Rhosin Suppressed Tumor Cell Metastasis through Inhibition of Rho/YAP Pathway and Expression of RHAMM and CXCR4 in Melanoma and Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blotting
2.3. Rho Pull-Down Assay
2.4. In Vitro Migration, Invasion, and Adhesion Assays
2.5. RNA Interference
2.6. Mice
2.7. Effects of Intraperitoneal Administration of Rhosin on Lung Metastasis of Tumor Cells
2.8. SurvExpress Analysis
2.9. Statistical Analysis
3. Results
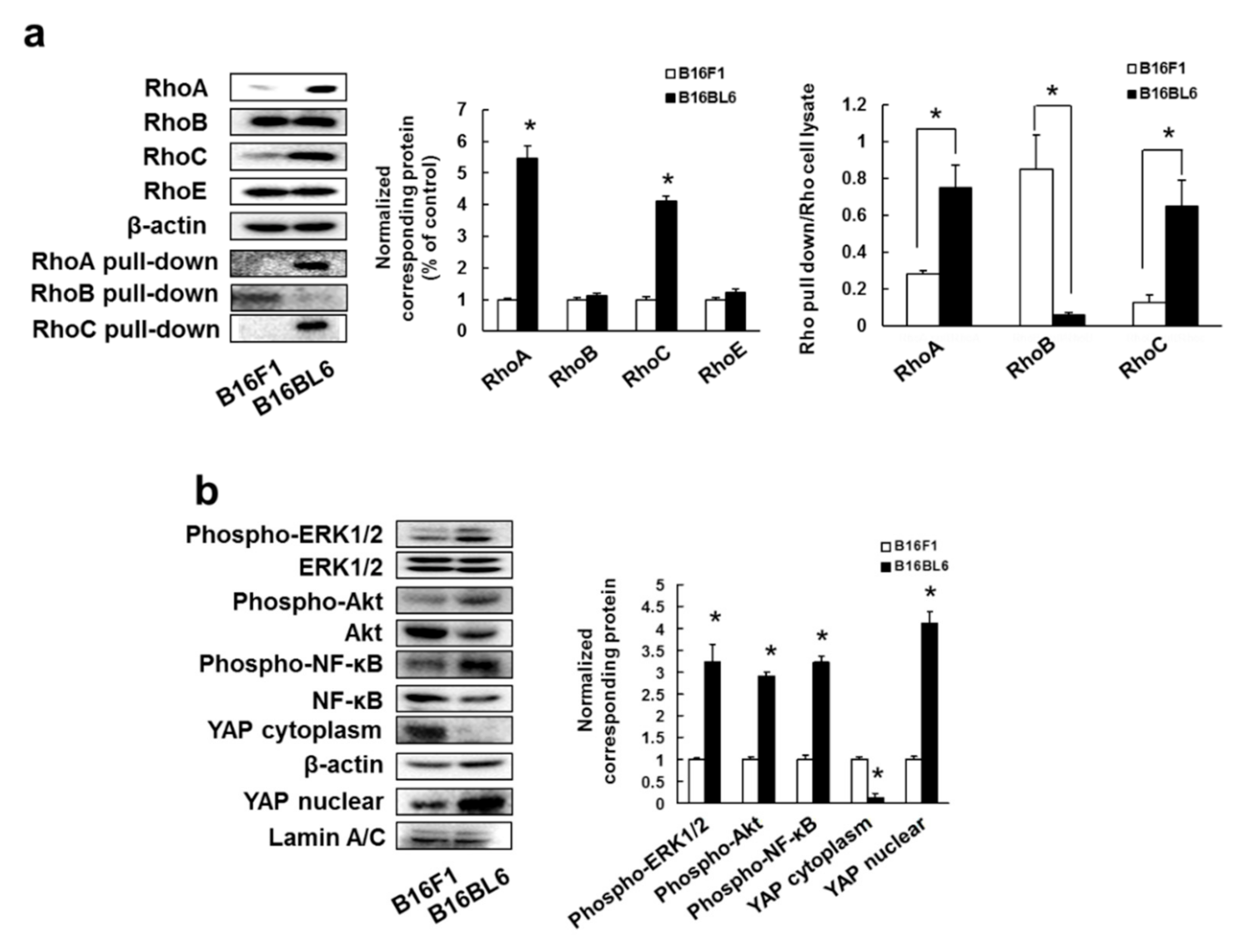
3.1. Overexpression and Activation of RhoA and RhoC, and Their Downstream Signaling Molecules Contribute to High Metastatic Potential
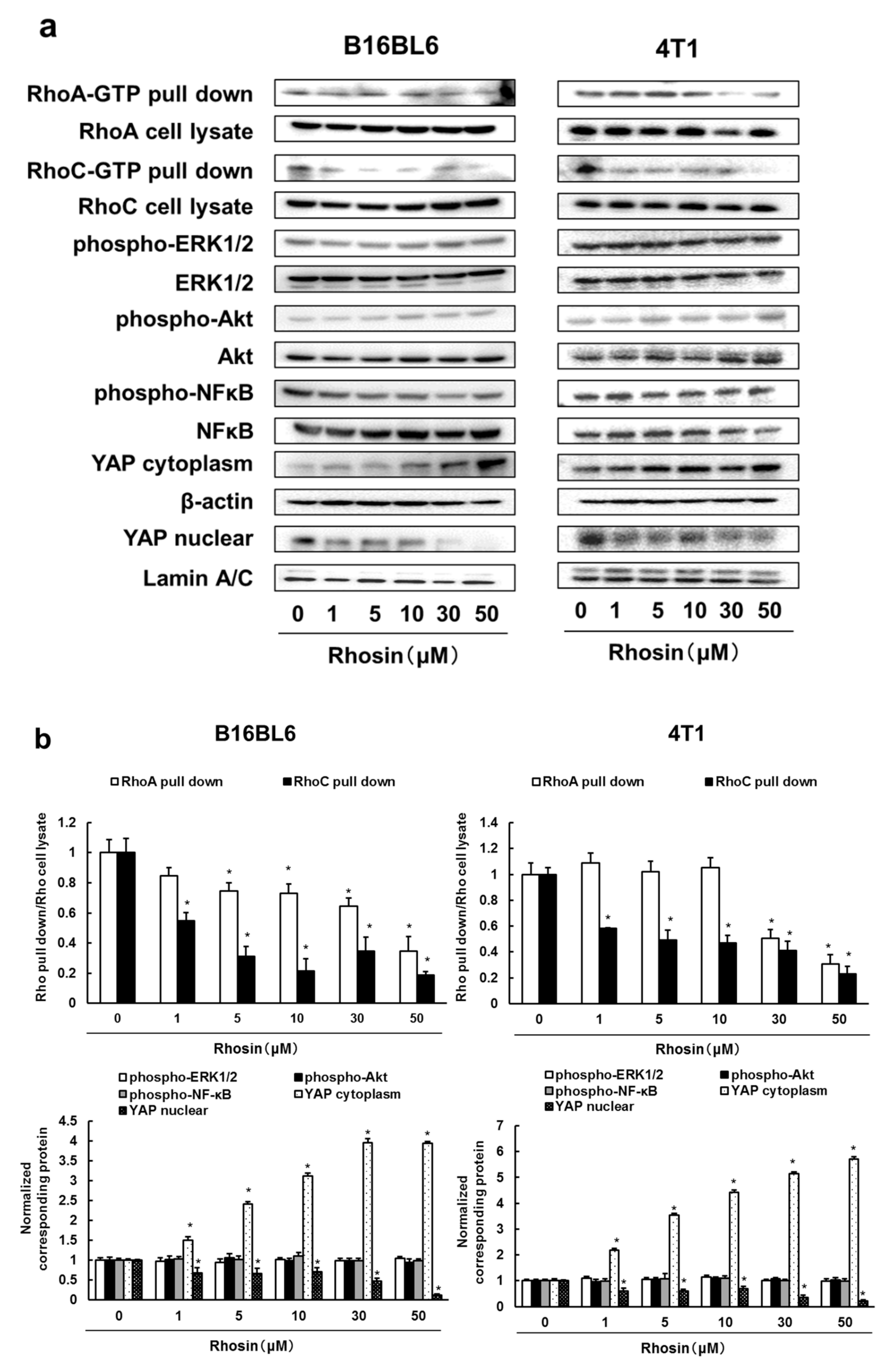
3.2. Rhosin Inhibits YAP Activation via Inhibition of RhoA and RhoC
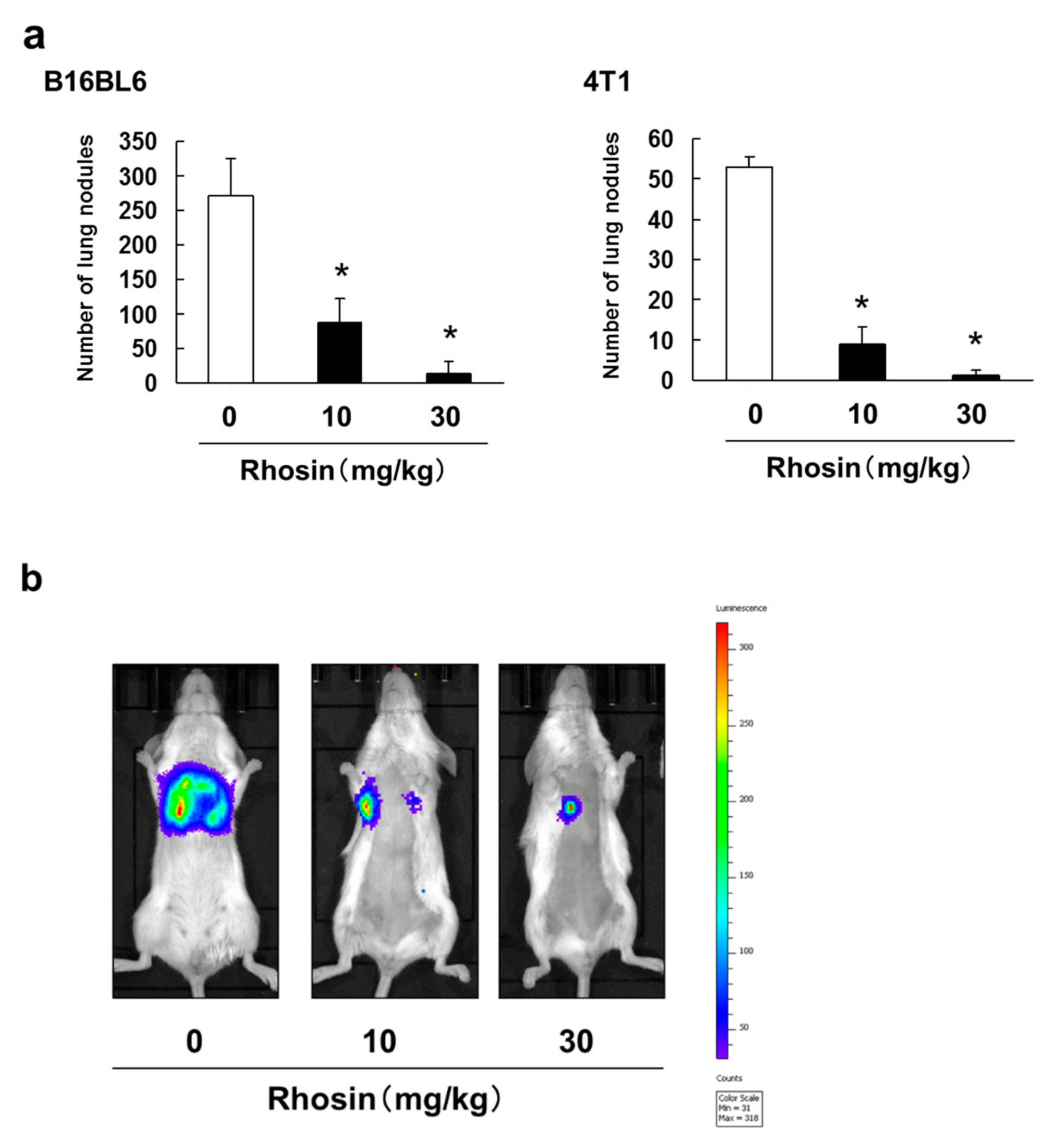
3.3. Inhibitory Effect of Rhosin on Lung Metastasis in Mice Injected with B16BL6 and 4T1 Cells
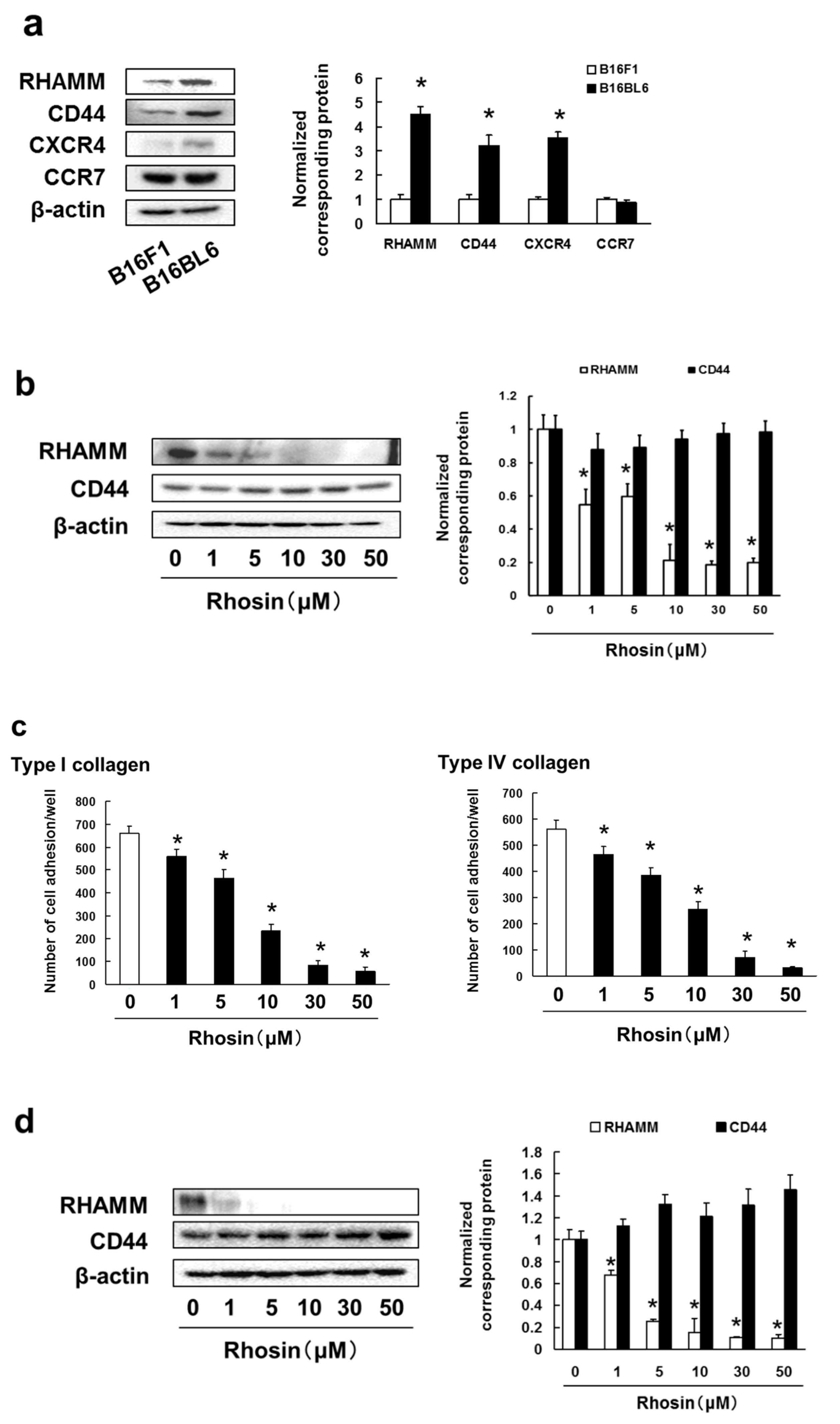
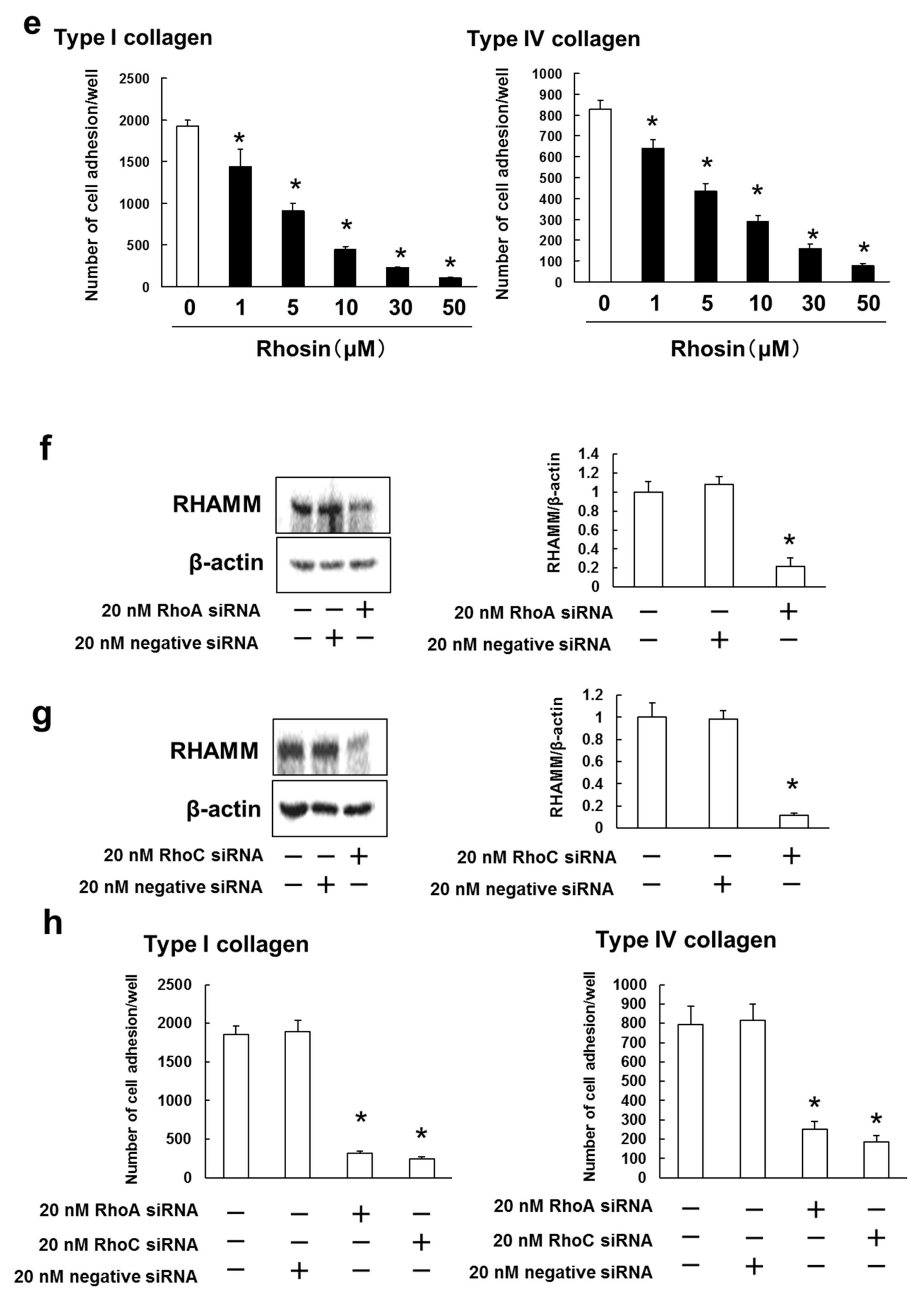
3.4. B16BL6 Cells Have Increased Expression of RHAMM, CD44 and CXCR4, and Rhosin Suppressed Cell Adhesion to Type I and IV Collagen via Inhibition of RHAMM Expression
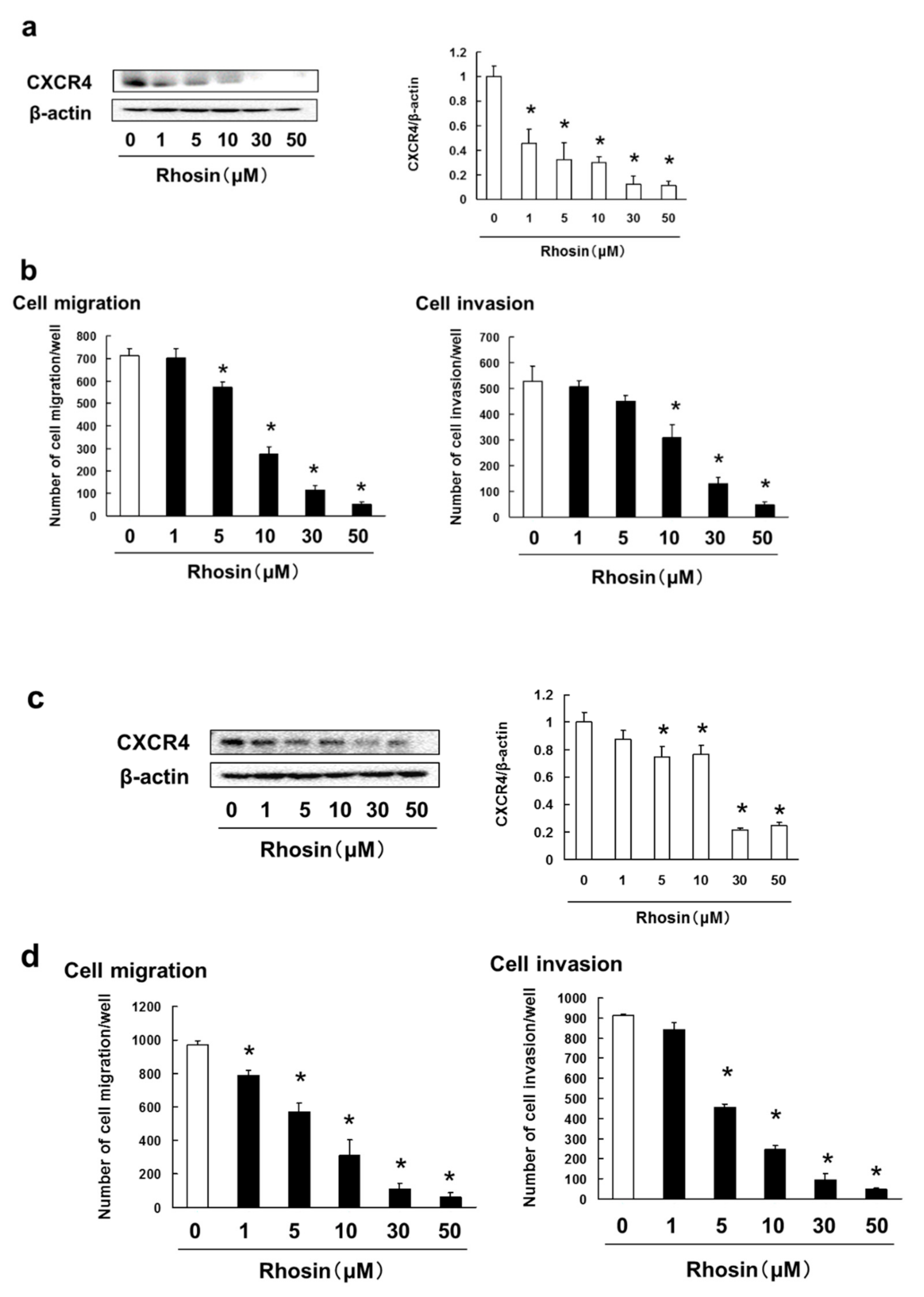
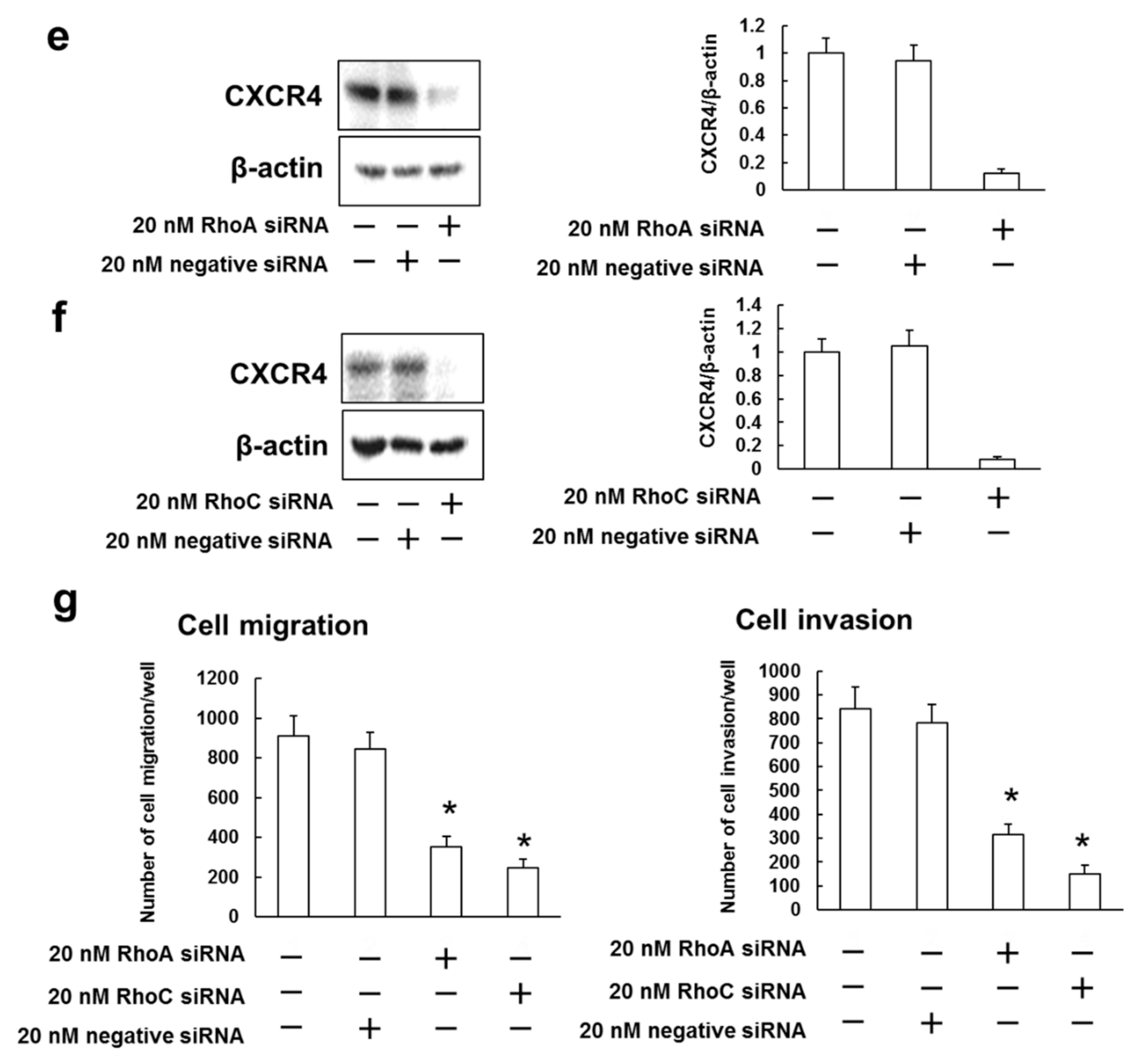
3.5. Rhosin Suppressed Cell Migration and Invasion via Inhibition of CXCR4 Expression
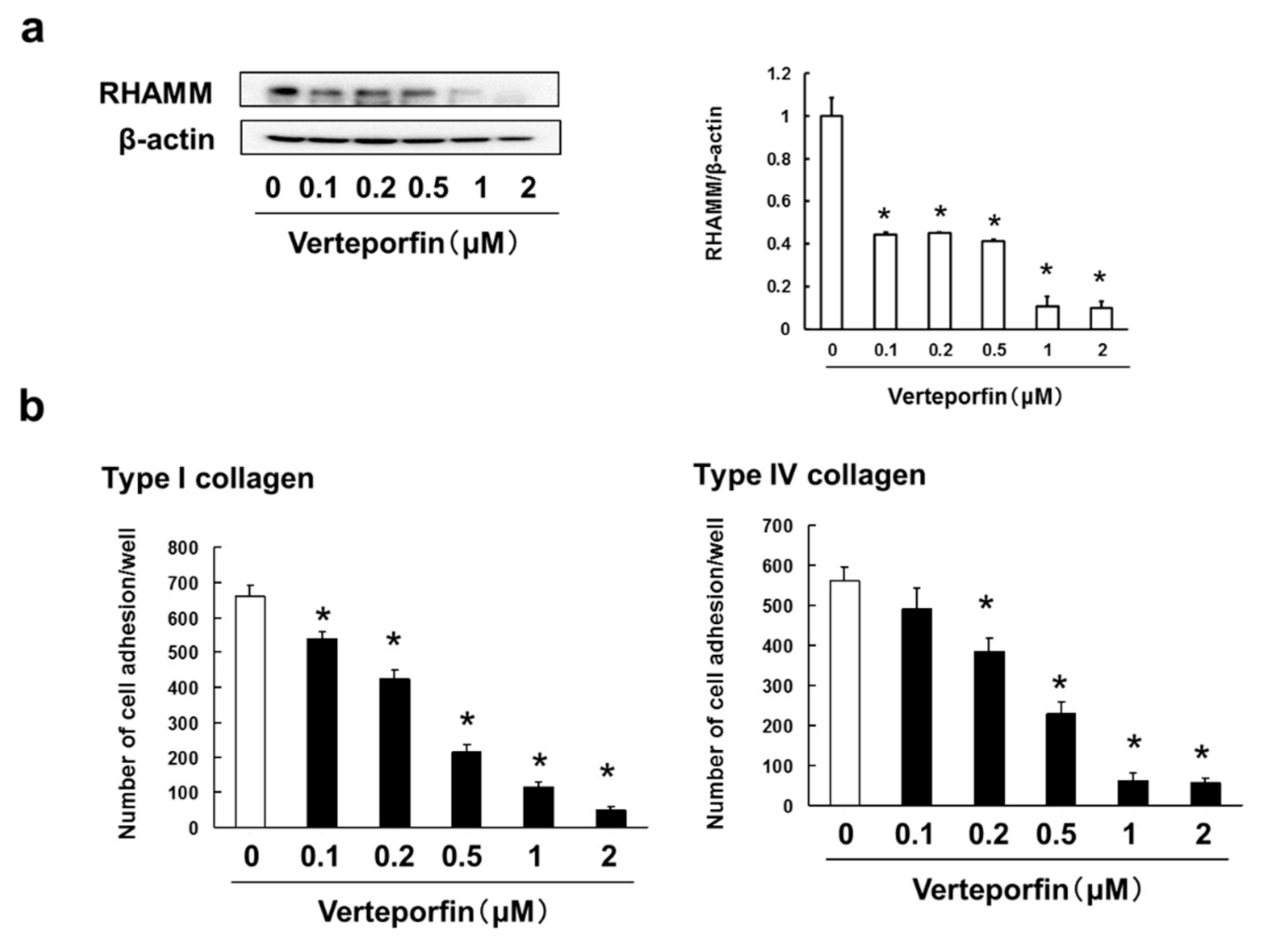
3.6. Verteporfin, a YAP Inhibitor, Suppressed the Cell Adhesion, Migration and Invasion via Inhibition of RHAMM and CXCR4 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.R.; Zhang, X.; Li, L.; Schaider, H.; Wells, J.W. Overcoming resistance to targeted therapy with immunotherapy and combination therapy for metastatic melanoma. Oncotarget 2017, 8, 75675–75686. [Google Scholar] [CrossRef]
- Young, G.J.; Bi, W.L.; Wu, W.W.; Johanns, T.M.; Dunn, G.P.; Dunn, I.F. Management of intracranial melanomas in the era of precision medicine. Oncotarget 2017, 8, 89326–89347. [Google Scholar] [CrossRef][Green Version]
- Prasad, V.; Kaestner, V. Nivolumab and pembrolizumab: Monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin. Oncol. 2017, 44, 132–135. [Google Scholar] [CrossRef]
- Li, J.; Kan, H.; Zhao, L.; Sun, Z.; Bai, C. Immune checkpoint inhibitors in advanced or metastatic mucosal melanoma: A systematic review. Ther. Adv. Med. Oncol. 2020, 12, 1758835920922028. [Google Scholar] [CrossRef]
- Olino, K.; Park, T.; Ahuja, N. Exposing Hidden Targets: Combining epigenetic and immunotherapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 114–122. [Google Scholar] [CrossRef]
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071. [Google Scholar] [CrossRef]
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Wong, G.; Earle, C.; Krueger, K.; Spevak, C.C. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J. Biol. Chem. 2010, 285, 36721–36735. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Liu, N.; Gu, Y.; Qiu, X.; Wang, E.H. PRL-3 facilitates angiogenesis and metastasis by increasing ERK phosphorylation and up-regulating the levels and activities of Rho-A/C in lung cancer. Pathology 2009, 41, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.J.; Li, J.A.; Bai, D.M.; Song, Y. miR-223-RhoB signaling pathway regulates the proliferation and apoptosis of colon adenocarcinoma. Chem. Biol. Interact. 2018, 289, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef]
- Ma, Y.; Gong, Y.; Cheng, Z.; Loganathan, S.; Kao, C.; Sarkaria, J.N.; Abel, T.W.; Wang, J. Critical functions of RhoB in support of glioblastoma tumorigenesis. Neuro Oncol. 2015, 17, 516–525. [Google Scholar] [CrossRef]
- Feng, B.; Li, K.; Zhong, H.; Ren, G.; Wang, H.; Shang, Y.; Bai, M.; Liang, J.; Wang, X.; Fan, D. RhoE promotes metastasis in gastric cancer through a mechanism dependent on enhanced expression of CXCR4. PLoS ONE 2013, 8, e81709. [Google Scholar] [CrossRef]
- Klein, R.M.; Aplin, A.E. Rnd3 regulation of the actin cytoskeleton promotes melanoma migration and invasive outgrowth in three dimensions. Cancer Res. 2009, 69, 2224–2233. [Google Scholar] [CrossRef]
- Hernández-Sánchez, M.; Poch, E.; Guasch, R.M.; Ortega, J.; López-Almela, I.; Palmero, I.; Pérez-Roger, I. RhoE is required for contact inhibition and negatively regulates tumor initiation and progression. Oncotarget 2015, 6, 17479–17490. [Google Scholar] [CrossRef]
- Bist, P.; Phua, Q.H.; Shu, S.; Yi, Y.; Anbalagan, D.; Lee, L.H.; Sethi, G.; Low, B.C.; Lim, L.H. Annexin-A1 controls an ERK-RhoA-NFκB activation loop in breast cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, S.; Wang, Z.; Wang, F.; Cao, X.; Wu, Q.; Zhao, C.; Ma, H.; Ye, F.; Wang, H.; et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J. Exp. Clin. Cancer Res. 2018, 37, 128. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Xiang, Z.; Yu, H.; Xu, L.; Guo, Y.; Tian, Y.; Shu, R.; Yang, X.; Xue, C.; et al. YAP regulates periodontal ligament cell differentiation into myofibroblast interacted with RhoA/ROCK pathway. J. Cell. Physiol. 2019, 234, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Xia, H.; Xu, H.; Ma, J.; Zhou, S.; Hou, W.; Tang, Q.; Gong, Q.; Nie, Y.; Bi, F. Standard CD44 modulates YAP1 through a positive feedback loop in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 103, 147–156. [Google Scholar] [CrossRef]
- Maolake, A.; Izumi, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Makino, T.; Naito, R.; Shigehara, K.; Kadono, Y.; Hiratsuka, K.; et al. Tumor necrosis factor-α induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 2018, 109, 1524–1531. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Cai, J.; Du, S.; Xin, B.; Wei, W.; Zhang, T.; Shen, X. Artemin regulates CXCR4 expression to induce migration and invasion in pancreatic cancer cells through activation of NF-κB signaling. Exp. Cell Res. 2018, 365, 12–23. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef]
- Luo, J.; Li, D.; Wei, D.; Wang, X.; Wang, L.; Zeng, X. RhoA and RhoC are involved in stromal cell-derived factor-1-induced cell migration by regulating F-actin redistribution and assembly. Mol. Cell. Biochem. 2017, 436, 13–21. [Google Scholar] [CrossRef]
- Shigeishi, H.; Higashikawa, K.; Takechi, M. Role of receptor for hyaluronan-mediated motility (RHAMM) in human head and neck cancers. J. Cancer Res. Clin. Oncol. 2014, 140, 1629–1640. [Google Scholar] [CrossRef]
- Yu, S.; Cai, X.; Wu, C.; Wu, L.; Wang, Y.; Liu, Y.; Yu, Z.; Qin, S.; Ma, F.; Thiery, J.P.; et al. Adhesion glycoprotein CD44 functions as an upstream regulator of a network connecting ERK, AKT and Hippo-YAP pathways in cancer progression. Oncotarget 2015, 6, 2951–2965. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Mu, G.; Ding, Q.; Li, H.; Zhang, L.; Zhang, L.; He, K.; Wu, L.; Deng, Y.; Yang, D.; Wu, L.; et al. Gastrin stimulates pancreatic cancer cell directional migration by activating the Gα12/13-RhoA-ROCK signaling pathway. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Nakajima, M.; Naito, M.; Muto, T.; Tsuruo, T. Identification of genes differentially expressed in B16 murine melanoma sublines with different metastatic potentials. Cancer Res. 1996, 56, 875–879. [Google Scholar] [PubMed]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Obata, N.; Kawashima, K.; Tabata, M.; Imano, M.; Satoh, T.; Nishida, S. Combination therapy with dacarbazine and statins improved the survival rate in mice with metastatic melanoma. J. Cell. Physiol. 2019, 234, 17975–17989. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef]
- Jönsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringnér, M.; Lillehaug, J.R.; Borg, A.; Lønning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Wirapati, P.; Lallemand, F.; Tutt, A.M.; Gillet, C.; Ellis, P.; Ryder, K.; Reid, J.F.; et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genom. 2008, 9, 239. [Google Scholar] [CrossRef]
- Schmidt, M.; Böhm, D.; von Törne, C.; Steiner, E.; Puhl, A.; Pilch, H.; Lehr, H.A.; Hengstler, J.G.; Kölbl, H.; Gehrmann, M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008, 68, 5405–5413. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Mouse models for tumor metastasis. Methods Mol. Biol. 2012, 928, 221–228. [Google Scholar]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2000, 39, 20.2.1–20.2.16. [Google Scholar] [CrossRef]
- Chung, T.W.; Choi, H.J.; Lee, J.Y.; Jeong, H.S.; Kim, C.H.; Joo, M.; Choi, J.Y.; Han, C.W.; Kim, S.Y.; Choi, J.S.; et al. Marine algal fucoxanthin inhibits the metastatic potential of cancer cells. Biochem. Biophys. Res. Commun. 2013, 439, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Y.; Yang, L.X.; Wang, Z.C.; Wang, L.Y.; Zhou, J.; Wang, X.Y.; Shi, G.M.; Ding, Z.B.; Ke, A.W.; Dai, Z.; et al. CC chemokine receptor-like 1 functions as a tumour suppressor by impairing CCR7-related chemotaxis in hepatocellular carcinoma. J. Pathol. 2015, 235, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Kouvidi, K.; Berdiaki, A.; Nikitovic, D.; Katonis, P.; Afratis, N.; Hascall, V.C.; Karamanos, N.K.; Tzanakakis, G.N. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J. Biol. Chem. 2011, 286, 38509–38520. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Hwang, D.; Lee, D.; Kim, J.H.; Kim, S.Y.; Lim, D.S. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017, 36, 520–535. [Google Scholar] [CrossRef]
- Wang, H.B.; Liu, X.P.; Liang, J.; Yang, K.; Sui, A.H.; Liu, Y.J. Expression of RhoA and RhoC in colorectal carcinoma and its relations with clinicopathological parameters. Clin. Chem. Lab. Med. 2009, 47, 811–817. [Google Scholar] [CrossRef][Green Version]
- Boone, B.; Van Gele, M.; Lambert, J.; Haspeslagh, M.; Brochez, L. The role of RhoC in growth and metastatic capacity of melanoma. J. Cutan. Pathol. 2009, 36, 629–636. [Google Scholar] [CrossRef]
- Kidera, Y.; Tsubaki, M.; Yamazoe, Y.; Shoji, K.; Nakamura, H.; Ogaki, M.; Satou, T.; Itoh, T.; Isozaki, M.; Kaneko, J.; et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 2010, 29, 127. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Kino, T.; Obata, N.; Itoh, T.; Imano, M.; Mashimo, K.; Fujiwara, D.; Sakaguchi, K.; Satou, T.; et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am. J. Cancer Res. 2015, 5, 3186–3197. [Google Scholar]
- Yang, H.; Zhou, J.; Mi, J.; Ma, K.; Fan, Y.; Ning, J.; Wang, C.; Wei, X.; Zhao, H.; Li, E. HOXD10 acts as a tumor-suppressive factor via inhibition of the RHOC/AKT/MAPK pathway in human cholangiocellular carcinoma. Oncol. Rep. 2015, 34, 1681–1691. [Google Scholar] [CrossRef]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (YAP) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal Transduct. Target Ther. 2018, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef] [PubMed]
- Nedvetzki, S.; Gonen, E.; Assayag, N.; Reich, R.; Williams, R.O.; Thurmond, R.L.; Huang, J.F.; Neudecker, B.A.; Wang, F.S.; Turley, E.A.; et al. RHAMM, a receptor for hyaluronan-mediated motility, compensates for CD44 in inflamed CD44-knockout mice: A different interpretation of redundancy. Proc. Natl. Acad. Sci. USA 2004, 101, 18081–18086. [Google Scholar] [CrossRef]
- Mele, V.; Sokol, L.; Kölzer, V.H.; Pfaff, D.; Muraro, M.G.; Keller, I.; Stefan, Z.; Centeno, I.; Terracciano, L.M.; Dawson, H.; et al. The hyaluronan-mediated motility receptor RHAMM promotes growth, invasiveness and dissemination of colorectal cancer. Oncotarget 2017, 8, 70617–70629. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Thor, A.D.; Moore, D.H., 2nd; Zhao, Y.; Kerschmann, R.; Stern, R.; Watson, P.H.; Turley, E.A. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin. Cancer Res. 1998, 4, 567–576. [Google Scholar] [PubMed]
- Saur, D.; Seidler, B.; Schneider, G.; Algül, H.; Beck, R.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Schmid, R.M. CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology 2005, 129, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef]
- Jansen, S.; Gosens, R.; Wieland, T.; Schmidt, M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018, 183, 1–21. [Google Scholar] [CrossRef]
- Xiang, J.; Hurchla, M.A.; Fontana, F.; Su, X.; Amend, S.R.; Esser, A.K.; Douglas, G.J.; Mudalagiriyappa, C.; Luker, K.E.; Pluard, T.; et al. CXCR4 Protein Epitope Mimetic Antagonist POL5551 Disrupts Metastasis and Enhances Chemotherapy Effect in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 2473–2485. [Google Scholar] [CrossRef] [PubMed]
- Zucchini, C.; Manara, M.C.; Cristalli, C.; Carrabotta, M.; Greco, S.; Pinca, R.S.; Ferrari, C.; Landuzzi, L.; Pasello, M.; Lollini, P.L.; et al. ROCK2 deprivation leads to the inhibition of tumor growth and metastatic potential in osteosarcoma cells through the modulation of YAP activity. J. Exp. Clin. Cancer Res. 2019, 38, 503. [Google Scholar] [CrossRef] [PubMed]
- García, P.; Rosa, L.; Vargas, S.; Weber, H.; Espinoza, J.A.; Suárez, F.; Romero-Calvo, I.; Elgueta, N.; Rivera, V.; Nervi, B.; et al. Hippo-YAP1 Is a Prognosis Marker and Potentially Targetable Pathway in Advanced Gallbladder Cancer. Cancers 2020, 12, 778. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, Y.; Li, X.; Yang, R.; Zhang, L.; Huangfu, L.; Zheng, N.; Zhao, X.; Lv, L.; Hong, Y.; et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co-factor TEAD. Cell Death Dis. 2018, 9, 464. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubaki, M.; Genno, S.; Takeda, T.; Matsuda, T.; Kimura, N.; Yamashita, Y.; Morii, Y.; Shimomura, K.; Nishida, S. Rhosin Suppressed Tumor Cell Metastasis through Inhibition of Rho/YAP Pathway and Expression of RHAMM and CXCR4 in Melanoma and Breast Cancer Cells. Biomedicines 2021, 9, 35. https://doi.org/10.3390/biomedicines9010035
Tsubaki M, Genno S, Takeda T, Matsuda T, Kimura N, Yamashita Y, Morii Y, Shimomura K, Nishida S. Rhosin Suppressed Tumor Cell Metastasis through Inhibition of Rho/YAP Pathway and Expression of RHAMM and CXCR4 in Melanoma and Breast Cancer Cells. Biomedicines. 2021; 9(1):35. https://doi.org/10.3390/biomedicines9010035
Chicago/Turabian StyleTsubaki, Masanobu, Shuuji Genno, Tomoya Takeda, Takuya Matsuda, Naoto Kimura, Yuuma Yamashita, Yuusuke Morii, Kazunori Shimomura, and Shozo Nishida. 2021. "Rhosin Suppressed Tumor Cell Metastasis through Inhibition of Rho/YAP Pathway and Expression of RHAMM and CXCR4 in Melanoma and Breast Cancer Cells" Biomedicines 9, no. 1: 35. https://doi.org/10.3390/biomedicines9010035
APA StyleTsubaki, M., Genno, S., Takeda, T., Matsuda, T., Kimura, N., Yamashita, Y., Morii, Y., Shimomura, K., & Nishida, S. (2021). Rhosin Suppressed Tumor Cell Metastasis through Inhibition of Rho/YAP Pathway and Expression of RHAMM and CXCR4 in Melanoma and Breast Cancer Cells. Biomedicines, 9(1), 35. https://doi.org/10.3390/biomedicines9010035