Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration

 , , ,
, , ,
Abstract
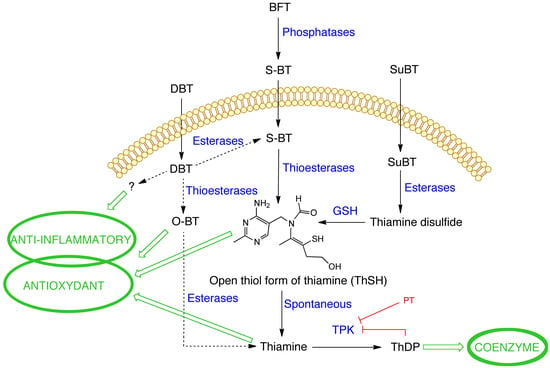
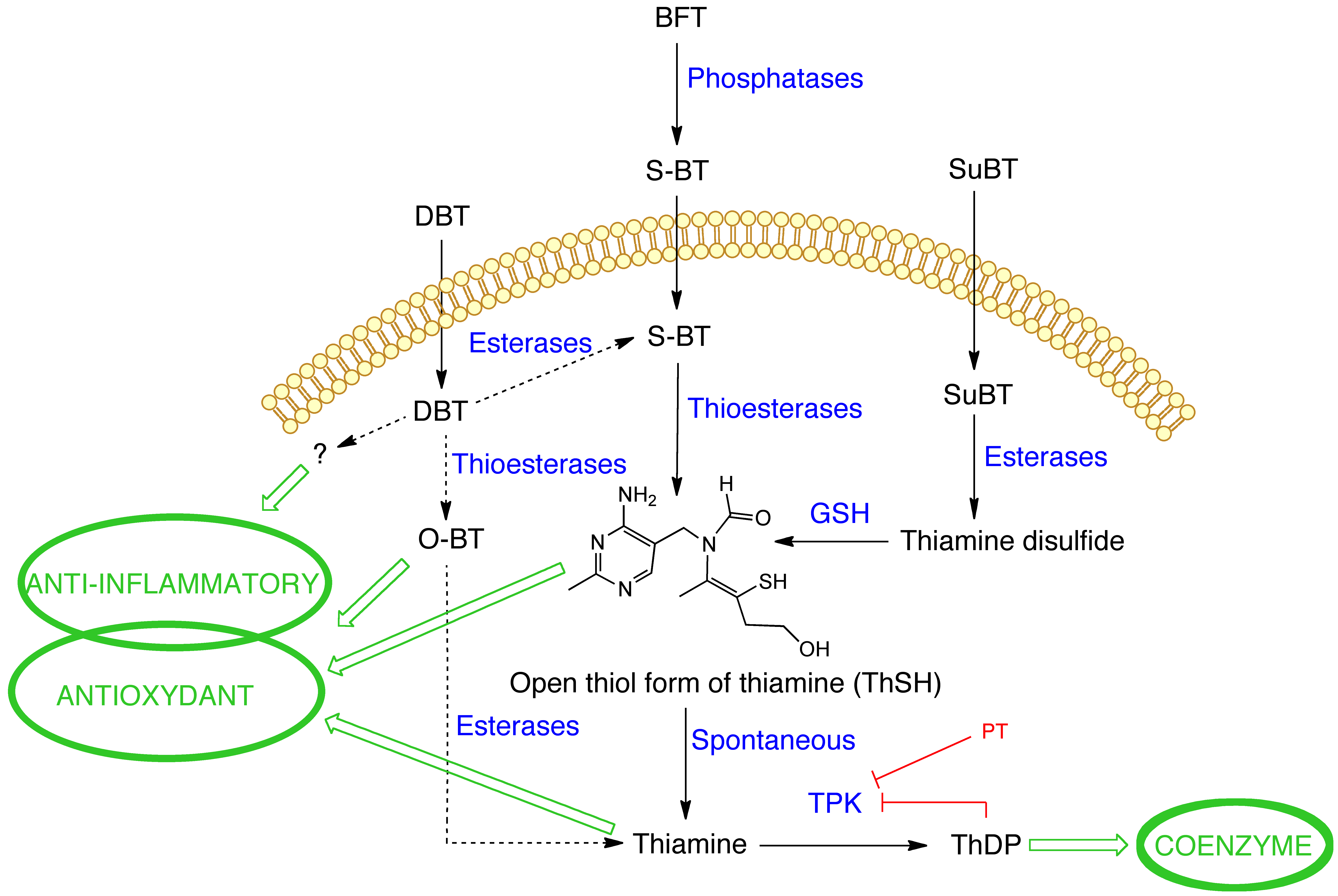{kind=link}
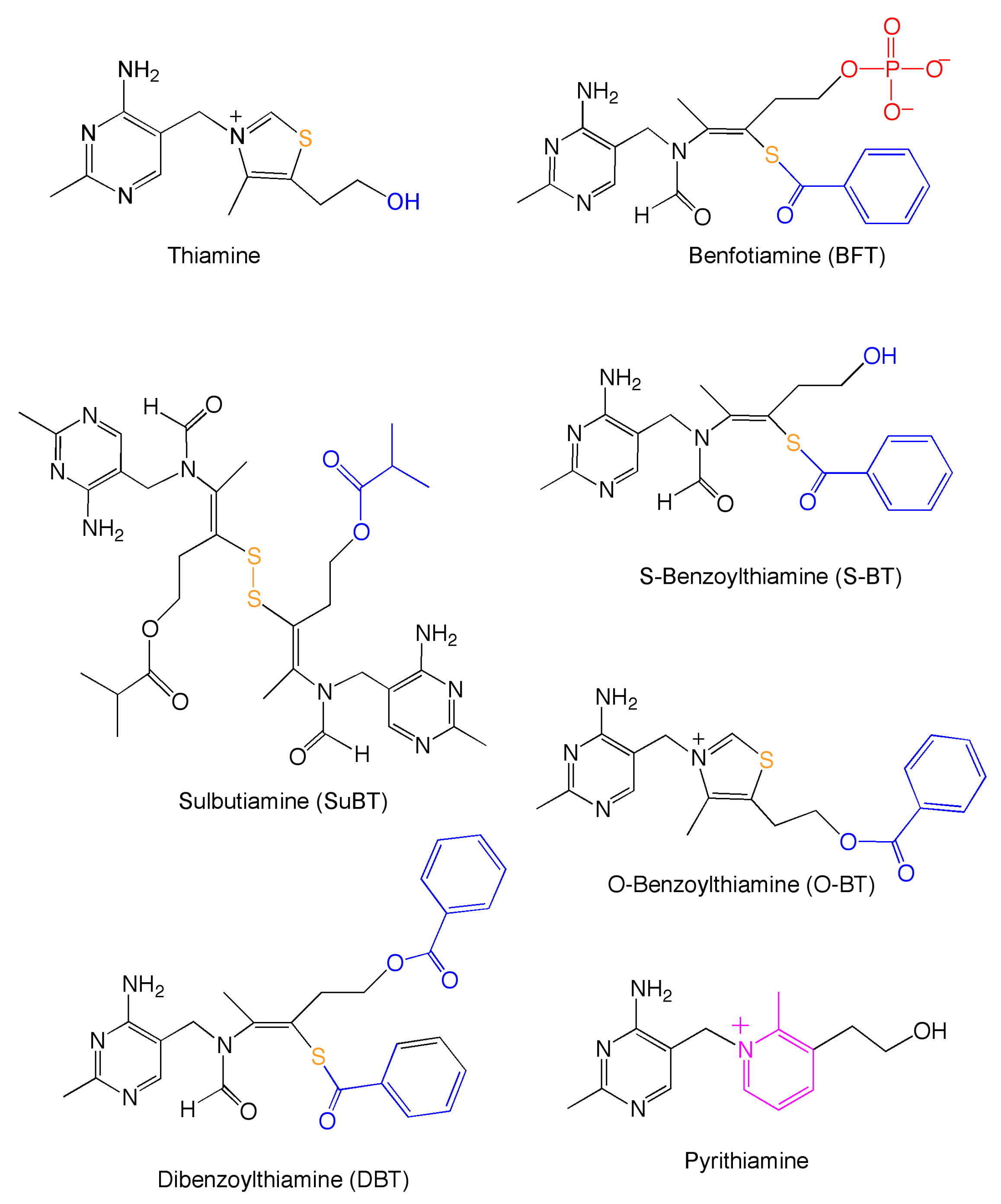{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Cells and Cell Culture
2.3. HPLC of Thiochrome Derivatives
2.4. Cell Viability Testing
2.5. UHPLC-MS Determination of Metabolites of DBT and Thiamine-d3
2.6. Protein Carbonyl Content
2.7. Thiol Quantification
2.8. Reduced Glutathione (GSH) Assay
2.9. NADPH Assay
2.10. Western Blotting
2.11. Quantification of TKT1 Expression by Real Time PCR
2.12. Assay of TKT1 Activity
2.13. Nitric Oxide Assay
2.14. TNFα Assay
2.15. SLC19A2 (ThTR1) Expression by RT-PCR
2.16. Silencing of Nrf2 Expression by siRNA
2.17. Pharmacokinetics in Mice
2.18. Study with ALS Mouse Model
2.19. Study with Ultrasound Model of Emotional Stress
2.20. Statistical Analysis
3. Results
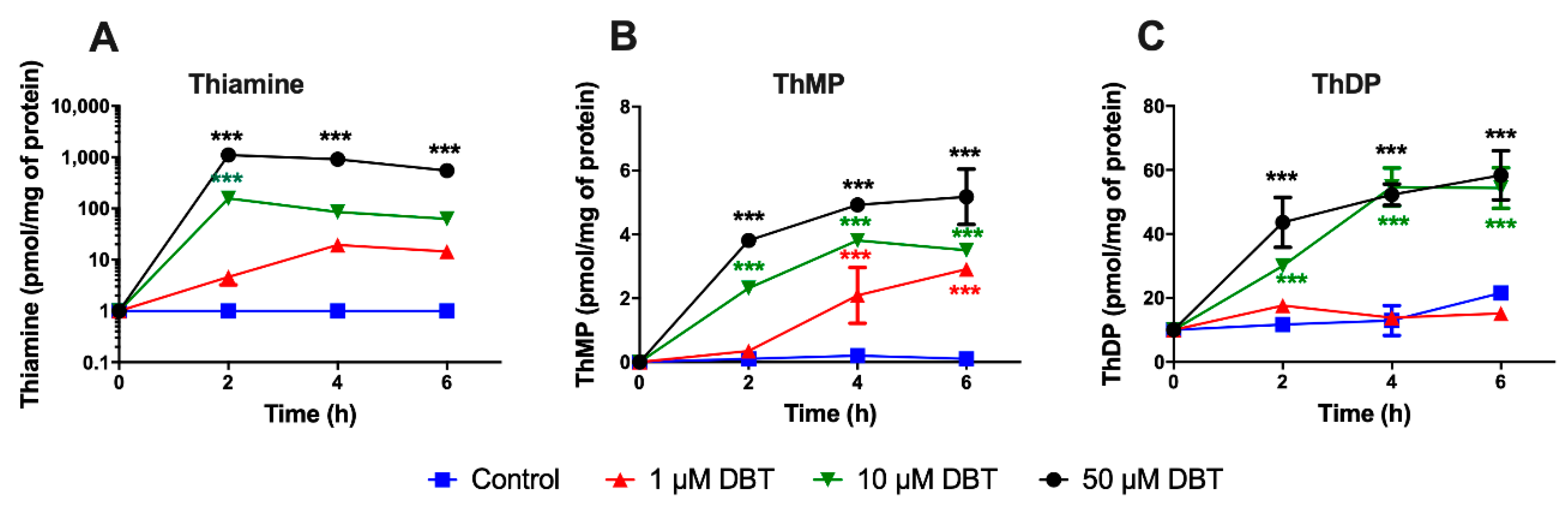
3.1. Treatment with DBT Induces a Rapid Accumulation of Thiamine in Neuro2a Cells Grown in Thiamine-Restricted Medium
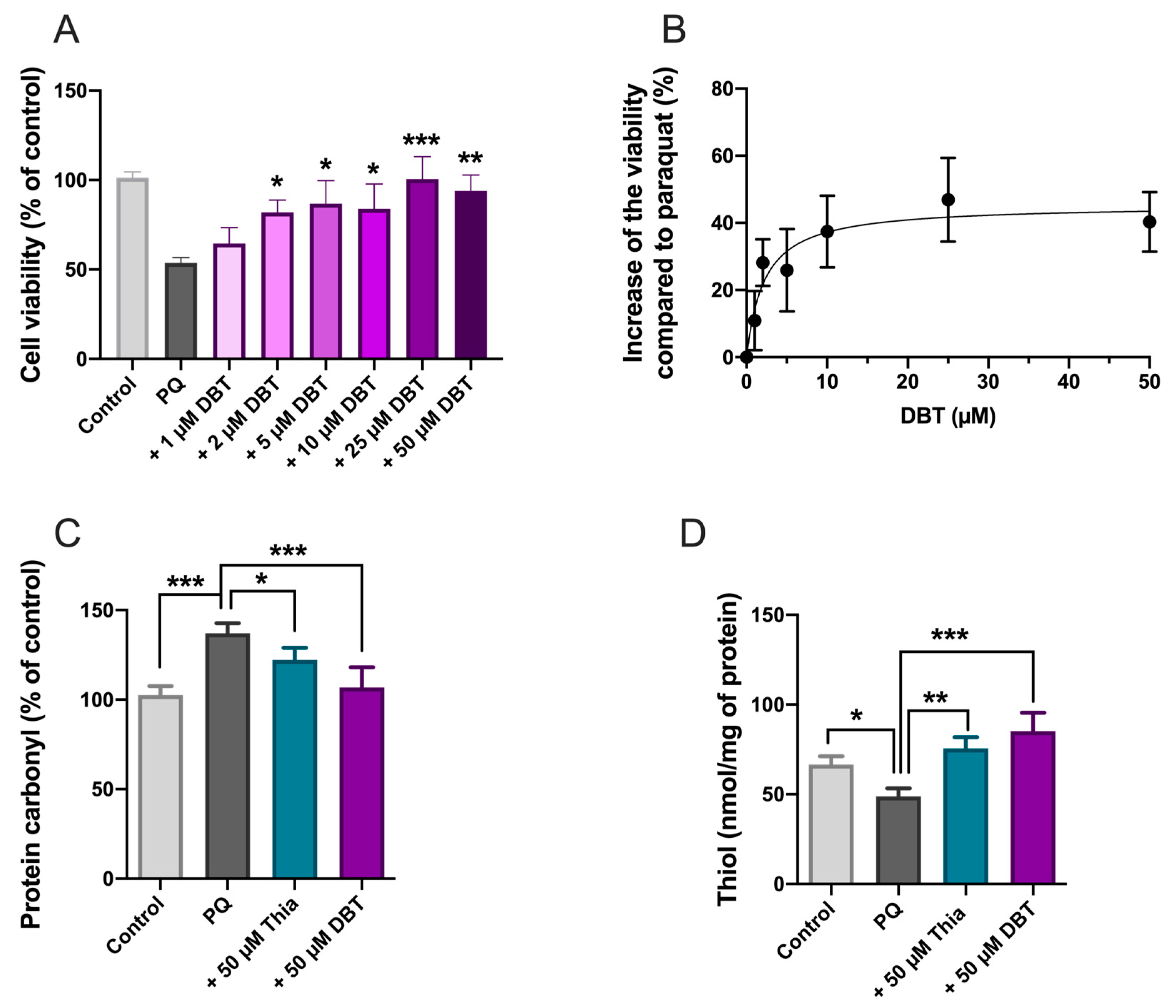
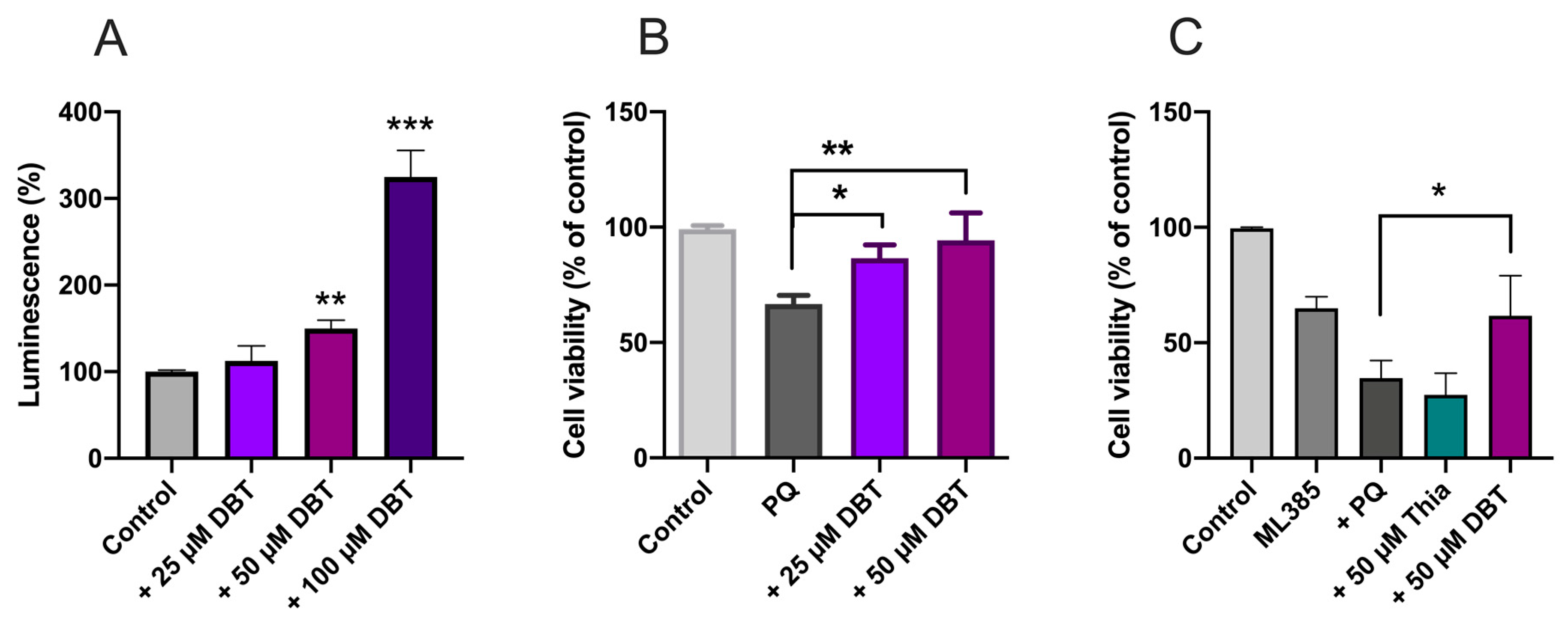
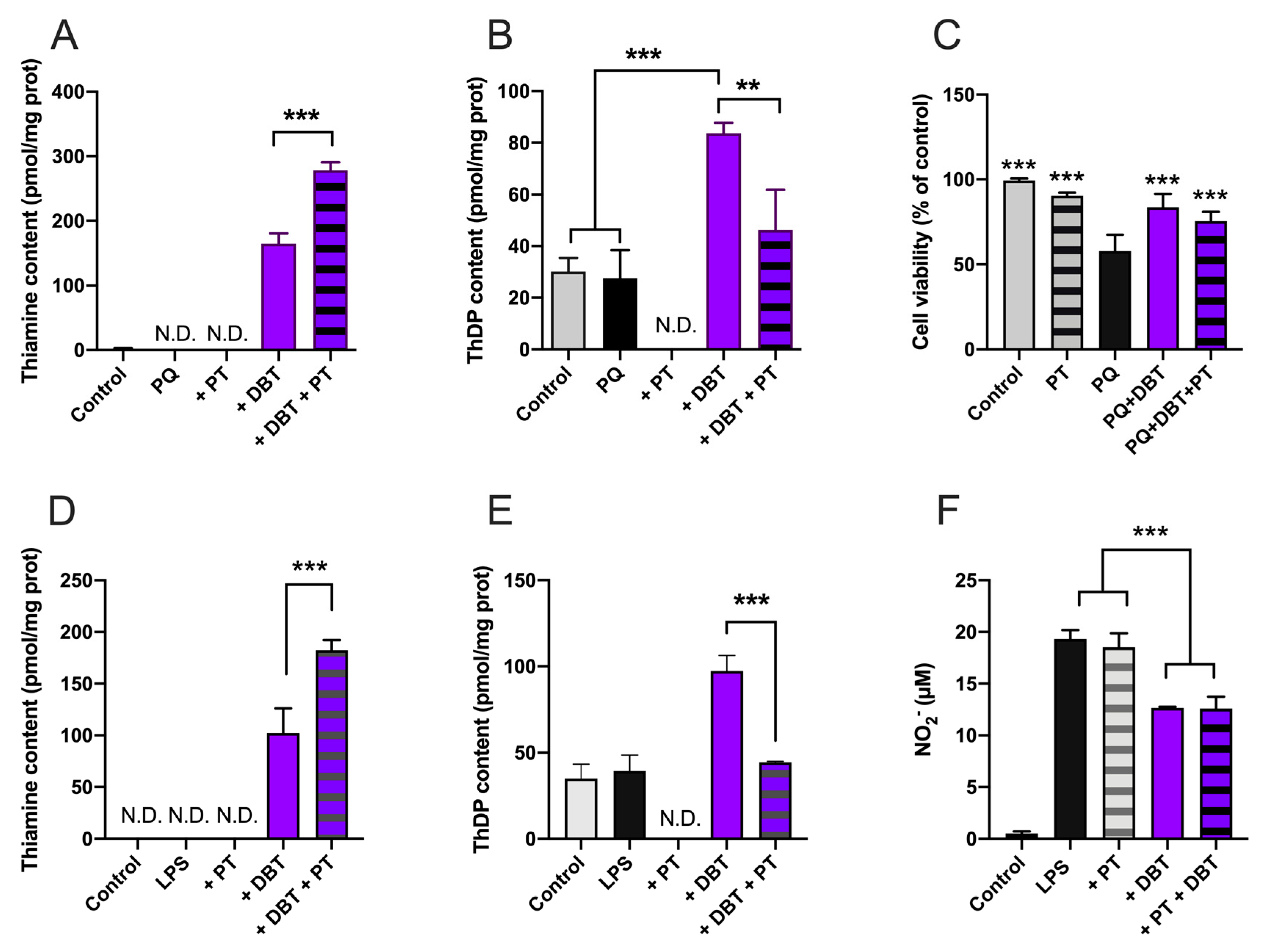
3.2. Preincubation with DBT Prevents PQ-Induced Toxicity and Oxidative Stress in Neuro2a Cells
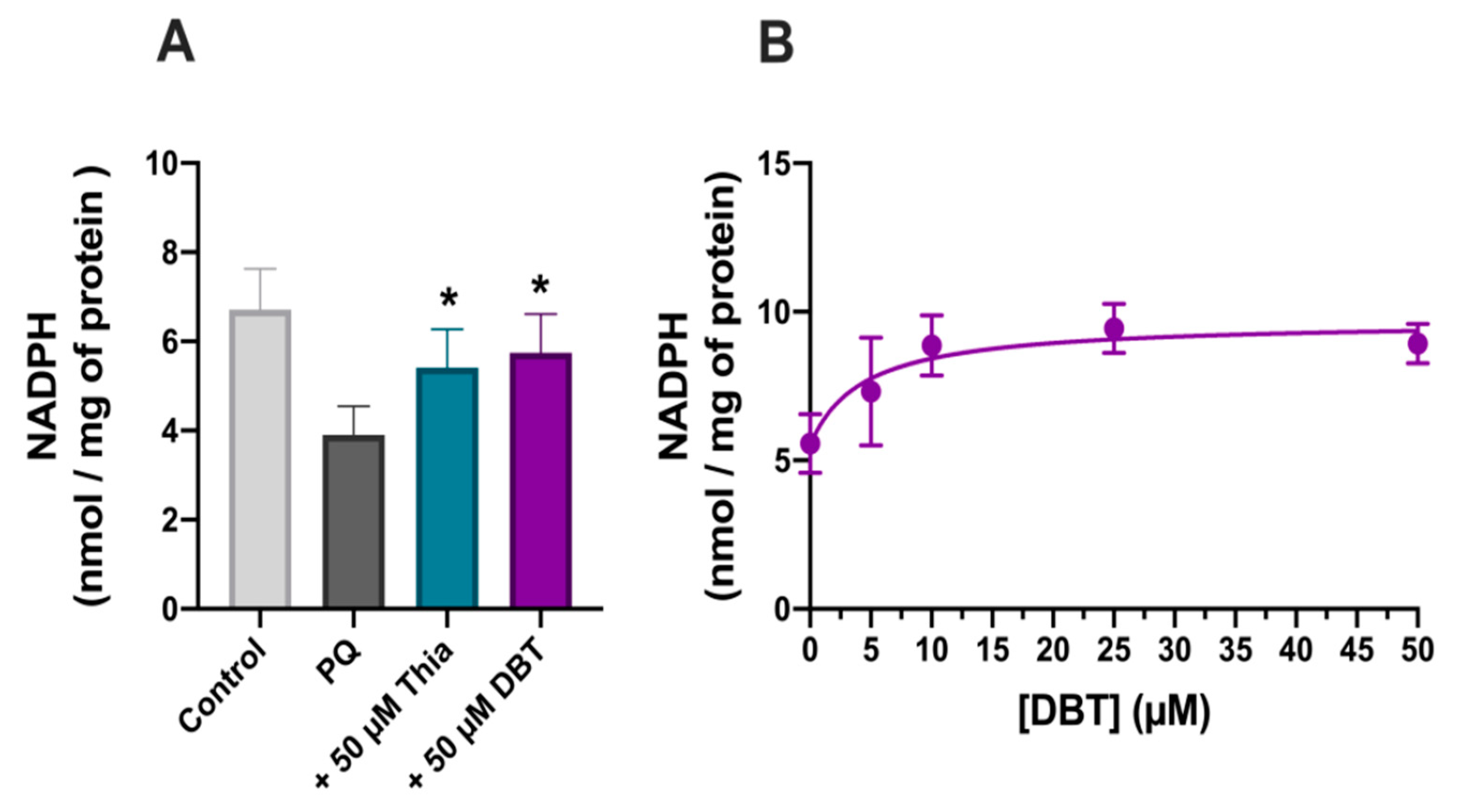
3.3. Possible Role of Glutathione and NADPH in the Antioxidant Effect of DBT
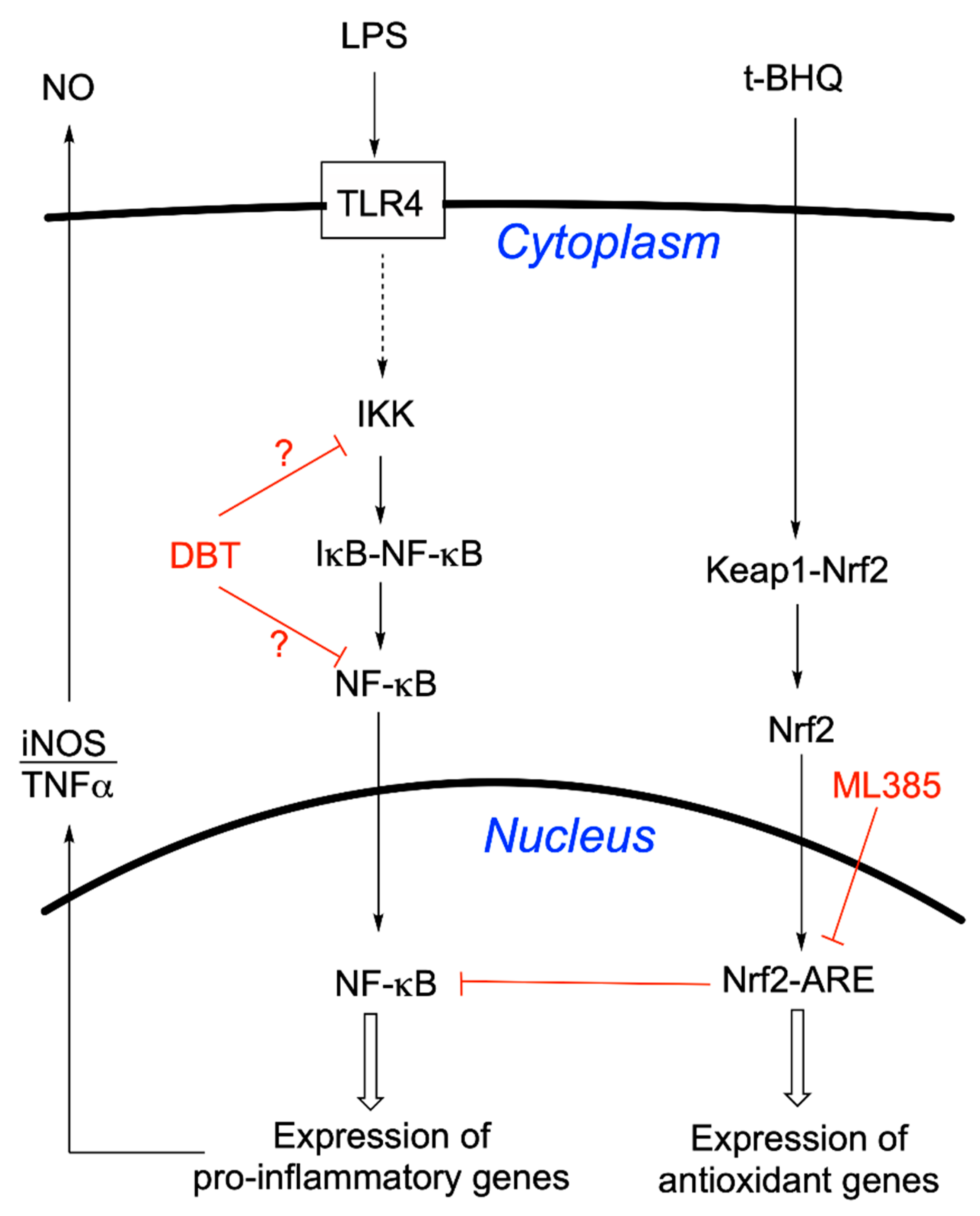
3.4. Protective Effects of DBT Do Not Involve the Activation of the Nrf2/ARE Pathway
3.5. Thiamine and ThDP Have No Direct Antioxidant Effect in Neuro2a Cells
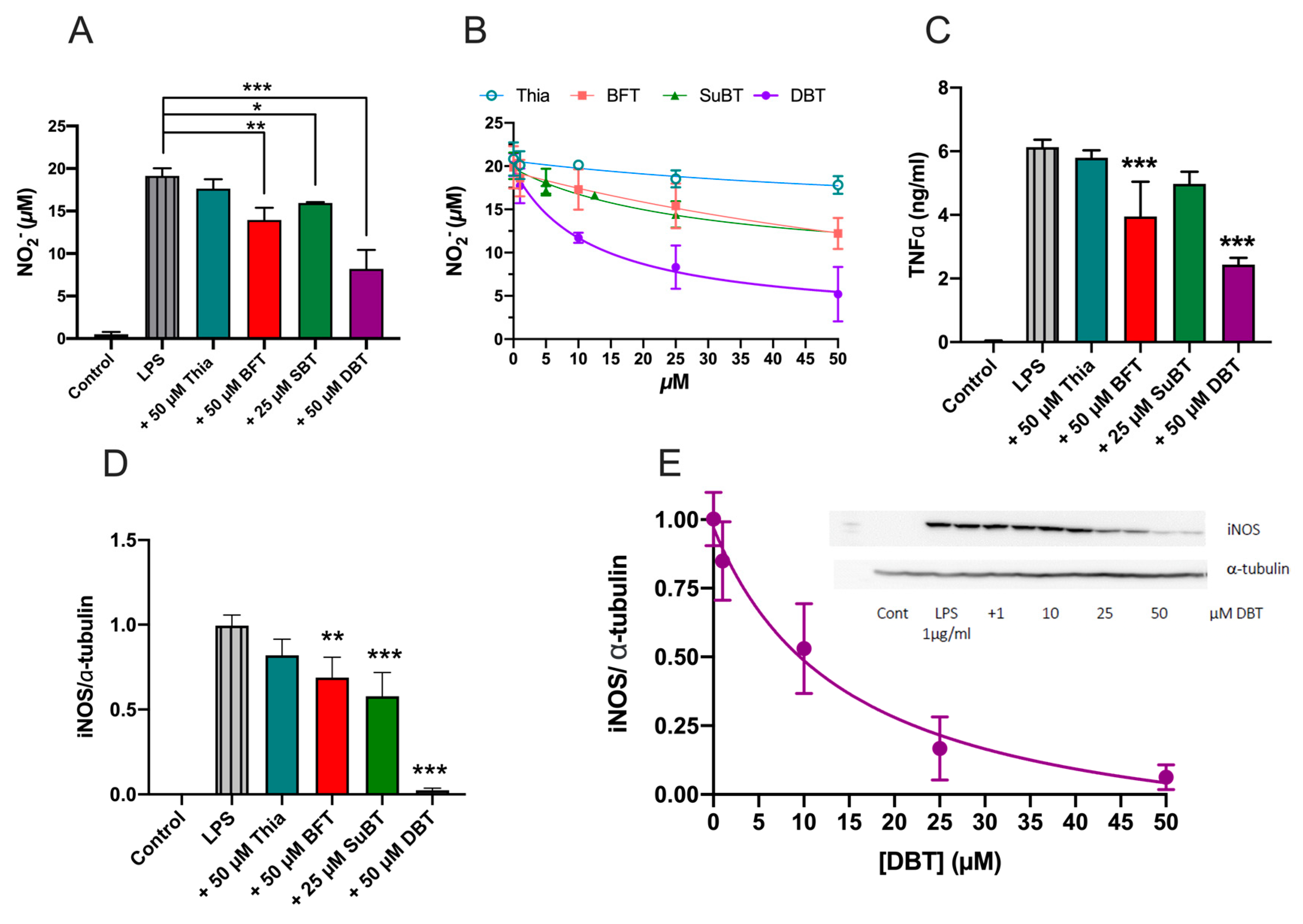
3.6. Prophylactic Anti-Inflammatory Effects of DBT in BV2 Cells Activated by LPS
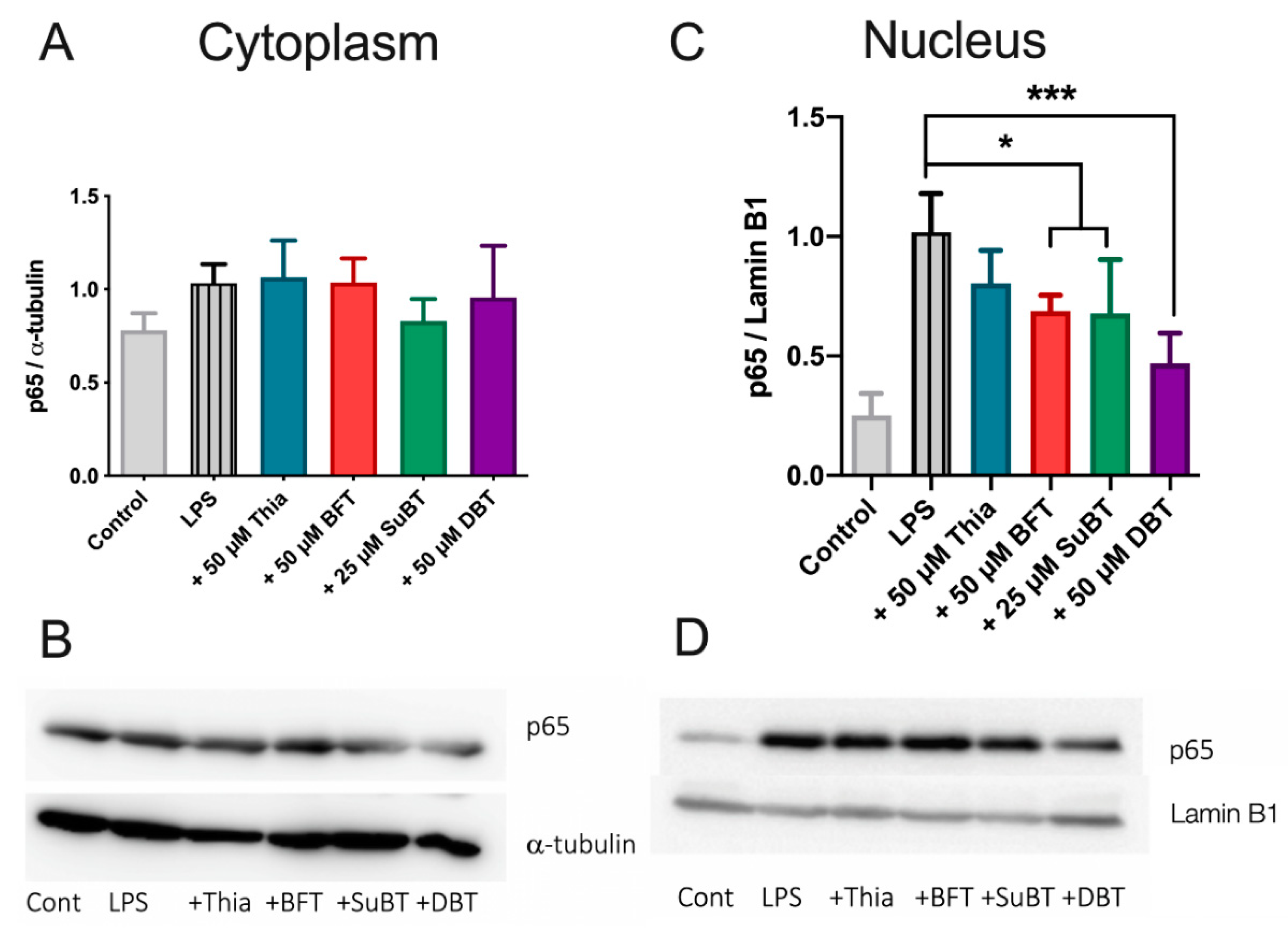
3.7. DBT Decreases the Translocation of NF-κB Induced by LPS into the Nucleus of BV2 Cells
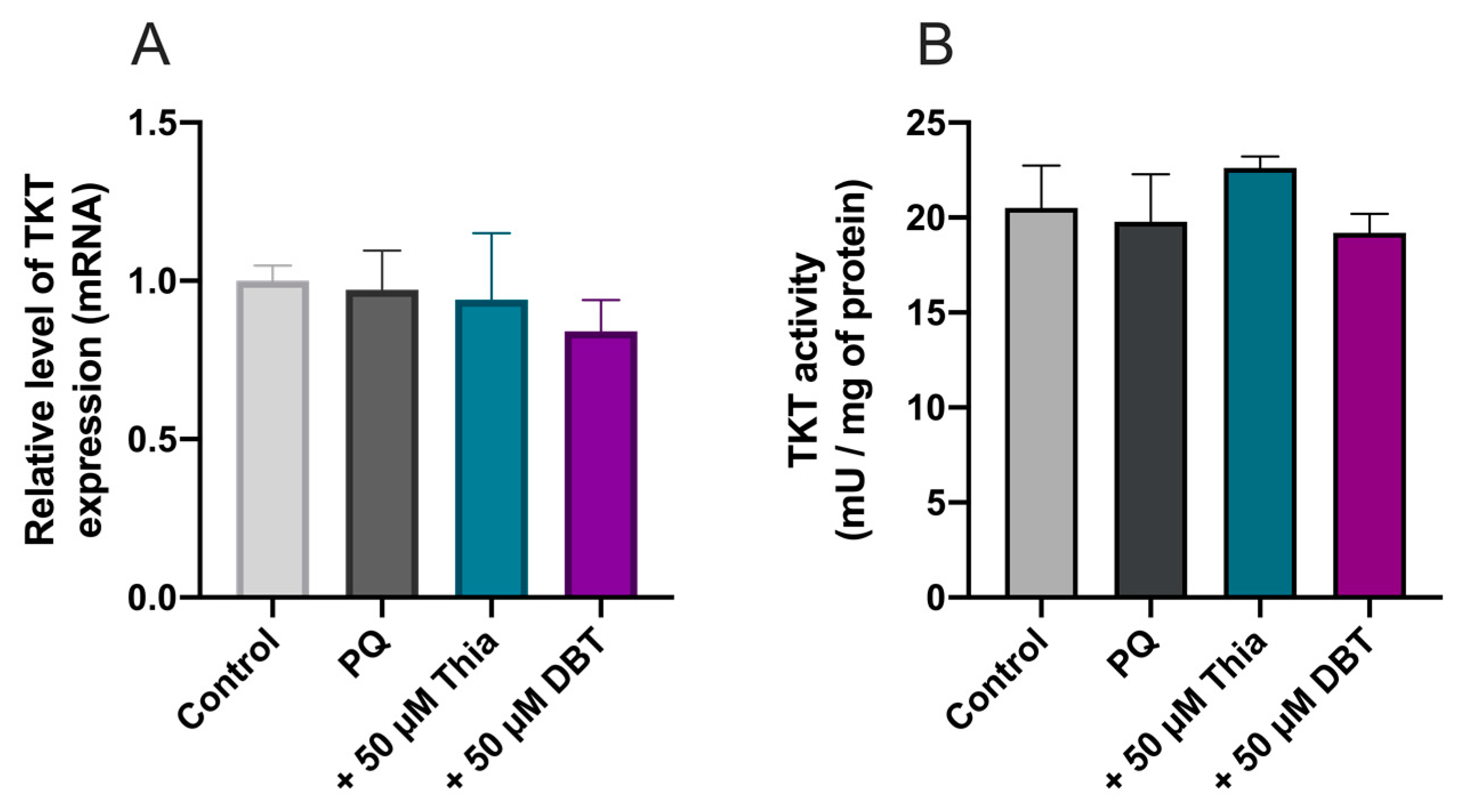
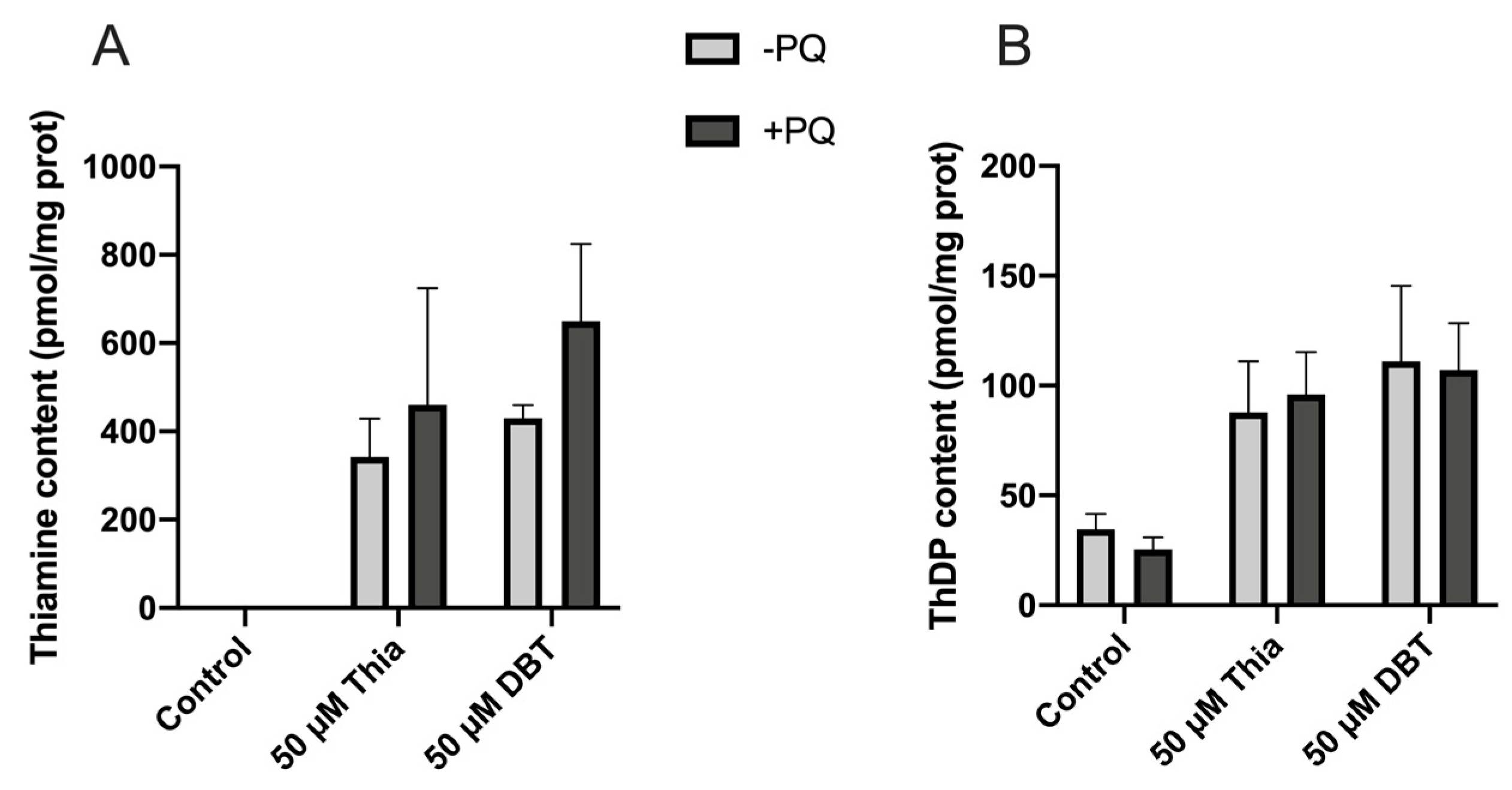
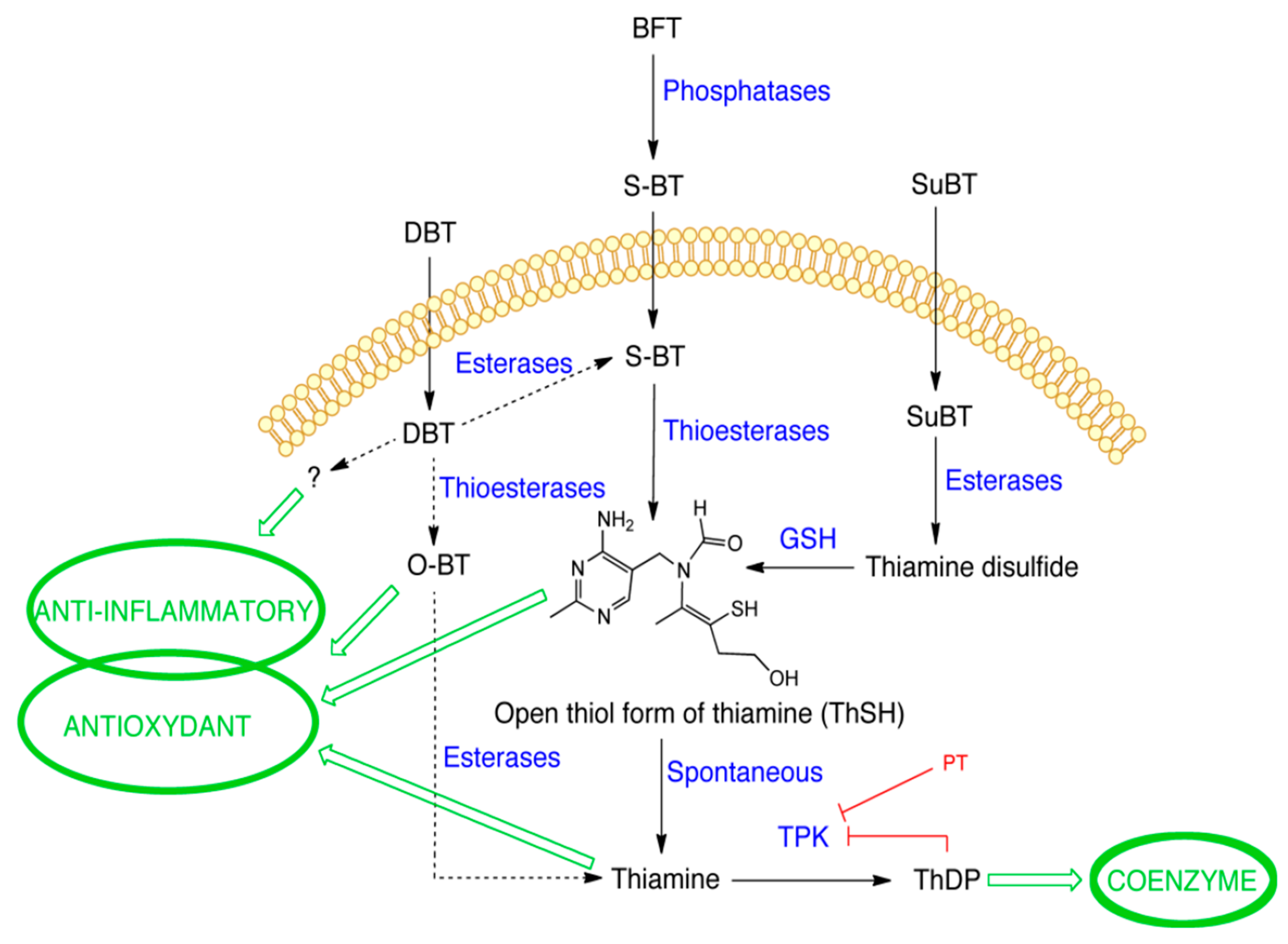
3.8. Antioxidant and Anti-Inflammatory Effects of DBT Are Not Linked to Increased Cellular Content of the Coenzyme ThDP
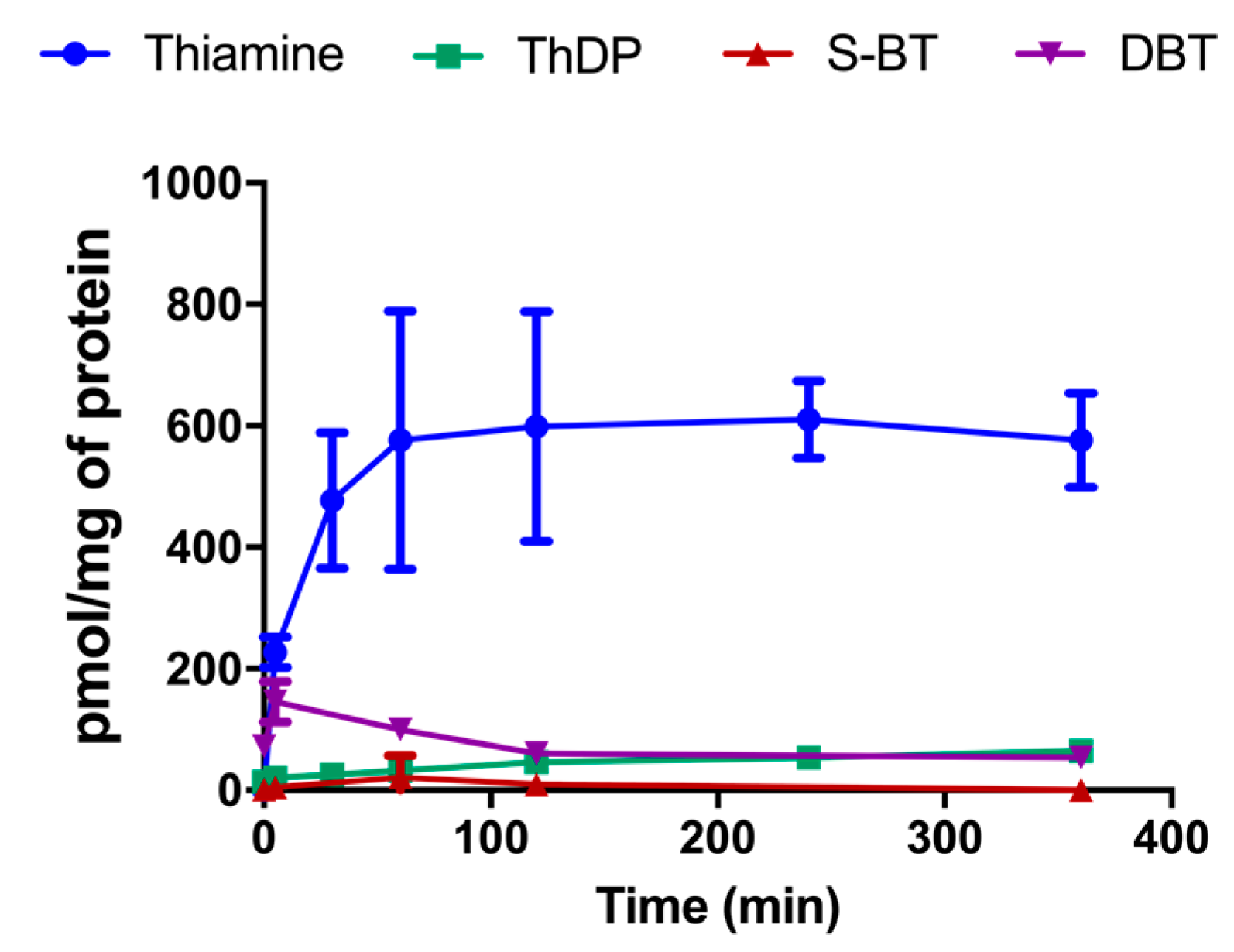
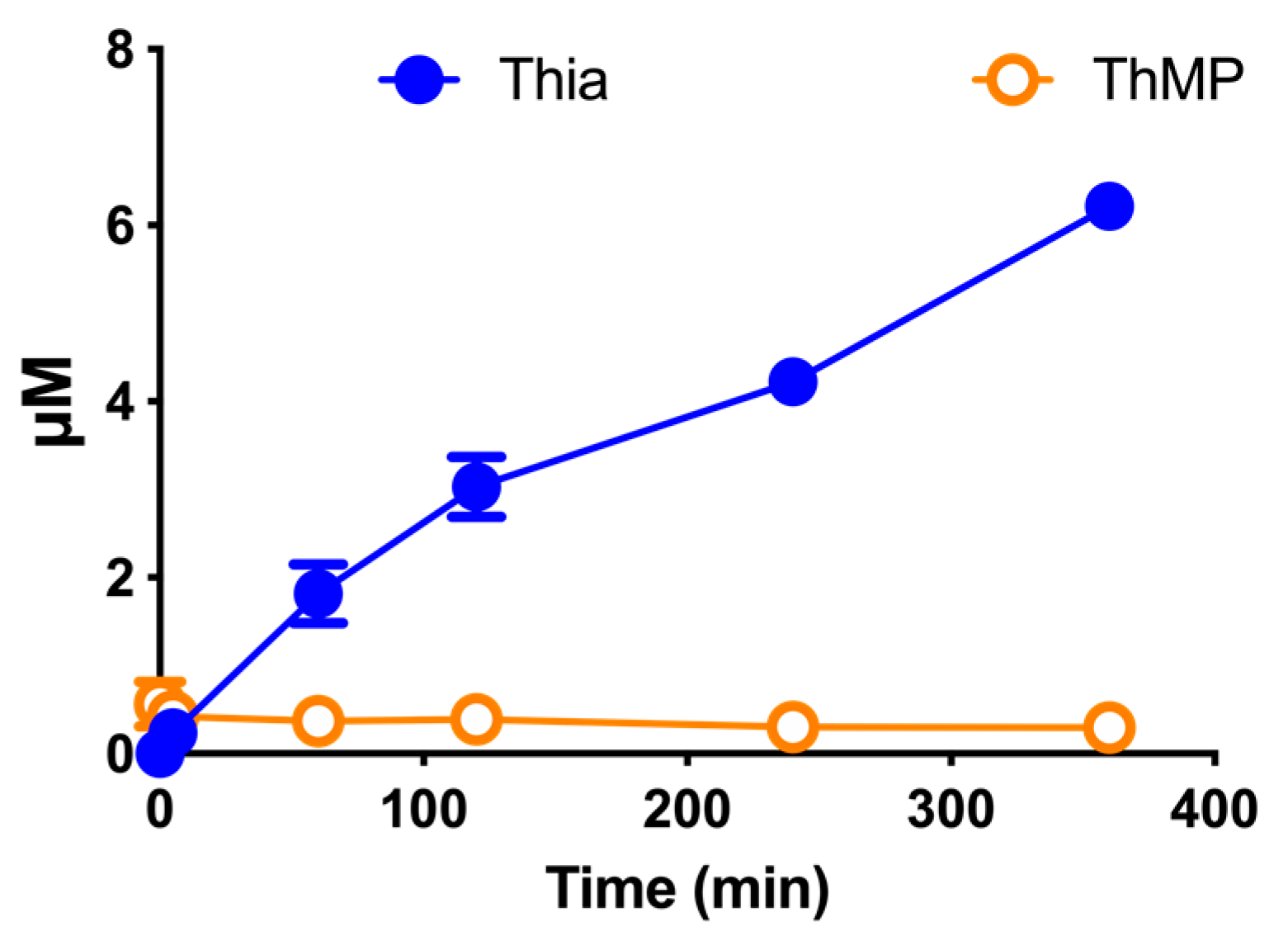
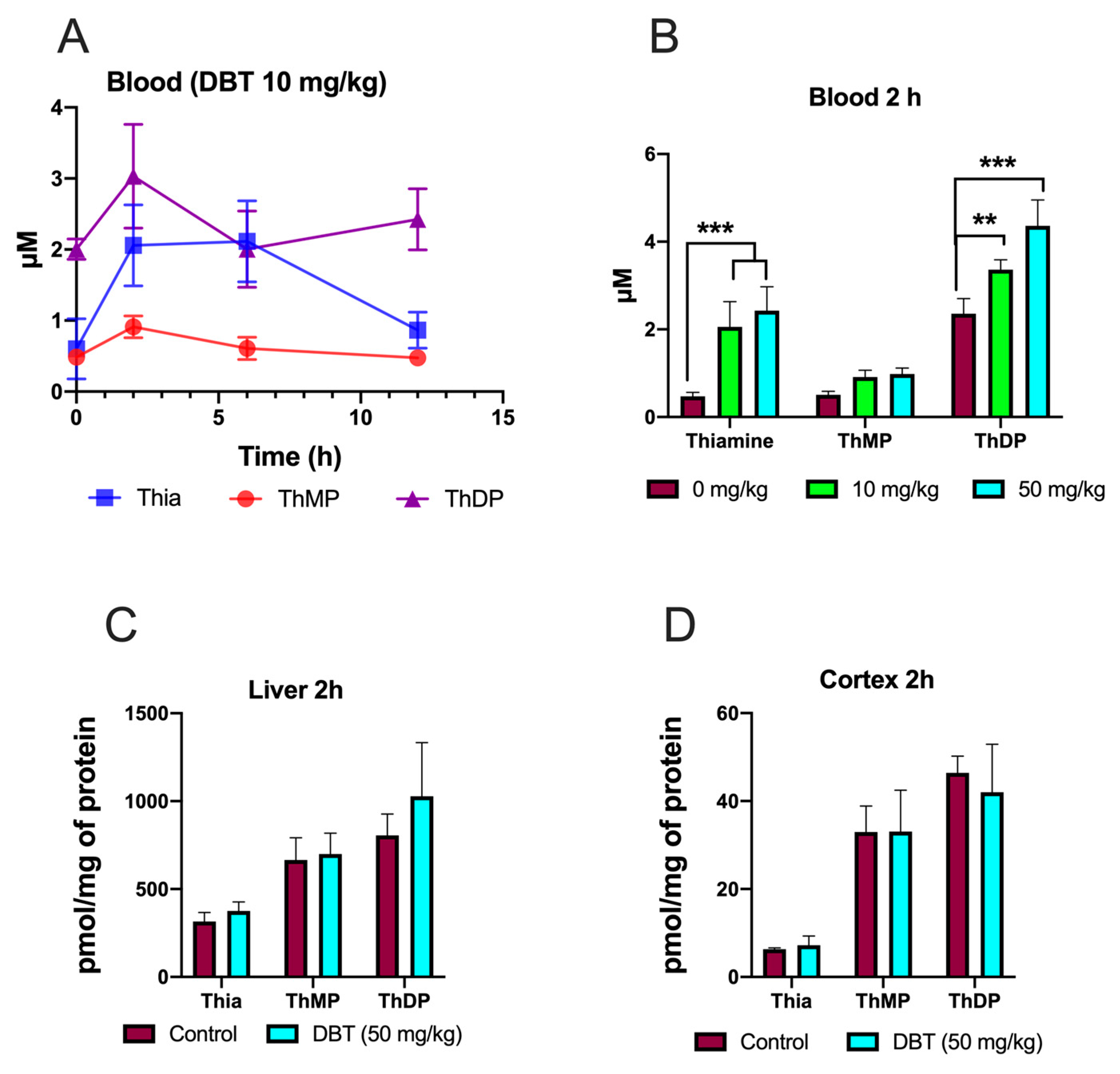
3.9. Effect of DBT Administration on the Content of Thiamine Derivatives in Mice
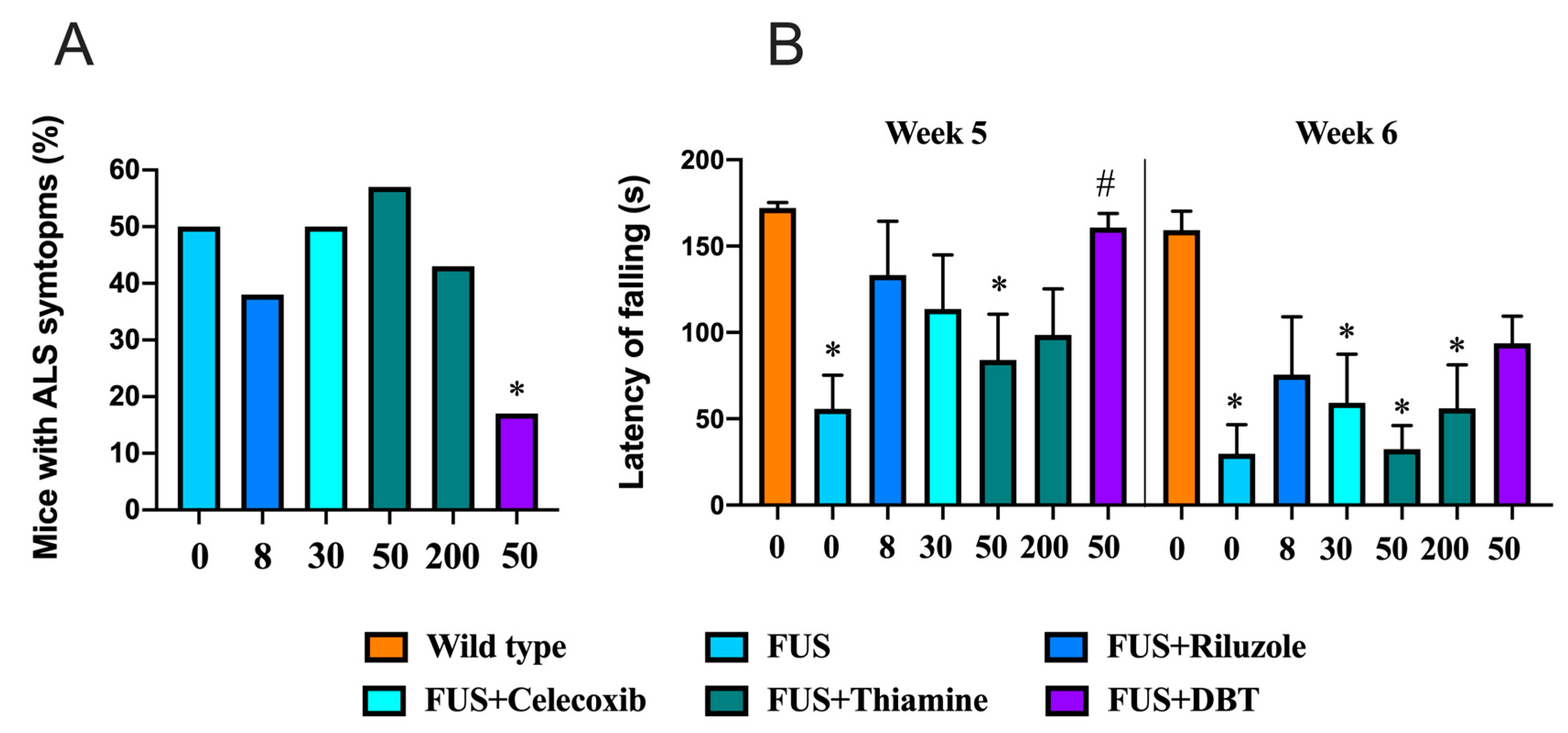
3.10. Administration of DBT but Not Thiamine Ameliorates ALS-Related Motor Dysfunctions
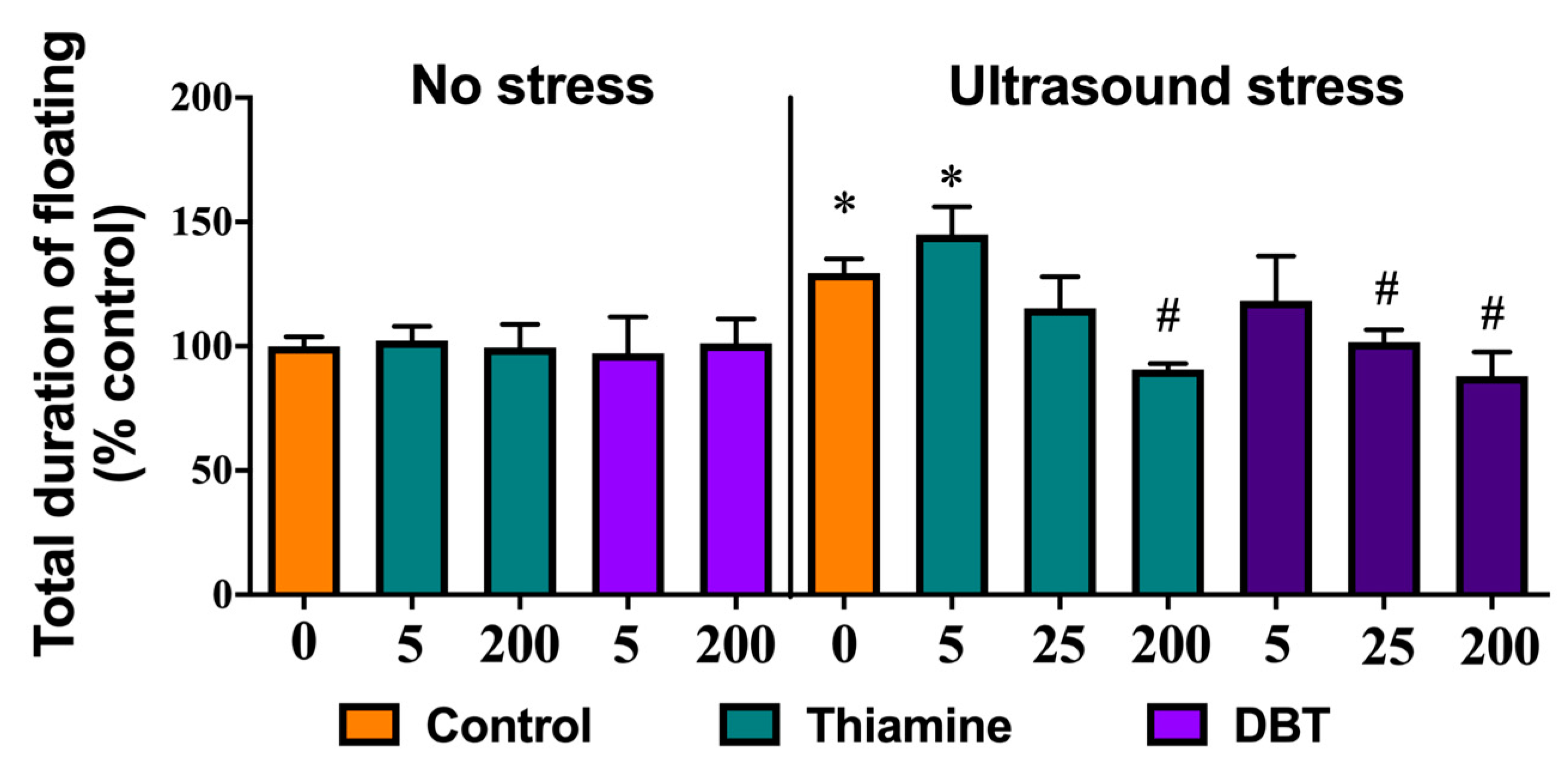
3.11. Greater Normalizing Effects of DBT than of Thiamine on Depressive-Like Behavior in the Ultrasound-Induced Stress Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ARE | antioxidant response element |
| BFT | benfotiamine |
| BSO | buthionine sulfoximine |
| DBT | dibenzoylthiamine |
| FBS | fetal bovine serum |
| GCLC | glutamate cysteine ligase catalytic subunit |
| GSH | reduced glutathione |
| LPS | lipopolysaccharides |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide tetrazolium |
| O-BT | O-benzoylthiamine |
| S-BT | S-benzoylthiamine |
| PQ | paraquat |
| PT | pyrithiamine |
| ROS | reactive oxygen species |
| SuBT | sulbutiamine |
| ThDP | thiamine diphosphate |
| ThMP | thiamine monophosphate |
| TKT1 | transketolase |
| TPK | thiamine pyrophosphokinase |
References
- Bettendorff, L. Thiamine. In Handbook of Vitamins; Zempleni, J., Suttie, J., Gregory, J.F., 3rd, Stover, P., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 268–323. [Google Scholar]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Luigi Di Salvo, M.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef] [PubMed]
- Vignisse, J.; Sambon, M.; Gorlova, A.; Pavlov, D.; Caron, N.; Malgrange, B.; Shevtsova, E.; Svistunov, A.; Anthony, D.C.; Markova, N.; et al. Thiamine and benfotiamine prevent stress-induced suppression of hippocampal neurogenesis in mice exposed to predation without affecting brain thiamine diphosphate levels. Mol. Cell. Neurosci. 2017, 82, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sambon, M.; Napp, A.; Demelenne, A.; Vignisse, J.; Wins, P.; Fillet, M.; Bettendorff, L. Thiamine and benfotiamine protect neuroblastoma cells against paraquat and β-amyloid toxicity by a coenzyme-independent mechanism. Heliyon 2019, 5, e01710. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of non-coenzyme action of thiamine: Protein targets and medical significance. Biochem. Mosc. 2019, 84, 829–850. [Google Scholar] [CrossRef]
- Whitfield, K.C.; Bourassa, M.W.; Adamolekun, B.; Bergeron, G.; Bettendorff, L.; Brown, K.H.; Cox, L.; Fattal-Valevski, A.; Fischer, P.R.; Frank, E.L.; et al. Thiamine deficiency disorders: Diagnosis, prevalence, and a roadmap for global control programs. Ann. N. Y. Acad. Sci. 2018, 1430, 3–43. [Google Scholar] [CrossRef]
- Starling-Soares, B.; Carrera-Bastos, P.; Bettendorff, L. Role of the synthetic B1 vitamin sulbutiamine on Health. J. Nutr. Metab. 2020, 2020, 9349063. [Google Scholar] [CrossRef]
- Lonsdale, D.; Shamberger, R.J.; Audhya, T. Treatment of autism spectrum children with thiamine tetrahydrofurfuryl disulfide: A pilot study. Neuroendocr. Lett. 2002, 23, 303–308. [Google Scholar]
- Volvert, M.L.; Seyen, S.; Piette, M.; Evrard, B.; Gangolf, M.; Plumier, J.C.; Bettendorff, L. Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives. BMC Pharmacol. 2008, 8, 10. [Google Scholar] [CrossRef]
- Stracke, H.; Hammes, H.P.; Werkmann, D.; Mavrakis, K.; Bitsch, I.; Netzel, M.; Geyer, J.; Kopcke, W.; Sauerland, C.; Bretzel, R.G.; et al. Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp. Clin. Endocrinol. Diabetes 2001, 109, 330–336. [Google Scholar] [CrossRef]
- Hammes, H.P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef]
- Markova, N.; Bazhenova, N.; Anthony, D.C.; Vignisse, J.; Svistunov, A.; Lesch, K.-P.; Bettendorff, L.; Strekalova, T. Thiamine and benfotiamine improve cognition and ameliorate GSK-3β-associated stress-induced behaviours in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, D.; Markova, N.; Bettendorff, L.; Chekhonin, V.; Pomytkin, I.; Lioudyno, V.; Svistunov, A.; Ponomarev, E.; Lesch, K.-P.; Strekalova, T. Elucidating the functions of brain GSK3α: Possible synergy with GSK3β upregulation and reversal by antidepressant treatment in a mouse model of depressive-like behaviour. Behav. Brain Res. 2017, 335, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, D.; Bettendorff, L.; Gorlova, A.; Olkhovik, A.; Kalueff, A.V.; Ponomarev, E.D.; Inozemtsev, A.; Chekhonin, V.; Lesch, K.-P.; Anthony, D.C.; et al. Neuroinflammation and aberrant hippocampal plasticity in a mouse model of emotional stress evoked by exposure to ultrasound of alternating frequencies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Gorlova, A.; Pavlov, D.; Anthony, D.C.; Ponomarev, E.; Sambon, M.; Proshin, A.; Shafarevich, I.; Babaevskaya, D.; Lesch, K.-P.; Bettendorff, L.; et al. Thiamine and benfotiamine counteract ultrasound-induced aggression, normalize AMPA receptor expression and plasticity markers, and reduce oxidative stress in mice. Neuropharmacology 2019, 156, 107543. [Google Scholar] [CrossRef]
- Pan, X.; Gong, N.; Zhao, J.; Yu, Z.; Gu, F.; Chen, J.; Sun, X.; Zhao, L.; Yu, M.; Xu, Z.; et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain 2010, 133, 1342–1351. [Google Scholar] [CrossRef]
- Tapias, V.; Jainuddin, S.; Ahuja, M.; Stack, C.; Elipenahli, C.; Vignisse, J.; Gerges, M.; Starkova, N.; Xu, H.; Starkov, A.A.; et al. Benfotiamine treatment activates the Nrf2/ARE Pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum. Mol. Genet. 2018, 27, 2874–2892. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Z.; Fei, G.; Pan, S.; Bao, W.; Ren, S.; Guan, Y.; Zhong, C. Long-term cognitive improvement after benfotiamine administration in patients with Alzheimer’s disease. Neurosci. Bull. 2016, 32, 591–596. [Google Scholar] [CrossRef]
- Sang, S.; Pan, X.; Chen, Z.; Zeng, F.; Pan, S.; Liu, H.; Jin, L.; Fei, G.; Wang, C.; Ren, S.; et al. Thiamine diphosphate reduction strongly correlates with brain glucose hypometabolism in Alzheimer’s disease, whereas amyloid deposition does not. Alzheimers Res. Ther. 2018, 10, 26. [Google Scholar] [CrossRef]
- Chen, Z.; Pan, X.; Fei, G.; Pan, S.; Bao, W.; Ren, S.; Guan, Y.; Zhong, C. Diagnostic value of blood thiamine metabolites in Alzheimer’s disease examined by 11C-PiB PET scanning. Future Sci. OA 2017, 3, FSO172. [Google Scholar] [CrossRef]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for Alzheimer’s disease diagnosis. EBioMedicine 2016, 3, 155–162. [Google Scholar] [CrossRef]
- Håglin, L.; Domellöf, M.; Bäckman, L.; Forsgren, L. Low plasma thiamine and phosphate in male patients with Parkinson’s disease is associated with mild cognitive impairment. Clin. Nutr. ESPEN 2020, 37, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Pala, M.I.; Compagnoni, L.; Colangeli, M. High-dose thiamine as initial treatment for Parkinson’s disease. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Luong, K.V.Q.; Nguyen, L.T.H. Thiamine and Parkinson’s disease. J. Neurol. Sci. 2012, 316, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef]
- Heywood, R.; Wood, J.D.; Majeed, S.K. Tumorigenic and toxic effect of O,S-dibenzoyl thiamine hydrochloride in prolonged dietary administration to rats. Toxicol. Lett. 1985, 26, 53–58. [Google Scholar] [CrossRef]
- Ketola, H.G.; Isaacs, G.R.; Robins, J.S.; Lloyd, R.C. Effectiveness and retention of thiamine and its analogs administered to steelhead and landlocked Atlantic salmon. J. Aquat. Anim. Health 2008, 20, 29–38. [Google Scholar] [CrossRef]
- Bettendorff, L.; Goessens, G.; Sluse, F.; Wins, P.; Bureau, M.; Laschet, J.; Grisar, T. Thiamine deficiency in cultured neuroblastoma cells: Effect on mitochondrial function and peripheral benzodiazepine receptors. J. Neurochem. 1995, 64, 2013–2021. [Google Scholar] [CrossRef]
- Bettendorff, L.; Peeters, M.; Jouan, C.; Wins, P.; Schoffeniels, E. Determination of thiamin and its phosphate esters in cultured neurons and astrocytes using an ion-pair reversed-phase high-performance liquid chromatographic method. Anal. Biochem. 1991, 198, 52–59. [Google Scholar] [CrossRef]
- Gangolf, M.; Czerniecki, J.; Radermecker, M.; Detry, O.; Nisolle, M.; Jouan, C.; Martin, D.; Chantraine, F.; Lakaye, B.; Wins, P.; et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS ONE 2010, 5, e13616. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar] [CrossRef]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Leuenberger, N.; Tan, M.J.; Yan, Y.W.; Chen, Y.; Kambadur, R.; Wahli, W.; Tan, N.S. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 2011, 60, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Matsui, K.; Iida, R.; Tsubota, E.; Matsuki, T. Time course of housekeeping gene expression changes in diffuse alveolar damage induced by hyperoxia exposure in mice. Leg. Med. 2009, 11 (Suppl. 1), S151–S154. [Google Scholar] [CrossRef] [PubMed]
- de Munter, J.P.J.M.; Shafarevich, I.; Liundup, A.; Pavlov, D.; Wolters, E.C.; Gorlova, A.; Veniaminova, E.; Umriukhin, A.; Kalueff, A.; Svistunov, A.; et al. Neuro-Cells therapy improves motor outcomes and suppresses inflammation during experimental syndrome of amyotrophic lateral sclerosis in mice. CNS Neurosci. Ther. 2020, 26, 504–517. [Google Scholar] [CrossRef]
- Costa-Nunes, J.P.; Cline, B.H.; Araújo-Correia, M.; Valença, A.; Markova, N.; Dolgov, O.; Kubatiev, A.; Yeritsyan, N.; Steinbusch, H.W.M.; Strekalova, T. Animal models of depression and drug delivery with food as an effective dosing method: Evidences from studies with celecoxib and dicholine succinate. Biomed Res. Int. 2015, 2015, 596126. [Google Scholar] [CrossRef]
- Veniaminova, E.; Oplatchikova, M.; Bettendorff, L.; Kotenkova, E.; Lysko, A.; Vasilevskaya, E.; Kalueff, A.V.; Fedulova, L.; Umriukhin, A.; Lesch, K.-P.; et al. Prefrontal cortex inflammation and liver pathologies accompany cognitive and motor deficits following Western diet consumption in non-obese female mice. Life Sci. 2020, 241, 117163. [Google Scholar] [CrossRef]
- Bettendorff, L.; Wins, P. Mechanism of thiamine transport in neuroblastoma cells. Inhibition of a high affinity carrier by sodium channel activators and dependence of thiamine uptake on membrane potential and intracellular ATP. J. Biol. Chem. 1994, 269, 14379–14385. [Google Scholar]
- Lei, S.; Zavala-Flores, L.; Garcia-Garcia, A.; Nandakumar, R.; Huang, Y.; Madayiputhiya, N.; Stanton, R.C.; Dodds, E.D.; Powers, R.; Franco, R. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 2014, 9, 2032–2048. [Google Scholar] [CrossRef]
- Piwien-Pilipuk, G.; Galigniana, M.D. Oxidative stress induced by L-buthionine-(S,R)-sulfoximine, a selective inhibitor of glutathione metabolism, abrogates mouse kidney mineralocorticoid receptor function. Biochim. Biophys. Acta 2000, 1495, 263–280. [Google Scholar] [CrossRef]
- Gadau, S.; Emanueli, C.; Van Linthout, S.; Graiani, G.; Todaro, M.; Meloni, M.; Campesi, I.; Invernici, G.; Spillmann, F.; Ward, K.; et al. Benfotiamine accelerates the healing of ischaemic diabetic limbs in mice through protein kinase B/Akt-mediated potentiation of angiogenesis and inhibition of apoptosis. Diabetologia 2006, 49, 405–420. [Google Scholar] [CrossRef]
- Saha, A.; Connelly, S.; Jiang, J.; Zhuang, S.; Amador, D.T.; Phan, T.; Pilz, R.B.; Boss, G.R. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol. Cell 2014, 55, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small molecule inhibitor of NRF2 Selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Lukienko, P.I.; Mel’nichenko, N.G.; Zverinskii, I.V.; Zabrodskaya, S.V. Antioxidant properties of thiamine. Bull. Exp. Biol. Med. 2000, 130, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fang, W.; Fan, S.; Liao, W.; Xiong, Y.; Liao, S.; Li, Y.; Xiao, S.; Liu, J. Neurotropin inhibits neuroinflammation via suppressing NF-κB and MAPKs signaling pathways in lipopolysaccharide-stimulated BV2 cells. J. Pharmacol. Sci. 2018, 136, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Savic, D.; Laketa, D.; Bjelobaba, I.; Milenkovic, I.; Pekovic, S.; Nedeljkovic, N.; Lavrnja, I. Benfotiamine attenuates inflammatory response in LPS stimulated BV-2 microglia. PLoS ONE 2015, 10, e0118372. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Liu, J.Y.; Timm, D.E.; Hurley, T.D. Pyrithiamine as a substrate for thiamine pyrophosphokinase. J. Biol. Chem. 2006, 281, 6601–6607. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Huang, C.-L.; Chao, C.-C.; Lee, Y.-C.; Lu, M.-K.; Cheng, J.-J.; Yang, Y.-C.; Wang, V.-C.; Chang, W.-C.; Huang, N.-K. Paraquat induces cell death through impairing mitochondrial membrane permeability. Mol. Neurobiol. 2016, 53, 2169–2188. [Google Scholar] [CrossRef]
- Fraser, D.A.; Hessvik, N.P.; Nikolic, N.; Aas, V.; Hanssen, K.F.; Bohn, S.K.; Thoresen, G.H.; Rustan, A.C. Benfotiamine increases glucose oxidation and downregulates NADPH oxidase 4 expression in cultured human myotubes exposed to both normal and high glucose concentrations. Genes Nutr. 2012, 7, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, I.M.-J.; Lai, R.K.-H.; Lin, S.-H.; Tse, A.P.-W.; Chiu, D.K.-C.; Koh, H.-Y.; Law, C.-T.; Wong, C.-M.; Cai, Z.; Wong, C.C.-L.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef] [PubMed]
- Benito, A.; Polat, I.H.; Noé, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef] [PubMed]
- Jonus, H.C.; Hanberry, B.S.; Khatu, S.; Kim, J.; Luesch, H.; Dang, L.H.; Bartlett, M.G.; Zastre, J.A. The adaptive regulation of thiamine pyrophosphokinase-1 facilitates malignant growth during supplemental thiamine conditions. Oncotarget 2018, 9, 35422–35438. [Google Scholar] [CrossRef]
- Yadav, U.C.S.; Kalariya, N.M.; Srivastava, S.K.; Ramana, K.V. Protective role of benfotiamine, a fat-soluble vitamin B1 analogue, in lipopolysaccharide-induced cytotoxic signals in murine macrophages. Free Radic. Biol. Med. 2010, 48, 1423–1434. [Google Scholar] [CrossRef]
- Walter, J. The Triggering receptor expressed on myeloid cells 2: A molecular link of neuroinflammation and neurodegenerative diseases. J. Biol. Chem. 2016, 291, 4334–4341. [Google Scholar] [CrossRef]
- Shindo, H.; Okamoto, K.; Totsu, J. Transport of organic compounds through biological membranes. I. Accumulative uptake os S-benzoylthiamine by human erythrocytes. Chem. Pharm. Bull. 1967, 15, 295–302. [Google Scholar] [CrossRef]
- Tominaga, F.; Furuno, K.; Shimakawa, K.; Suzuoki, Z. Metabolic fate of the benzoic acid moiety of OS-dibenzoyl thiamine in rats. Vitamins 1976, 50, 415–420. [Google Scholar]
- Hurt, J.K.; Coleman, J.L.; Fitzpatrick, B.J.; Taylor-Blake, B.; Bridges, A.S.; Vihko, P.; Zylka, M.J. Prostatic Acid phosphatase is required for the antinociceptive effects of thiamine and benfotiamine. PLoS ONE 2012, 7, e48562. [Google Scholar] [CrossRef]
- Inoue, K.; Itokawa, Y. Metabolism of O-benzoylthiamine in animal body. Biochem. Med. 1973, 8, 450–463. [Google Scholar] [CrossRef]
- Kohno, K.; Noda, K.; Mizobe, M.; Utsumi, I. Enzymatic reduction of disulfide-type thiamine derivatives. Biochem. Pharmacol. 1969, 18, 1685–1692. [Google Scholar] [PubMed]
- Loew, D. Pharmacokinetics of thiamine derivatives especially of benfotiamine. Int. J. Clin. Pharmacol. Ther. 1996, 34, 47–50. [Google Scholar] [PubMed]
- Hopmann, R.F.W. The alkali-induced transformations of thiamin. Ann. N. Y. Acad. Sci. 1982, 378, 32–50. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sambon, M.; Gorlova, A.; Demelenne, A.; Alhama-Riba, J.; Coumans, B.; Lakaye, B.; Wins, P.; Fillet, M.; Anthony, D.C.; Strekalova, T.; et al. Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration. Biomedicines 2020, 8, 361. https://doi.org/10.3390/biomedicines8090361
Sambon M, Gorlova A, Demelenne A, Alhama-Riba J, Coumans B, Lakaye B, Wins P, Fillet M, Anthony DC, Strekalova T, et al. Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration. Biomedicines. 2020; 8(9):361. https://doi.org/10.3390/biomedicines8090361
Chicago/Turabian StyleSambon, Margaux, Anna Gorlova, Alice Demelenne, Judit Alhama-Riba, Bernard Coumans, Bernard Lakaye, Pierre Wins, Marianne Fillet, Daniel C. Anthony, Tatyana Strekalova, and et al. 2020. "Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration" Biomedicines 8, no. 9: 361. https://doi.org/10.3390/biomedicines8090361
APA StyleSambon, M., Gorlova, A., Demelenne, A., Alhama-Riba, J., Coumans, B., Lakaye, B., Wins, P., Fillet, M., Anthony, D. C., Strekalova, T., & Bettendorff, L. (2020). Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration. Biomedicines, 8(9), 361. https://doi.org/10.3390/biomedicines8090361