Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-κB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability
2.4. Western Blotting
2.5. Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.6. Proteasome Activity Assay
2.7. Autophagy Assay
2.8. Luminex Assay
2.9. Gene Expression Omnibus (GEO) Data Set
2.10. Statistical Analysis
3. Results
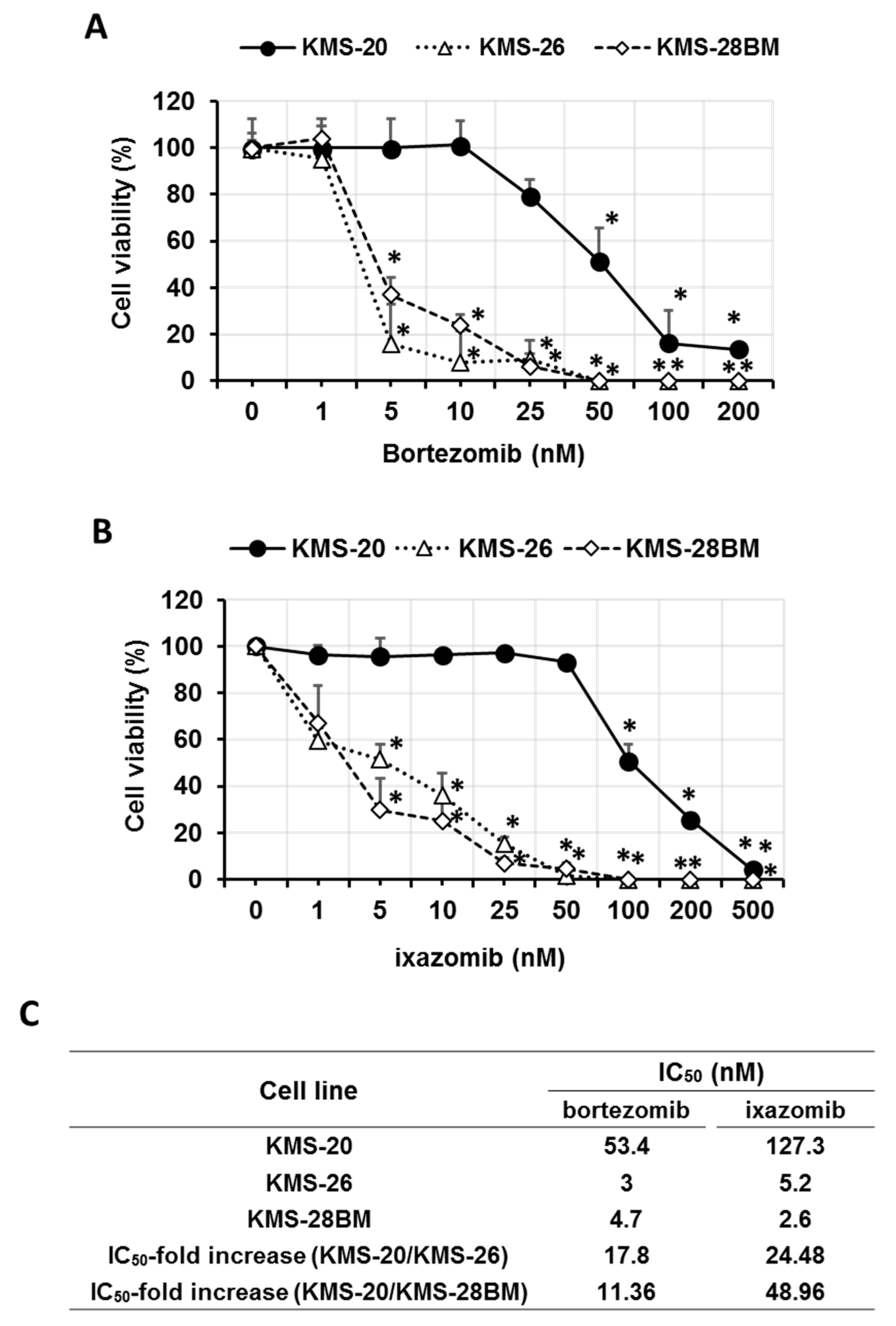
3.1. Sensitivity of Multiple Myeloma (MM) Cells to Bortezomib and Ixazomib
3.2. Expression and Activity of Proteasome β5 Subunit and Effect of Autophagy on Bortezomib- and Ixazomib-Treated Multiple Myeloma (MM) Cells
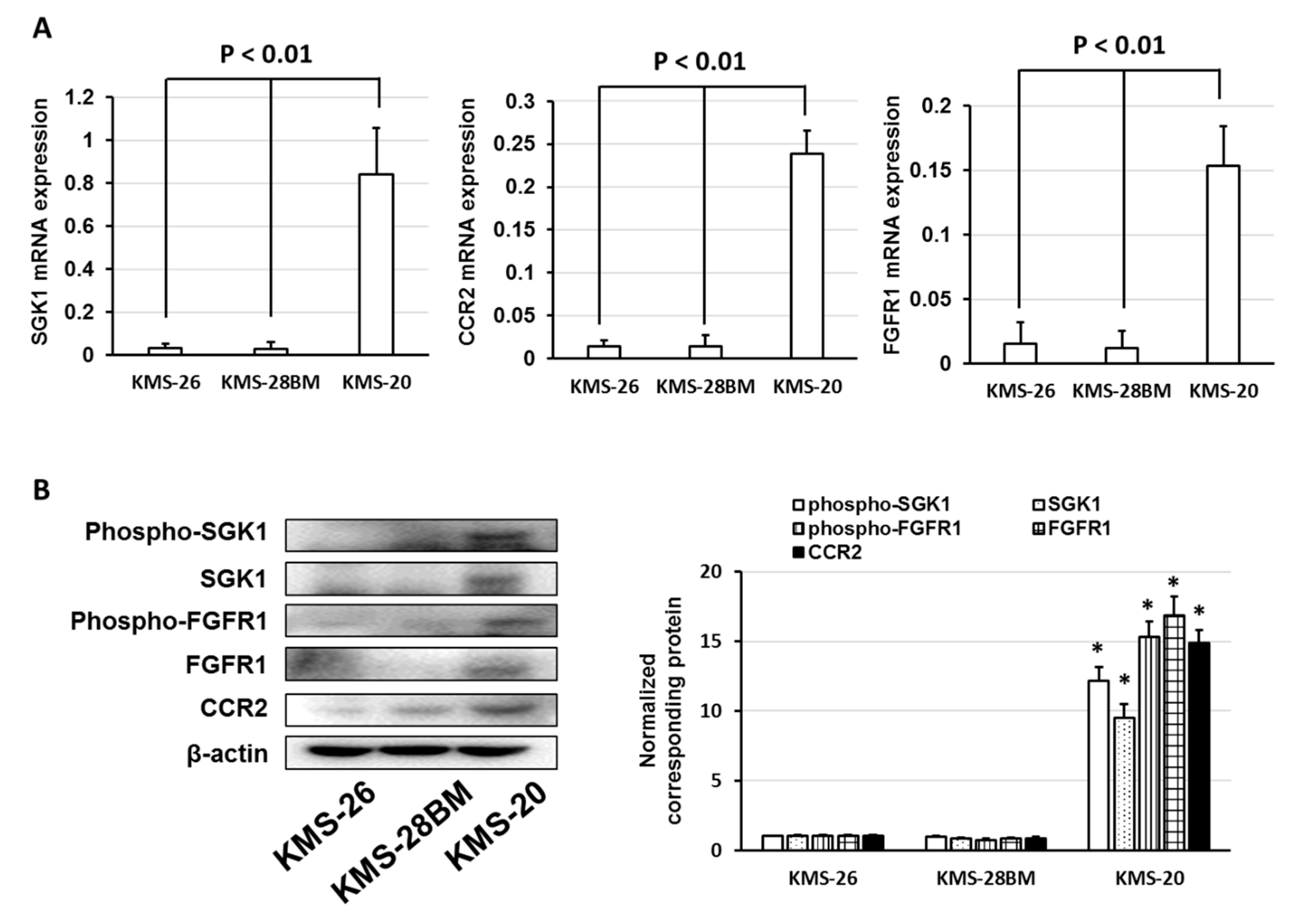
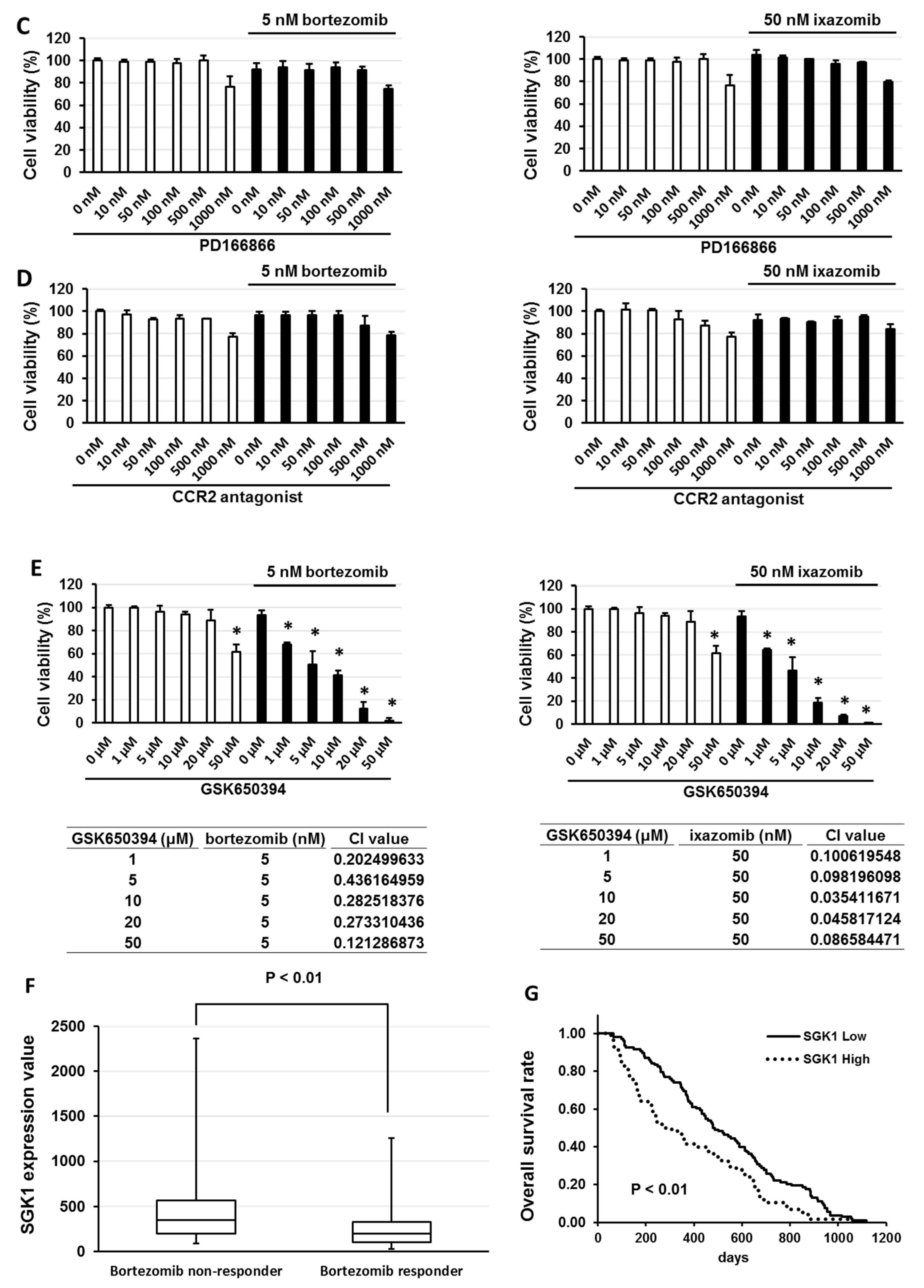
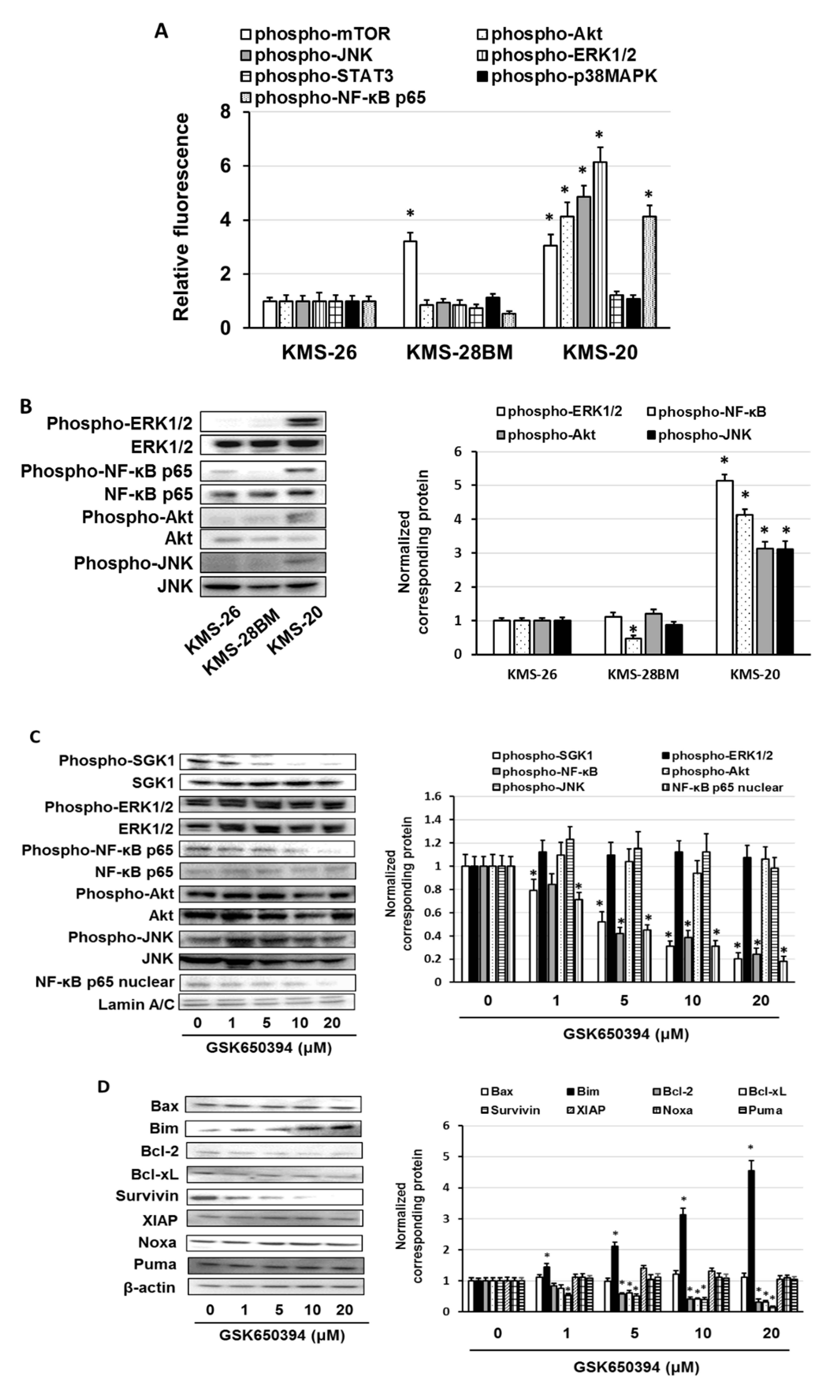
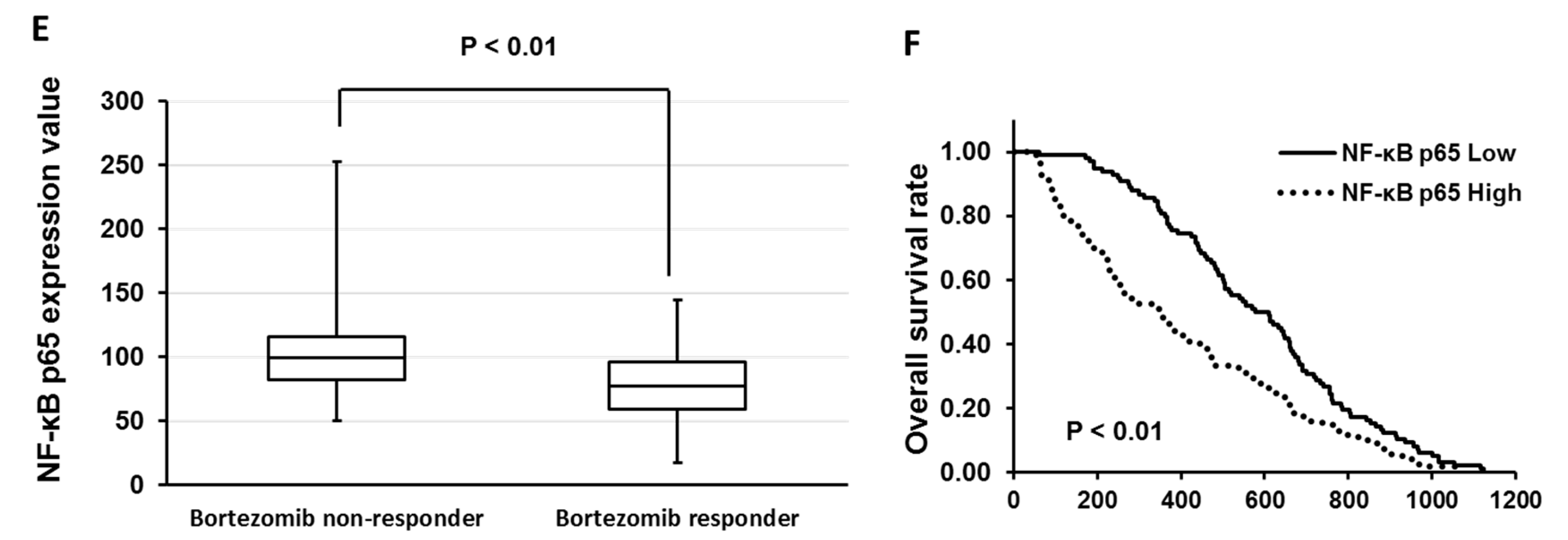
3.3. Overexpression of Anti-Phospho-Serum and Glucocorticoid-Regulated Kinase (SGK1) Correlated with Low Sensitivity of Bortezomib and Ixazomib in Multiple Myeloma (MM) Cells
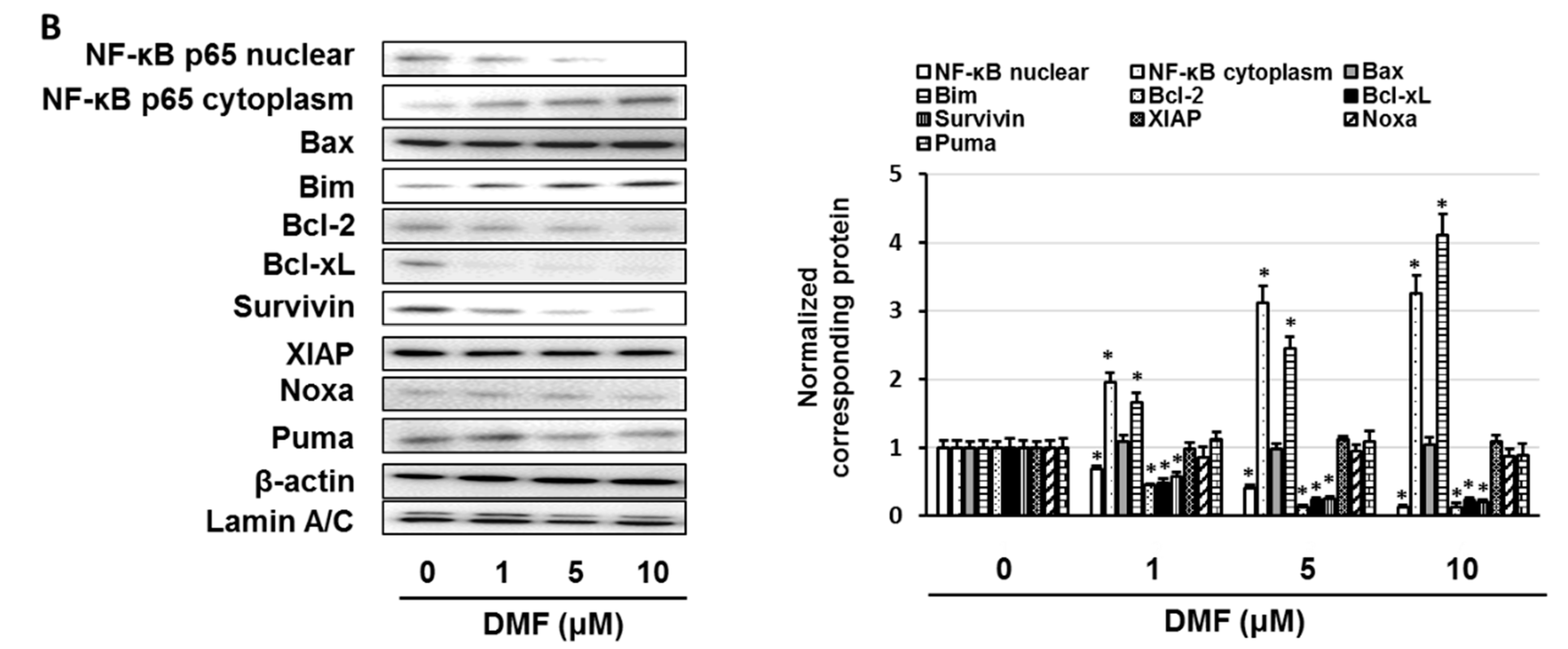
3.4. GSK650394 Inhibited Nuclear Factor (NF)-κB Activation and Regulated the Expression of Cell Survival-Related Molecules
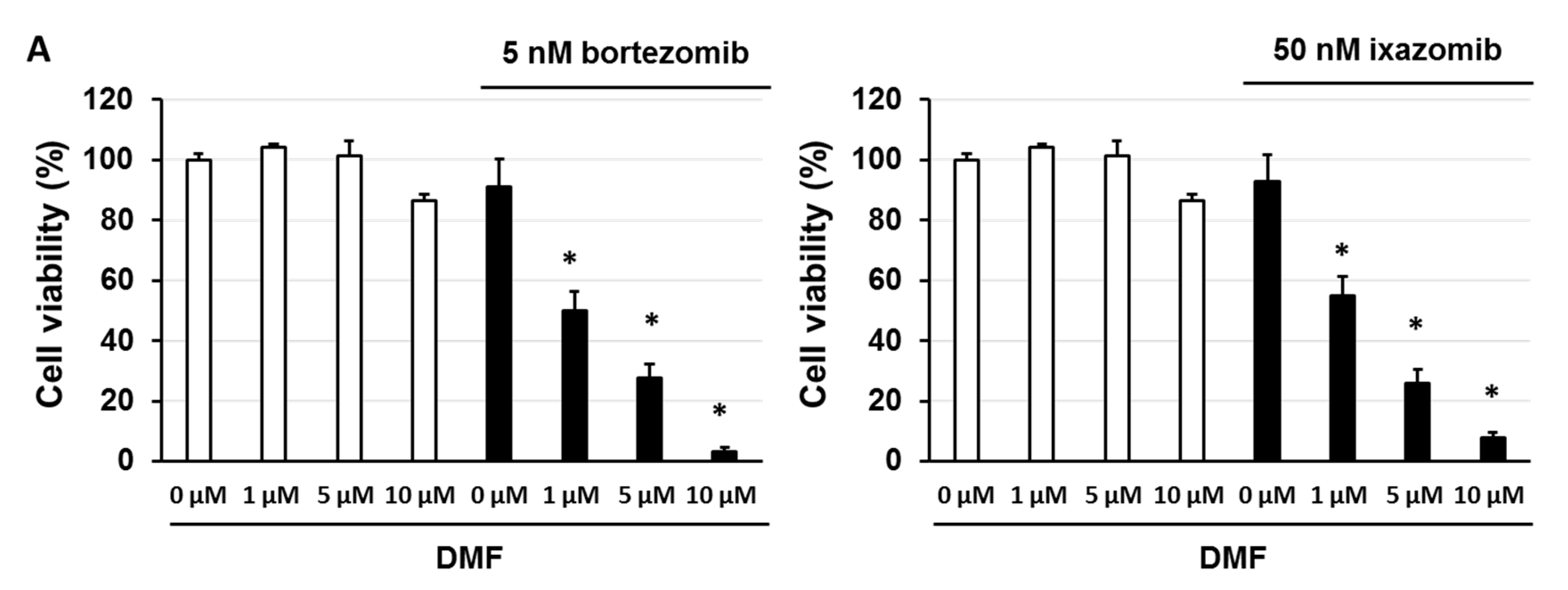
3.5. Dimethyl Fumarate (DMF), a Nuclear Factor (NF)-κB Inhibitor, Enhanced the Sensitivity of KMS-20 Cells to Bortezomib and Ixazomib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Myeloma today: Disease definitions and treatment advances. Am. J. Hematol. 2016, 91, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.; Baughn, L.; Kumar, S.; Van Ness, B. The future of myeloma precision medicine: Integrating the compendium of known drug resistance mechanisms with emerging tumor profiling technologies. Leukemia 2019, 33, 863–883. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef]
- Reddy, N.; Czuczman, M.S. Enhancing activity and overcoming chemoresistance in hematologic malignancies with bortezomib: Preclinical mechanistic studies. Ann. Oncol. 2010, 21, 1756–1764. [Google Scholar] [CrossRef]
- Terpos, E.; Ramasamy, K.; Maouche, N.; Minarik, J.; Ntanasis-Stathopoulos, I.; Katodritou, E.; Jenner, M.W.; Plonkova, H.; Gavriatopoulou, M.; Vallance, G.D.; et al. Real-world effectiveness and safety of ixazomib-lenalidomide-dexamethasone in relapsed/refractory multiple myeloma. Ann. Hematol. 2020, 99, 1049–1061. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Bahlis, N.J.; Chng, W.J.; Masszi, T.; Viterbo, L.; Pour, L.; Ganly, P.; Palumbo, A.; Cavo, M.; Langer, C.; et al. Ixazomib significantly prolongs progression-free survival in high-risk relapsed/refractory myeloma patients. Blood 2017, 130, 2610–2618. [Google Scholar] [CrossRef]
- Kumar, S.K.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012, 26, 149–157. [Google Scholar] [CrossRef]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Brünnert, D.; Kraus, M.; Stühmer, T.; Kirner, S.; Heiden, R.; Goyal, P.; Driessen, C.; Bargou, R.C.; Chatterjee, M. Novel cell line models to study mechanisms and overcoming strategies of proteasome inhibitor resistance in multiple myeloma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1666–1676. [Google Scholar] [CrossRef]
- Balsas, P.; Galán-Malo, P.; Marzo, I.; Naval, J. Bortezomib resistance in a myeloma cell line is associated to PSMβ5 overexpression and polyploidy. Leuk. Res. 2012, 36, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Niewerth, D.; Jansen, G.; Riethoff, L.F.; van Meerloo, J.; Kale, A.J.; Moore, B.S.; Assaraf, Y.G.; Anderl, J.L.; Zweegman, S.; Kaspers, G.J.; et al. Antileukemic activity and mechanism of drug resistance to the marine Salinispora tropica proteasome inhibitor salinosporamide A (Marizomib). Mol. Pharmacol. 2014, 86, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef]
- Lang, F.; Voelkl, J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin. Investig. Drugs 2013, 22, 701–714. [Google Scholar] [CrossRef]
- Fagerli, U.M.; Ullrich, K.; Stühmer, T.; Holien, T.; Köchert, K.; Holt, R.U.; Bruland, O.; Chatterjee, M.; Nogai, H.; Lenz, G.; et al. Serum/glucocorticoid-regulated kinase 1 (SGK1) is a prominent target gene of the transcriptional response to cytokines in multiple myeloma and supports the growth of myeloma cells. Oncogene 2011, 30, 3198–3206. [Google Scholar] [CrossRef]
- Hoang, B.; Shi, Y.; Frost, P.J.; Mysore, V.; Bardeleben, C.; Lichtenstein, A. SGK Kinase Activity in Multiple Myeloma Cells Protects against ER Stress Apoptosis via a SEK-Dependent Mechanism. Mol. Cancer Res. 2016, 14, 397–407. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Noguchi, M.; Jinushi, M.; Seki, S.; Morii, Y.; Shimomura, K.; Imano, M.; Satou, T.; Nishida, S. Overactivation of Akt Contributes to MEK Inhibitor Primary and Acquired Resistance in Colorectal Cancer Cells. Cancers 2019, 11, 1866. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Tomonari, Y.; Koumoto, Y.I.; Imano, M.; Satou, T.; Nishida, S. Overexpression of HIF-1α contributes to melphalan resistance in multiple myeloma cells by activation of ERK1/2, Akt, and NF-κB. Lab. Investig. 2019, 99, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Tomonari, Y.; Mashimo, K.; Koumoto, Y.I.; Hoshida, S.; Itoh, T.; Imano, M.; Satou, T.; Sakaguchi, K.; et al. The MIP-1α autocrine loop contributes to decreased sensitivity to anticancer drugs. J. Cell. Physiol. 2018, 233, 4258–4271. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Kino, T.; Sakai, K.; Itoh, T.; Imano, M.; Nakayama, T.; Nishio, K.; Satou, T.; Nishida, S. Contributions of MET activation to BCR-ABL1 tyrosine kinase inhibitor resistance in chronic myeloid leukemia cells. Oncotarget 2017, 8, 38717–38730. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Mashimo, K.; Takeda, T.; Kino, T.; Fujita, A.; Itoh, T.; Imano, M.; Sakaguchi, K.; Satou, T.; Nishida, S. Statins inhibited the MIP-1α expression via inhibition of Ras/ERK and Ras/Akt pathways in myeloma cells. Biomed. Pharmacother. 2016, 78, 23–29. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Ogawa, N.; Sakamoto, K.; Shimaoka, H.; Fujita, A.; Itoh, T.; Imano, M.; Ishizaka, T.; Satou, T.; et al. Overexpression of survivin via activation of ERK1/2, Akt, and NF-κB plays a central role in vincristine resistance in multiple myeloma cells. Leuk. Res. 2015, 39, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Komai, M.; Nishinobo, M.; Yamashita, M.; Yanae, M.; Yamazoe, Y.; Nishida, S. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk. Res. 2012, 36, 1315–1322. [Google Scholar] [CrossRef]
- Fujiwara, D.; Tsubaki, M.; Takeda, T.; Tomonari, Y.; Koumoto, Y.I.; Sakaguchi, K.; Nishida, S. Statins induce apoptosis through inhibition of Ras signaling pathways and enhancement of Bim and p27 expression in human hematopoietic tumor cells. Tumour Biol. 2017, 39, 1010428317734947. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Boer, J.M.; Besselink, N.J.M.; Koudijs, M.J.; Pieters, R.; Boer, M.L.D. Fibroblast growth factor receptor signaling in pediatric B-cell precursor acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 1875. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Zhu, L.; Somlo, G.; Hughes, A.; Zhou, B.; Yen, Y. Bortezomib therapeutic effect is associated with expression of FGFR3 in multiple myeloma cells. Anticancer Res. 2009, 29, 1–9. [Google Scholar] [PubMed]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019, 110, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Fang, W.; Smart, C.; Hu, Q.; Huang, S.; Alvarez, N.; Fields, P.; Cheng, N. CCR2 Chemokine Receptors Enhance Growth and Cell-Cycle Progression of Breast Cancer Cells through SRC and PKC Activation. Mol. Cancer Res. 2019, 17, 604–617. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Yin, L.; Hou, L.; Lan, J.; Zhu, X. TCRP1 induces tamoxifen resistance by promoting the activation of SGK1 in MCF-7 cells. Oncol. Rep. 2020, 43, 2017–2027. [Google Scholar] [CrossRef]
- Ueki, S.; Fujishima, F.; Kumagai, T.; Ishida, H.; Okamoto, H.; Takaya, K.; Sato, C.; Taniyma, Y.; Kamei, T.; Sasano, H. GR, Sgk1, and NDRG1 in esophageal squamous cell carcinoma: Their correlation with therapeutic outcome of neoadjuvant chemotherapy. BMC Cancer 2020, 20, 161. [Google Scholar] [CrossRef]
- Costa, C.; Bosch, A. The Strategy of PIKing a Target: What Is AKTually Most Effective? Clin. Cancer Res. 2018, 24, 2029–2031. [Google Scholar] [CrossRef]
- Iqbal, S.; Howard, S.; LoGrasso, P.V. Serum- and Glucocorticoid-Inducible Kinase 1 Confers Protection in Cell-Based and in In Vivo Neurotoxin Models via the c-Jun N-Terminal Kinase Signaling Pathway. Mol. Cell. Biol. 2015, 35, 1992–2006. [Google Scholar] [CrossRef]
- Lang, F.; Stournaras, C.; Zacharopoulou, N.; Voelkl, J.; Alesutan, I. Serum- and glucocorticoid-inducible kinase 1 and the response to cell stress. Cell Stress 2018, 3, 1–8. [Google Scholar] [CrossRef]
- You, H.; Jang, Y.; You-Ten, A.I.; Okada, H.; Liepa, J.; Wakeham, A.; Zaugg, K.; Mak, T.W. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc. Natl. Acad. Sci. USA 2004, 101, 14057–14062. [Google Scholar] [CrossRef]
- Lang, F.; Böhmer, C.; Palmada, M.; Seebohm, G.; Strutz-Seebohm, N.; Vallon, V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 2006, 86, 1151–1178. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bonafé, P.; Tortosa, A.; Pérez-Tomás, R. Overcoming drug resistance by enhancing apoptosis of tumor cells. Curr. Cancer Drug Targets 2009, 9, 320–340. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yu, T.T.; Young, K.H. Cross-talk between Myc and p53 in B-cell lymphomas. Chronic Dis. Transl. Med. 2019, 5, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, J.; Liu, Z.; Lu, Y.; Zheng, Y.; Li, H.; Xu, J.; Liu, H.; Qian, J.; Orlowski, R.Z.; et al. Anti-β₂-microglobulin monoclonal antibodies overcome bortezomib resistance in multiple myeloma by inhibiting autophagy. Oncotarget 2015, 6, 8567–8578. [Google Scholar] [CrossRef]
- Vad-Nielsen, J.; Gammelgaard, K.R.; Daugaard, T.F.; Nielsen, A.L. Cause-and-Effect relationship between FGFR1 expression and epithelial-mesenchymal transition in EGFR-mutated non-small cell lung cancer cells. Lung Cancer 2019, 132, 132–140. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef]
- Kim, S.H.; Ryu, H.; Ock, C.Y.; Suh, K.J.; Lee, J.Y.; Kim, J.W.; Lee, J.O.; Kim, J.W.; Kim, Y.J.; Lee, K.W.; et al. BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression. Int. J. Mol. Sci. 2018, 19, 3164. [Google Scholar] [CrossRef]
- Sommer, E.M.; Dry, H.; Cross, D.; Guichard, S.; Davies, B.R.; Alessi, D.R. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem. J. 2013, 452, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, L.; Dattilo, V.; Catalogna, G.; Scumaci, D.; Fiumara, C.V.; Musumeci, F.; Perrotti, G.; Schenone, S.; Tallerico, R.; Spoleti, C.B.; et al. In Preclinical Model of Ovarian Cancer, the SGK1 Inhibitor SI113 Counteracts the Development of Paclitaxel Resistance and Restores Drug Sensitivity. Transl. Oncol. 2019, 12, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A. SGK1: The Dark Side of PI3K Signaling. Curr. Top. Dev. Biol. 2017, 123, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Tsubaki, M.; Tomonari, Y.; Kawashima, K.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Bavachin induces the apoptosis of multiple myeloma cell lines by inhibiting the activation of nuclear factor kappa B and signal transducer and activator of transcription 3. Biomed. Pharmacother. 2018, 100, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Tsubaki, M.; Kino, T.; Kawamura, A.; Isoyama, S.; Itoh, T.; Imano, M.; Tanabe, G.; Muraoka, O.; Matsuda, H.; et al. Mangiferin enhances the sensitivity of human multiple myeloma cells to anticancer drugs through suppression of the nuclear factor κB pathway. Int. J. Oncol. 2016, 48, 2704–2712. [Google Scholar] [CrossRef]
- Takeda, T.; Tsubaki, M.; Kino, T.; Yamagishi, M.; Iida, M.; Itoh, T.; Imano, M.; Tanabe, G.; Muraoka, O.; Satou, T.; et al. Mangiferin induces apoptosis in multiple myeloma cell lines by suppressing the activation of nuclear factor kappa B-inducing kinase. Chem. Biol. Interact. 2016, 251, 26–33. [Google Scholar] [CrossRef]
- Tsubaki, M.; Ogawa, N.; Takeda, T.; Sakamoto, K.; Shimaoka, H.; Fujita, A.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Dimethyl fumarate induces apoptosis of hematopoietic tumor cells via inhibition of NF-κB nuclear translocation and down-regulation of Bcl-xL and XIAP. Biomed. Pharmacother. 2014, 68, 999–1005. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; Takeda, T.; et al. Inhibition of the tumour necrosis factor-alpha autocrine loop enhances the sensitivity of multiple myeloma cells to anticancer drugs. Eur. J. Cancer 2013, 49, 3708–3717. [Google Scholar] [CrossRef]
- Sevilla-Movilla, S.; Arellano-Sánchez, N.; Martínez-Moreno, M.; Gajate, C.; Sánchez-Vencells, A.; Valcárcel, L.V.; Agirre, X.; Valeri, A.; Martínez-López, J.; Prósper, F.; et al. Upregulated expression and function of the α4β1 integrin in multiple myeloma cells resistant to bortezomib. J. Pathol. 2020, 252, 29–40. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Xie, M.J.; Ma, Y.H.; Miao, L.; Wang, Y.; Wang, H.Z.; Xing, Y.Y.; Xi, T.; Lu, Y.Y. Emodin-provoked oxidative stress induces apoptosis in human colon cancer HCT116 cells through a p53-mitochondrial apoptotic pathway. Asian Pac. J. Cancer Prev. 2014, 15, 5201–5205. [Google Scholar] [CrossRef]
- Poulsen, R.C.; Knowles, H.J.; Carr, A.J.; Hulley, P.A. Cell differentiation versus cell death: Extracellular glucose is a key determinant of cell fate following oxidative stress exposure. Cell Death Dis. 2014, 5, e1074. [Google Scholar] [CrossRef] [PubMed]
- Jullien, M.; Gomez-Bougie, P.; Chiron, D.; Touzeau, C. Restoring Apoptosis with BH3 Mimetics in Mature B-Cell Malignancies. Cells 2020, 9, 717. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, A.; Mascitti, M.; Lo Russo, L.; Sartini, D.; Troiano, G.; Emanuelli, M.; Lo Muzio, L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 971. [Google Scholar] [CrossRef]
- Ailawadhi, S.; Miecznikowski, J.; Gaile, D.P.; Wang, D.; Sher, T.; Mulligan, G.; Bryant, B.; Wilding, G.E.; Mashtare, T.; Stein, L.; et al. Bortezomib mitigates adverse prognosis conferred by Bcl-2 overexpression in patients with relapsed/refractory multiple myeloma. Leuk. Lymphoma 2012, 53, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Remily-Wood, E.R.; Oliveira, V.; Yarde, D.; He, L.; Cheng, J.Q.; Mathews, L.; Boucher, K.; Cubitt, C.; Perez, L.; et al. Monitoring a nuclear factor-κB signature of drug resistance in multiple myeloma. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y.; Zhou, L.; Leng, Y.; Lin, H.; Kmieciak, M.; Pei, X.Y.; Jones, R.; Orlowski, R.Z.; Dai, Y.; et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014, 124, 2687–2697. [Google Scholar] [CrossRef]
- Duvefelt, C.F.; Lub, S.; Agarwal, P.; Arngården, L.; Hammarberg, A.; Maes, K.; Van Valckenborgh, E.; Vanderkerken, K.; Jernberg Wiklund, H. Increased resistance to proteasome inhibitors in multiple myeloma mediated by cIAP2--implications for a combinatorial treatment. Oncotarget 2015, 6, 20621–20635. [Google Scholar] [CrossRef]
- Turner, J.G.; Kashyap, T.; Dawson, J.L.; Gomez, J.; Bauer, A.A.; Grant, S.; Dai, Y.; Shain, K.H.; Meads, M.; Landesman, Y.; et al. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget 2016, 7, 78896–78909. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | logFC | p Value |
|---|---|---|---|
| CFH | complement factor H | 6.16 | 0.0000457 |
| CRIP1 | Cysteine-rich protein 1 | 5.71 | 0.0000437 |
| GPX1 | glutathione peroxidase 1 | 4.56 | 0.0001157 |
| VCX2 | variable charge, X-linked 2 | 4.51 | 0.0001059 |
| CCR2 | C-C motif chemokine receptor 2 | 4.5 | 0.0001194 |
| VCX2 | variable charge, X-linked 2 | 4.24 | 0.0001347 |
| PHLDA2 | pleckstrin homology-like domain family A member 2 | 4.21 | 0.0002172 |
| CCR2 | C-C motif chemokine receptor 2 | 4.14 | 0.0001985 |
| SGK1 | serum/glucocorticoid regulated kinase 1 | 4.08 | 0.0001536 |
| CD52 | CD52 molecule | 4.08 | 0.0001583 |
| CALML4 | Calmodulin-like 4 | 4.08 | 0.0005235 |
| S100A4 | S100 calcium binding protein A4 | 3.99 | 0.0011388 |
| COL21A1 | collagen type XXI alpha 1 chain | 3.92 | 0.0002591 |
| IFITM2 | Interferon-induced transmembrane protein 2 | 3.91 | 0.0004026 |
| SCN3A | sodium voltage-gated channel alpha subunit 3 | 3.86 | 0.0003191 |
| BHLHE41 | basic helix-loop-helix family member e41 | 3.78 | 0.0012547 |
| SORL1 | Sortilin-related receptor 1 | 3.75 | 0.0002626 |
| MOXD1 | monooxygenase DBH-like 1 | 3.74 | 0.0011837 |
| CALML4 | Calmodulin-like 4 | 3.68 | 0.0007518 |
| EPCAM | epithelial cell adhesion molecule | 3.64 | 0.0003476 |
| MEST | Mesoderm-specific transcript | 3.62 | 0.0002583 |
| SLC25A21 | solute carrier family 25 member 21 | 3.58 | 0.0002612 |
| ARMCX2 | armadillo repeat containing, X-linked 2 | 3.56 | 0.0003608 |
| PLS3 | plastin 3 | 3.49 | 0.0003084 |
| NLGN4X | neuroligin 4, X-linked | 3.47 | 0.0003288 |
| RFTN1 | raftlin, lipid raft linker 1 | 3.46 | 0.0004193 |
| IFITM1 | Interferon-induced transmembrane protein 1 | 3.46 | 0.0005397 |
| ACTN1 | actinin alpha 1 | 3.44 | 0.0028258 |
| TUSC3 | tumor suppressor candidate 3 | 3.42 | 0.0003053 |
| GAGE3 | G antigen 3 | 3.41 | 0.0004645 |
| RNASET2 | ribonuclease T2 | 3.41 | 0.0006196 |
| THY1 | Thy-1 cell surface antigen | 3.39 | 0.0003143 |
| PTN | pleiotrophin | 3.37 | 0.0003203 |
| CD37 | CD37 molecule | 3.3 | 0.0013067 |
| HHLA2 | HERV-H LTR-associating 2 | 3.29 | 0.0004471 |
| RNASET2 | ribonuclease T2 | 3.09 | 0.0004523 |
| SLC2A3 | solute carrier family 2 member 3 | 3.05 | 0.0011833 |
| PLAC1 | placenta specific 1 | 2.98 | 0.0013038 |
| FGFR1 | fibroblast growth factor receptor 1 | 2.95 | 0.0005482 |
| KYNU | kynureninase | 2.92 | 0.0006091 |
| FCRL2 | Fc receptor-like 2 | 2.9 | 0.0006978 |
| NPDC1 | neural proliferation, differentiation, and control 1 | 2.84 | 0.0015714 |
| SGPP1 | sphingosine-1-phosphate phosphatase 1 | 2.84 | 0.0019385 |
| PSTPIP1 | proline-serine-threonine phosphatase interacting protein 1 | 2.81 | 0.0015156 |
| NLRP2 | NLR family pyrin domain containing 2 | 2.78 | 0.00072 |
| ANXA3 | annexin A3 | 2.74 | 0.0007048 |
| IDUA | iduronidase, alpha-L- | 2.74 | 0.0030465 |
| SPARC | secreted protein acidic and cysteine-rich | 2.72 | 0.0007207 |
| TSPYL5 | TSPY-like 5 | 2.71 | 0.0007929 |
| SORL1 | Sortilin-related receptor 1 | 2.7 | 0.0012272 |
| SEMA4A | semaphorin 4A | 2.68 | 0.0009015 |
| TUSC3 | tumor suppressor candidate 3 | 2.67 | 0.0007865 |
| LCP2 | lymphocyte cytosolic protein 2 | 2.63 | 0.0009132 |
| FMO3 | Flavin-containing monooxygenase 3 | 2.61 | 0.000845 |
| TPTE | transmembrane phosphatase with tensin homology | 2.61 | 0.0010097 |
| ABCC1 | ATP-binding cassette subfamily C member 1 | 2.58 | 0.0013177 |
| TNIK | TRAF2- and NCK-interacting kinase | 2.53 | 0.0009599 |
| THY1 | Thy-1 cell surface antigen | 2.52 | 0.0010731 |
| SERPINE2 | serpin family E member 2 | 2.52 | 0.0027214 |
| BASP1 | brain abundant membrane attached signal protein 1 | 2.52 | 0.0049738 |
| ALOX5AP | arachidonate 5-lipoxygenase-activating protein | 2.51 | 0.0049432 |
| C1orf54 | chromosome 1 open reading frame 54 | 2.5 | 0.0013496 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubaki, M.; Takeda, T.; Matsuda, T.; Seki, S.; Tomonari, Y.; Koizumi, S.; Nagatakiya, M.; Katsuyama, M.; Yamamoto, Y.; Tsurushima, K.; et al. Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-κB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells. Biomedicines 2021, 9, 33. https://doi.org/10.3390/biomedicines9010033
Tsubaki M, Takeda T, Matsuda T, Seki S, Tomonari Y, Koizumi S, Nagatakiya M, Katsuyama M, Yamamoto Y, Tsurushima K, et al. Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-κB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells. Biomedicines. 2021; 9(1):33. https://doi.org/10.3390/biomedicines9010033
Chicago/Turabian StyleTsubaki, Masanobu, Tomoya Takeda, Takuya Matsuda, Shiori Seki, Yoshika Tomonari, Shoutaro Koizumi, Miki Nagatakiya, Mai Katsuyama, Yuuta Yamamoto, Katsumasa Tsurushima, and et al. 2021. "Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-κB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells" Biomedicines 9, no. 1: 33. https://doi.org/10.3390/biomedicines9010033
APA StyleTsubaki, M., Takeda, T., Matsuda, T., Seki, S., Tomonari, Y., Koizumi, S., Nagatakiya, M., Katsuyama, M., Yamamoto, Y., Tsurushima, K., Ishizaka, T., & Nishida, S. (2021). Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-κB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells. Biomedicines, 9(1), 33. https://doi.org/10.3390/biomedicines9010033