycin with DNA. Evidence that mithramycin binds to DNA as a dimer in a right-handed screw conformation. JBIC J. Biol. Inorg. Chem. 1991, 198, 333–338. [Google Scholar] [CrossRef]
- Cons, B.M.; Fox, K.R. Interaction of mithramycin with metal ions and DNA. Biochem. Biophys. Res. Commun. 1989, 160, 517–524. [Google Scholar] [CrossRef]
- Carpenter, M.L.; Cassidy, S.A.; Fox, K.R. Interaction of mithramycin with isolated GC and CG sites. J. Mol. Recognit. 1994, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Aich, P.; Dasgupta, D. Role of Mg++ in the mithramycin-DNA interaction: Evidence for two types of mithramycin-Mg++ complex. Biochem. Biophys. Res. Commun. 1990, 173, 689–696. [Google Scholar] [CrossRef]
- Albertini, V.; Jain, A.; Vignati, S.; Napoli, S.; Rinaldi, A.; Kwee, I.; Nur-e-Alam, M.; Bergant, J.; Bertoni, F.; Carbone, G.M.; et al. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithra-mycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: Implications for cancer therapy. Nucleic Acids Res. 2006, 34, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Guizán, A.; Mansilla, S.; Barceló, F.; Vizcaíno, C.; Núñez, L.-E.; Morís, F.; González, L.E.N.; Portugal, J. The activity of a novel mithramycin analog is related to its binding to DNA, cellular accumulation, and inhibition of Sp1-driven gene transcription. Chem. Interactions 2014, 219, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Méndez, C.; González-Sabín, J.; Morís, F.; Salas, J.A. Expanding the Chemical Diversity of the Antitumoral Compound Mithramycin by Combinatorial Bio-synthesis and Biocatalysis: The Quest for Mithralogs with Improved Therapeutic Window. Planta Med. 2015, 81, 1326–1338. [Google Scholar] [PubMed]
- Núñez, L.E.; Nybo, S.E.; González-Sabín, J.; Pérez, M.; Menéndez, N.; Braña, A.F.; Shaaban, K.A.; He, M.; Morís, F.; Salas, J.A.; et al. A Novel Mithramycin Analogue with High Antitumor Activity and Less Toxicity Generated by Com-binatorial Biosynthesis. J. Med. Chem. 2012, 55, 5813–5825. [Google Scholar] [CrossRef]
- Previdi, S.; Malek, A.; Albertini, V.; Riva, C.; Capella, C.; Broggini, M.; Carbone, G.M.; Rohr, J.; Catapano, C.V. Inhibition of Sp1-dependent transcription and antitumor activity of the new aureolic acid analogues mithramycin SDK and SK in human ovarian cancer xenografts. Gynecol. Oncol. 2010, 118, 182–188. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef]
- Rao, M.; Atay, S.M.; Shukla, V.; Hong, Y.; Upham, T.; Ripley, R.T.; Hong, J.A.; Zhang, M.; Reardon, E.S.; A Fetsch, P.; et al. Mithramycin Depletes Specificity Protein 1 and Activates p53 to Mediate Senescence and Apoptosis of Malignant Pleural Mesothelioma Cells. Clin. Cancer Res. 2016, 22, 1197–1210. [Google Scholar] [CrossRef]
- Ohgami, T.; Kato, K.; Kobayashi, H.; Sonoda, K.; Inoue, T.; Yoneda, T.; Wake, N.; Yamaguchi, S.-I. Low-dose mithramycin exerts its anticancer effect via the p53 signaling pathway and synergizes with nutlin-3 in gynecologic cancers. Cancer Sci. 2010, 101, 1387–1395. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.A.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [PubMed]
- Kadonaga, J.T.; Carner, K.R.; Masiarz, F.R.; Tjian, R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 1987, 51, 1079–1090. [Google Scholar] [CrossRef]
- Kadonaga, J.; Courey, A.; Ladika, J.; Tjian, R.; Sager, R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science 1988, 242, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Kadonaga, J.T.; Jones, K.A.; Tjian, R. Promoter-specific activation of RNA polymerase II transcription by Sp1. Trends Biochem. Sci. 1986, 11, 20–23. [Google Scholar] [CrossRef]
- Chuang, J.-Y.; Wang, S.-A.; Yang, W.-B.; Yang, H.-C.; Hung, C.-Y.; Su, T.-P.; Chang, W.-C.; Hung, J.-J. Sp1 phosphorylation by cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding activity during mitosis in cancer cells. Oncogene 2012, 31, 4946–4959. [Google Scholar] [CrossRef]
- Grinstein, E.; Jundt, F.; Weinert, I.; Wernet, P.; Royer, H.-D. Sp1 as G1 cell cycle phase specific transcription factor in epithelial cells. Oncogene 2002, 21, 1485–1492. [Google Scholar] [CrossRef]
- Mansilla, S.; Portugal, J. Sp1 transcription factor as a target for anthracyclines: Effects on gene transcription. Biochimie 2008, 90, 976–987. [Google Scholar] [CrossRef]
- Tan, N.Y.; Khachigian, L.M. Sp1 Phosphorylation and Its Regulation of Gene Transcription. Mol. Cell. Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, P.; Huang, J.; Ge, S.; Lu, J.; Qian, G. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem. Biophys. Res. Commun. 2007, 356, 286–292. [Google Scholar] [CrossRef]
- Portugal, J. Challenging transcription by DNA-binding antitumor drugs. Biochem. Pharmacol. 2018, 155, 336–345. [Google Scholar] [CrossRef]
- Vellingiri, B.; Iyer, M.; Subramaniam, M.D.; Jayaramayya, K.; Siama, Z.; Giridharan, B.; Narayanasamy, A.; Dayem, A.A. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: From Theory to Practice. Int. J. Mol. Sci. 2020, 21, 1153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ye, W.; Wu, J.; Liu, L.; Yang, L.; Gao, L.; Chen, B.; Zhang, F.; Yang, H.; Li, Y. Sp1-CD147 positive feedback loop promotes the invasion ability of ovarian cancer. Oncol. Rep. 2015, 34, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Geng, J.; Li, X.; Qiao, C.; Luo, L.; Feng, J.; Dong, X.; Lv, M. SP1 promotes tumor angiogenesis and invasion by activating VEGF expression in an acquired trastuzumab-resistant ovarian cancer model. Oncol. Rep. 2017, 38, 2677–2684. [Google Scholar] [CrossRef]
- Xu, F.; Zhu, X.; Han, T.; You, X.; Liu, F.; Ye, L.; Zhang, X.; Wang, X.; Yao, Y. The oncoprotein hepatitis B X-interacting protein promotes the migration of ovarian cancer cells through the upregulation of S-phase kinase-associated protein 2 by Sp1. Int. J. Oncol. 2014, 45, 255–263. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Sun, S.; Cai, J.; Cao, J. Sp1 promotes ovarian cancer cell migration through repressing miR-335 expression. Biochem. Biophys. Res. Commun. 2020, 524, 211–216. [Google Scholar] [CrossRef]
- Sankpal, U.T.; Ingersoll, S.B.; Ahmad, S.; Holloway, R.W.; Bhat, V.B.; Simecka, J.W.; Daniel, L.; Kariali, E.; Vishwanatha, J.K.; Basha, R. Association of Sp1 and survivin in epithelial ovarian cancer: Sp1 inhibitor and cisplatin, a novel combination for inhibiting epithelial ovarian cancer cell proliferation. Tumor Biol. 2016, 37, 14259–14269. [Google Scholar] [CrossRef]
- Knappskog, S.; Bjørnslett, M.; Myklebust, L.M.; Huijts, P.E.; Vreeswijk, M.P.; Edvardsen, H.; Guo, Y.; Zhang, X.; Yang, M.; Ylisaukko-Oja, S.K.; et al. The MDM2 Promoter SNP285C/309G Haplotype Diminishes Sp1 Transcription Factor Binding and Reduces Risk for Breast and Ovarian Cancer in Caucasians. Cancer Cell 2011, 19, 273–282. [Google Scholar] [CrossRef]
- Mahalaxmi, I.; Santhy, K. Role and hallmarks of Sp1 in promoting ovarian cancer. J. Oncol. Sci. 2018, 4, 102–105. [Google Scholar] [CrossRef]
- Zhang, Q.; Yan, G.; Lei, J.; Chen, Y.; Wang, T.; Gong, J.; Zhou, Y.; Zhao, H.; Chen, H.; Zhou, Y.; et al. The SP1-12LOX axis promotes chemoresistance and metastasis of ovarian cancer. Mol. Med. 2020, 26, 39. [Google Scholar] [CrossRef]
- Miyata, K.; Yotsumoto, F.; Nam, S.O.; Odawara, T.; Manabe, S.; Ishikawa, T.; Itamochi, H.; Kigawa, J.; Takada, S.; Asahara, H.; et al. Contribution of transcription factor, SP 1, to the promotion of HB - EGF expression in defense mechanism against the treatment of irinotecan in ovarian clear cell carcinoma. Cancer Med. 2014, 3, 1159–1169. [Google Scholar] [CrossRef]
- Xia, B.; Hou, Y.; Chen, H.; Yang, S.; Liu, T.; Lin, M.; Lou, G. Long non-coding RNA ZFAS1 interacts with miR-150-5p to regulate Sp1 expression and ovarian cancer cell malignancy. Oncotarget 2017, 8, 19534–19546. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, C.; Núñez, L.-E.; Morís, F.; Portugal, J. Genome-Wide Modulation of Gene Transcription in Ovarian Carcinoma Cells by a New Mithramycin Analogue. PLoS ONE 2014, 9, e104687. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Wang, L.; McDonald, J.F. Identification of inhibitors of ovarian cancer stem-like cells by high-throughput screening. J. Ovarian Res. 2012, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.C.; Young, R.C.; McKoy, W.M.; Grotzinger, K.R.; Green, J.A.; Chu, E.W.; Whang-Peng, J.; Rogan, A.M.; Green, W.R.; Ozlos, R.F. Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and es-trogen receptors. Cancer Res. 1983, 43, 5379–5389. [Google Scholar]
- Sakhare, S.S.; Rao, G.G.; Mandape, S.; Pratap, S. Transcriptome profile of OVCAR3 cisplatin-resistant ovarian cancer cell line. BMC Bioinform. 2014, 15, P21. [Google Scholar] [CrossRef]
- Fernández-Guizán, A.; López-Soto, A.; Acebes-Huerta, A.; Huergo-Zapico, L.; Villa-Álvarez, M.; Núñez, L.-E.; Morís, F.; Gonzales, S. Pleiotropic Anti-Angiogenic and Anti-Oncogenic Activities of the Novel Mithralog Demycarosyl-3D-β-d-Digitoxosyl-Mithramycin SK (EC-8042). PLoS ONE 2015, 10, e0140786. [Google Scholar] [CrossRef]
- Saha, S.; Mukherjee, S.; Mazumdar, M.; Manna, A.; Khan, P.; Adhikary, A.; Kajal, K.; Jana, D.; Sa, G.; Mukherjee, S.; et al. Mithramycin A sensitizes therapy-resistant breast cancer stem cells toward genotoxic drug doxorubicin. Transl. Res. 2015, 165, 558–577. [Google Scholar] [CrossRef]
- Sachrajda, I.; Ratajewski, M. Mithramycin A suppresses expression of the human melanoma-associated gene ABCB8. Mol. Genet. Genom. 2010, 285, 57–65. [Google Scholar] [CrossRef]
- Taylor, D.J.; E Parsons, C.; Han, H.; Jayaraman, A.; Rege, K. Parallel screening of FDA-approved antineoplastic drugs for identifying sensitizers of TRAIL-induced apoptosis in cancer cells. BMC Cancer 2011, 11, 470. [Google Scholar] [CrossRef]
- Ragheb, R.; Venton, G.; Chelbi, R.; Bonnet, N.; Le Treut, T.; Ivanov, V.; Mercier, C.; Poulin, P.; Beaufils, N.; Gabert, J.; et al. Vorinostat and Mithramycin A in combination therapy as an interesting strategy for the treatment of Sézary T lymphoma: A transcriptomic approach. Arch. Dermatol. Res. 2017, 309, 611–623. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, L.; Zhou, W.; Xie, X.; Wu, M.; Chen, Y.; Hu, Y.; Du, J.; He, Y.; Li, Y. EPS8-mediated regulation of multiple myeloma cell growth and survival. Am. J. Cancer Res. 2019, 9, 1622–1634. [Google Scholar]
- Nair, R.; Piktel, D.; Geldenhuys, W.J.; Gibson, L.F. Combination of cabazitaxel and plicamycin induces cell death in drug resistant B-cell acute lymphoblastic leukemia. Leuk. Res. 2018, 72, 59–66. [Google Scholar] [CrossRef]
- Gao, Y.; Jia, Z.; Kong, X.; Li, Q.; Chang, D.Z.; Wei, D.; Le, X.; Suyun, H.; Huang, S.; Wang, L.; et al. Combining betulinic acid and mithramycin a effectively suppresses pancreatic cancer by inhibiting prolif-eration, invasion, and angiogenesis. Cancer Res. 2011, 71, 5182–5193. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, J.; Wei, D.; Wang, L.; Yuan, P.; Le, X.; Li, Q.; Yao, J.; Xie, K. Molecular Basis of the Synergistic Antiangiogenic Activity of Bevacizumab and Mithramycin A. Cancer Res. 2007, 67, 4878–4885. [Google Scholar] [CrossRef]
- Hou, C.; Mandal, A.; Rohr, J.; Tsodikov, O. Allosteric Interference in Oncogenic FLI1 and ERG Transactions by Mithramycins. Structure 2020. [Google Scholar] [CrossRef]
- Remsing, L.L.; González, A.M.; Nur-e-Alam, M.; Fernández-Lozano, M.J.; Braña, A.F.; Rix, U.; Oliveira, M.A.; Méndez, C.; Salas, J.A.; Rohr, J. Mithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosyl-mithramycin SK: Three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesis. J. Am. Chem. Soc. 2003, 125, 5745–5753. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Chen, J.-M.; Bae, Y.; Rohr, J. Semi-synthetic mithramycin SA derivatives with improved anticancer activity. Chem. Biol. Drug Des. 2013, 81, 615–624. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Núñez, L.-E.; Méndez, C.; Salas, J.A.; Morís, F.; Portugal, J. Novel mithramycins abrogate the involvement of protein factors in the transcription of cell cycle control genes. Biochem. Pharmacol. 2012, 84, 1133–1142. [Google Scholar] [CrossRef]
- Mitra, P.; Eckenrode, J.M.; Mandal, A.; Jha, A.M.; Salem, S.M.; Leggas, M.; Rohr, J. Development of Mithramycin Analogues with Increased Selectivity toward ETS Transcription Factor Ex-pressing Cancers. J. Med. Chem. 2018, 61, 8001–8016. [Google Scholar] [CrossRef]
- Liu, Y.; Eckenrode, J.M.; Zhang, Y.; Zhang, J.; Hayden, R.C.; Kyomuhangi, A.; Ponomareva, L.V.; Cui, Z.; Rohr, J.; Tsodikov, O.V.; et al. Mithramycin 2’-Oximes with Improved Selectivity, Pharmacokinetics, and Ewing Sarcoma Antitumor Effi-cacy. J. Med. Chem. 2020, 63, 14067–14086. [Google Scholar] [CrossRef]
- Federico, A.; Steinfass, T.; Larribére, L.; Novak, D.; Morís, F.; Núñez, L.-E.; Umansky, V.; Utikal, J. Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Ma-lignant Melanoma. Mol. Ther. Oncolytics 2020, 18, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Tornin, J.; Martinez-Cruzado, L.; Santos, L.; Rodriguez, A.; Núñez, L.-E.; Oro, P.; Hermosilla, M.A.; Allonca, E.; Fernández-García, M.T.; Astudillo, A.; et al. Inhibition of SP1 by the mithramycin analog EC-8042 efficiently targets tumor initiating cells in sarcoma. Oncotarget 2016, 7, 30935–30950. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.; Núñez, L.-E.; Magistri, M.; Brambilla, L.; Jović, S.; Carbone, G.M.; Morís, F.; Catapano, C.V. Modulation of the Activity of Sp Transcription Factors by Mithramycin Analogues as a New Strategy for Treatment of Metastatic Prostate Cancer. PLoS ONE 2012, 7, e35130. [Google Scholar] [CrossRef] [PubMed]
- Pandiella, A.; Morís, F.; Ocaña, A.; Núñez, L.-E.; Montero, J.C. Antitumoral activity of the mithralog EC-8042 in triple negative breast cancer linked to cell cycle arrest in G2. Oncotarget 2015, 6, 32856–32867. [Google Scholar] [CrossRef]
- Cohen-Sela, E.; Teitlboim, S.; Chorny, M.; Koroukhov, N.; Danenberg, H.D.; Gao, J.; Golomb, G. Single and Double Emulsion Manufacturing Techniques of an Amphiphilic Drug in PLGA Nanoparticles: Formulations of Mithramycin and Bioactivity. J. Pharm. Sci. 2009, 98, 1452–1462. [Google Scholar] [CrossRef]
- Liu, X.J.; Li, L.; Li, Y.; Zhao, C.-Y.; Wang, R.-Q.; Zhen, Y.-S. Mithramycin-loaded mPEG-PLGA nanoparticles exert potent antitumor efficacy against pancreatic carci-noma. Int. J. Nanomed. 2017, 12, 5255–5269. [Google Scholar] [CrossRef]
- Scott, D.; Rohr, J.; Bae, Y. Nanoparticulate formulations of mithramycin analogs for enhanced cytotoxicity. Int. J. Nanomed. 2011, 6, 2757–2767. [Google Scholar] [CrossRef]
- van Driel, W.J.; Koole, S.N.; Sikorska, K.; van Leeuwen, J.H.S.; Schreyder, H.W.R.; Hermans, R.H.M.; de Hingh, I.H.J.T.; van der Velden, J.; Arts, H.; Massuger, L.F.A.G.; et al. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N. Engl. J. Med. 2018, 378, 230–240. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schweer, D.; McCorkle, J.R.; Rohr, J.; Tsodikov, O.V.; Ueland, F.; Kolesar, J. Mithramycin and Analogs for Overcoming Cisplatin Resistance in Ovarian Cancer. Biomedicines 2021, 9, 70. https://doi.org/10.3390/biomedicines9010070
Schweer D, McCorkle JR, Rohr J, Tsodikov OV, Ueland F, Kolesar J. Mithramycin and Analogs for Overcoming Cisplatin Resistance in Ovarian Cancer. Biomedicines. 2021; 9(1):70. https://doi.org/10.3390/biomedicines9010070
Chicago/Turabian StyleSchweer, David, J. Robert McCorkle, Jurgen Rohr, Oleg V. Tsodikov, Frederick Ueland, and Jill Kolesar. 2021. "Mithramycin and Analogs for Overcoming Cisplatin Resistance in Ovarian Cancer" Biomedicines 9, no. 1: 70. https://doi.org/10.3390/biomedicines9010070
APA StyleSchweer, D., McCorkle, J. R., Rohr, J., Tsodikov, O. V., Ueland, F., & Kolesar, J. (2021). Mithramycin and Analogs for Overcoming Cisplatin Resistance in Ovarian Cancer. Biomedicines, 9(1), 70. https://doi.org/10.3390/biomedicines9010070