HDX-MS: An Analytical Tool to Capture Protein Motion in Action


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
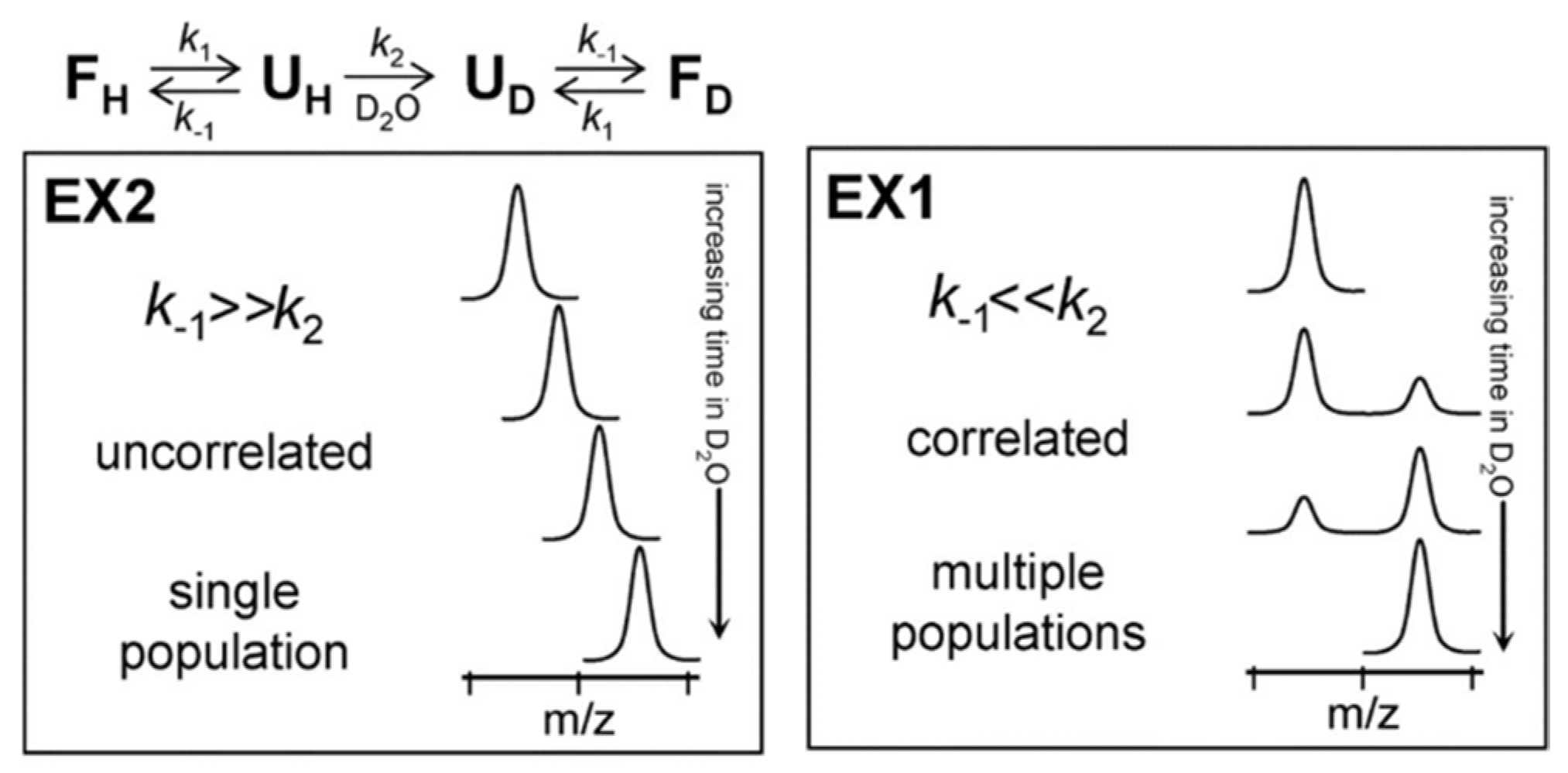
Underlying Theory
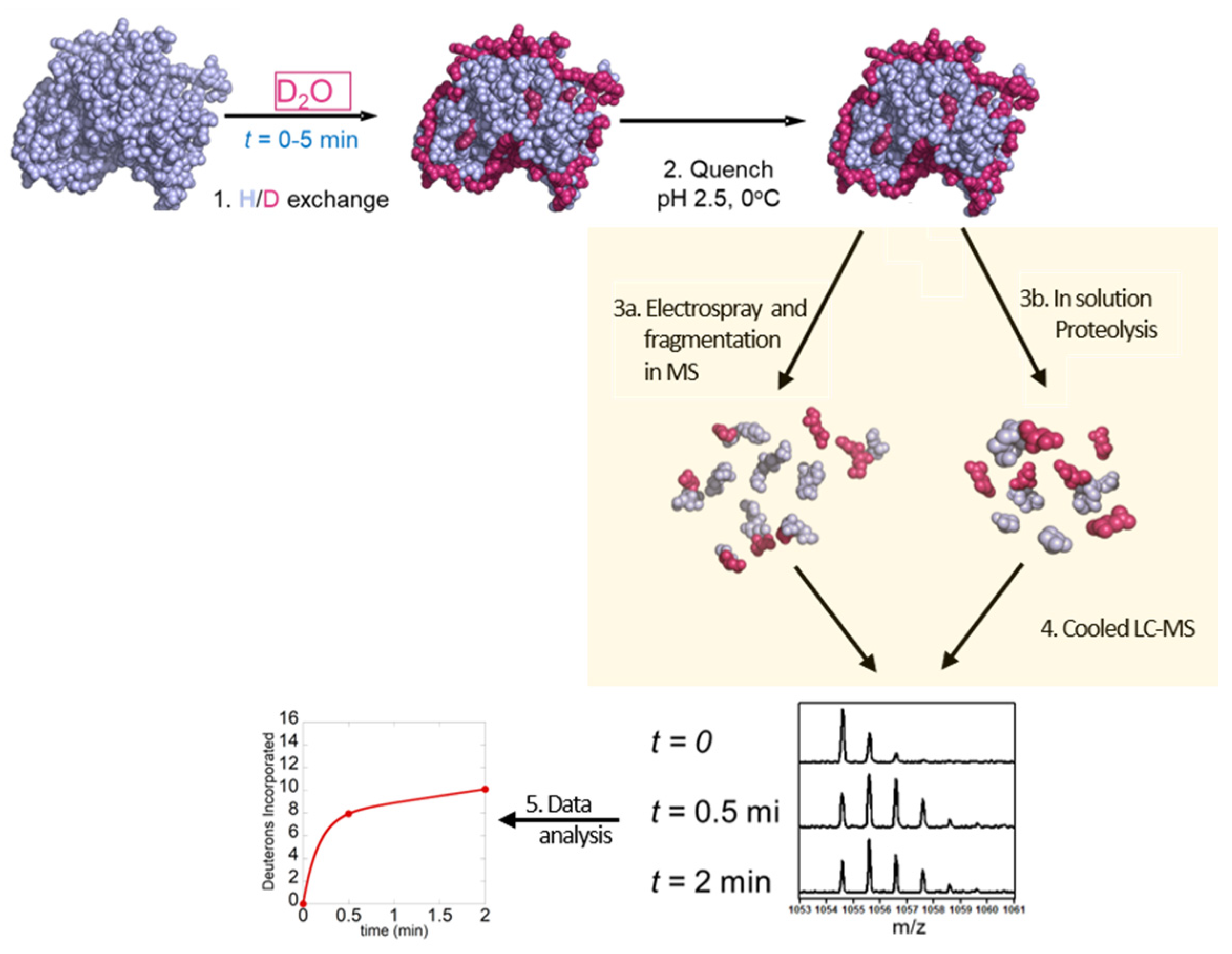
2. HDX-MS Methodology
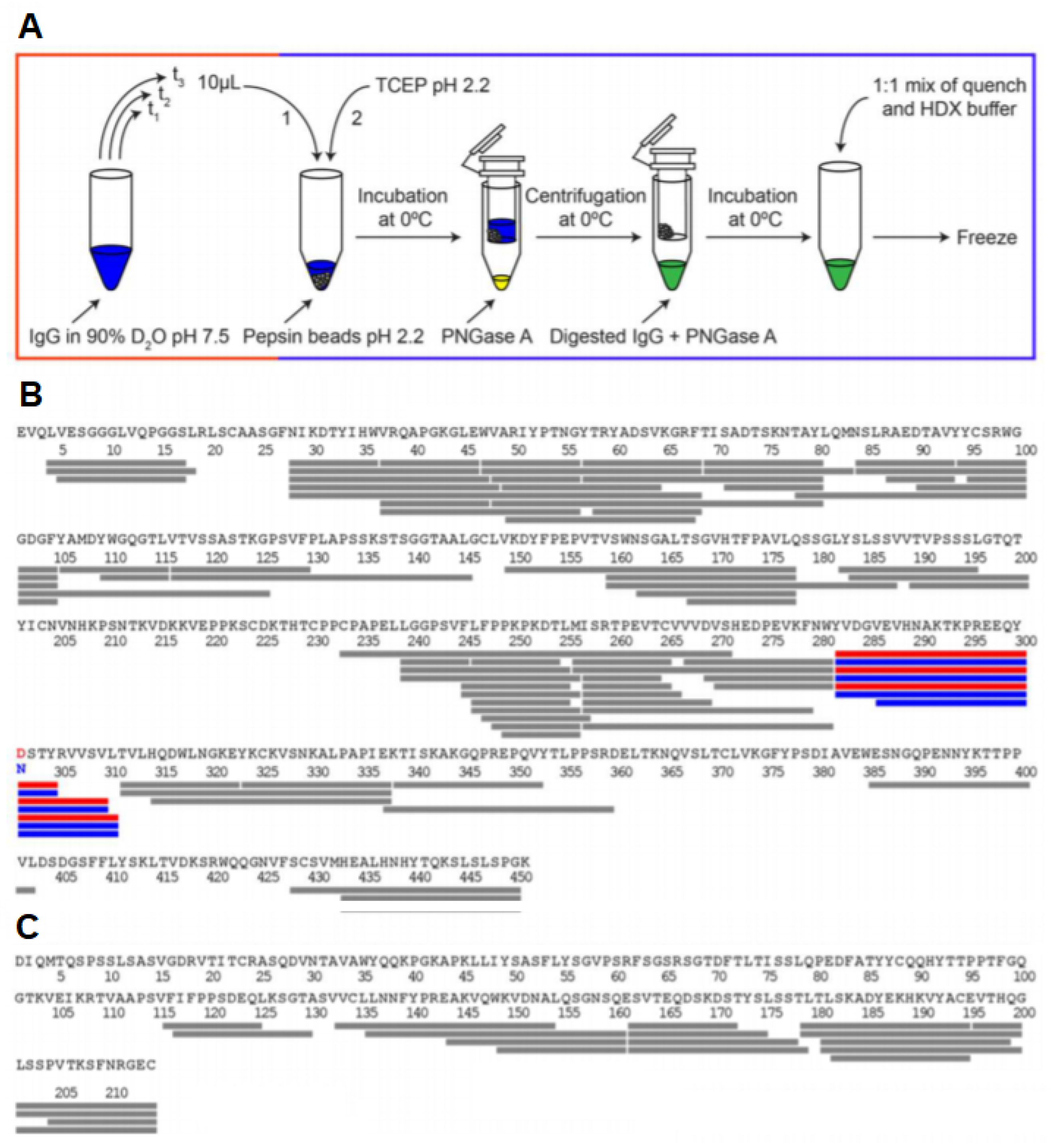
2.1. Labeling of Protein
2.2. Quenching and Digestion
2.3. Separation of Peptides and Mass Spectrometry
2.4. Data Analysis
2.5. Millisecond Bottom-Up HDX-MS
2.6. Top-Down HDX-MS
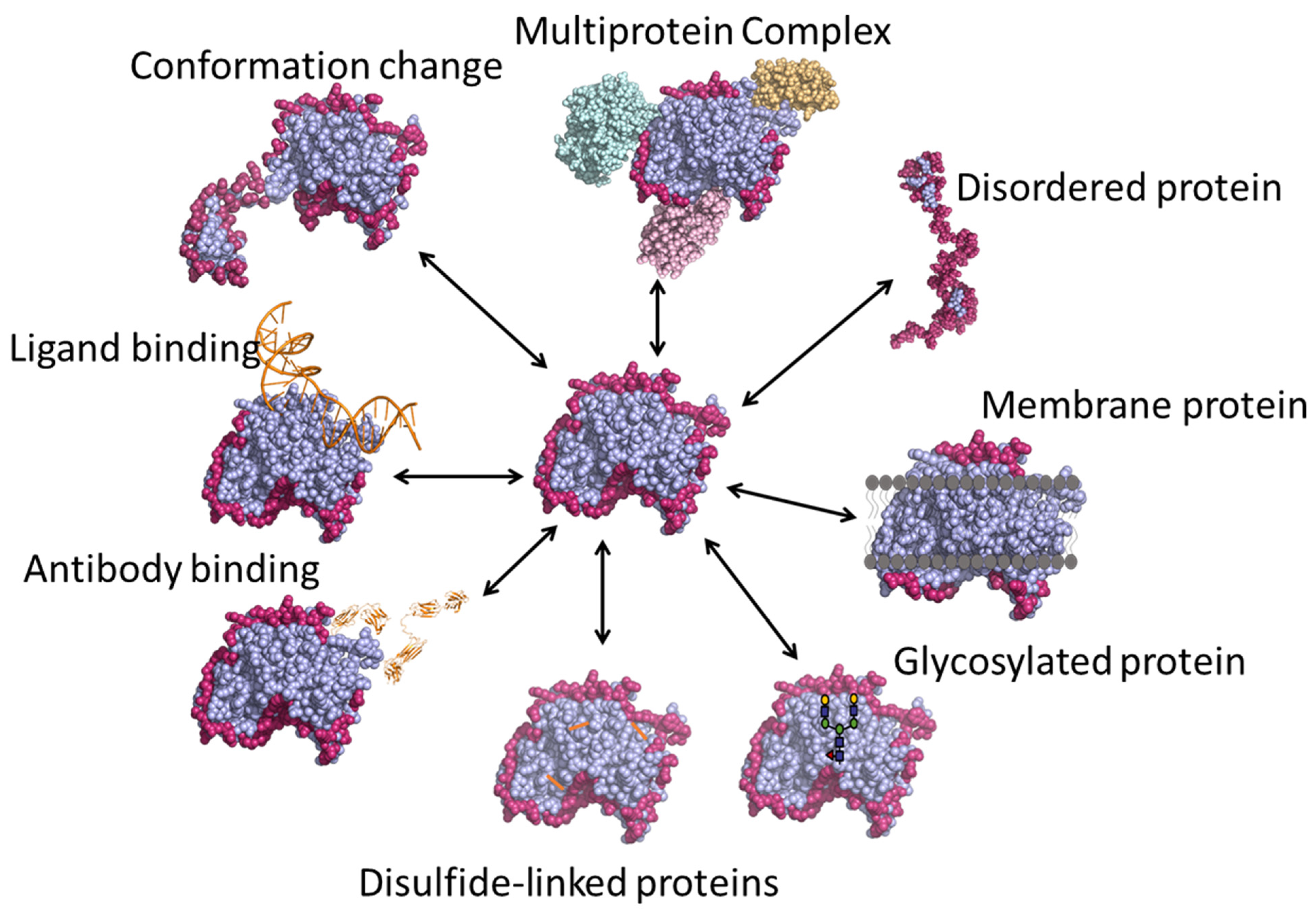
3. Applications of HDX-MS
3.1. Protein Conformation and Comparability
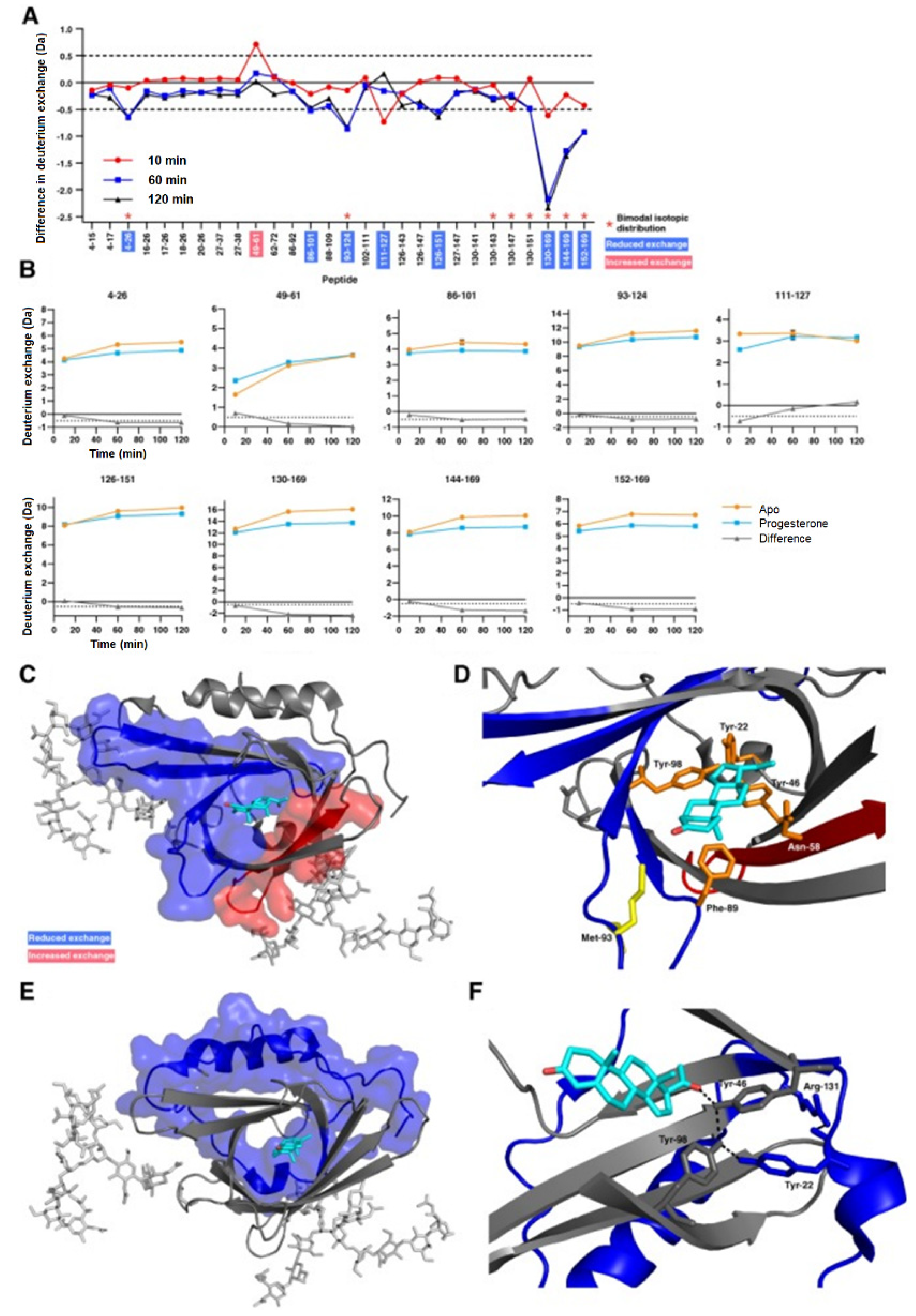
3.2. Protein–Small Molecule Binding
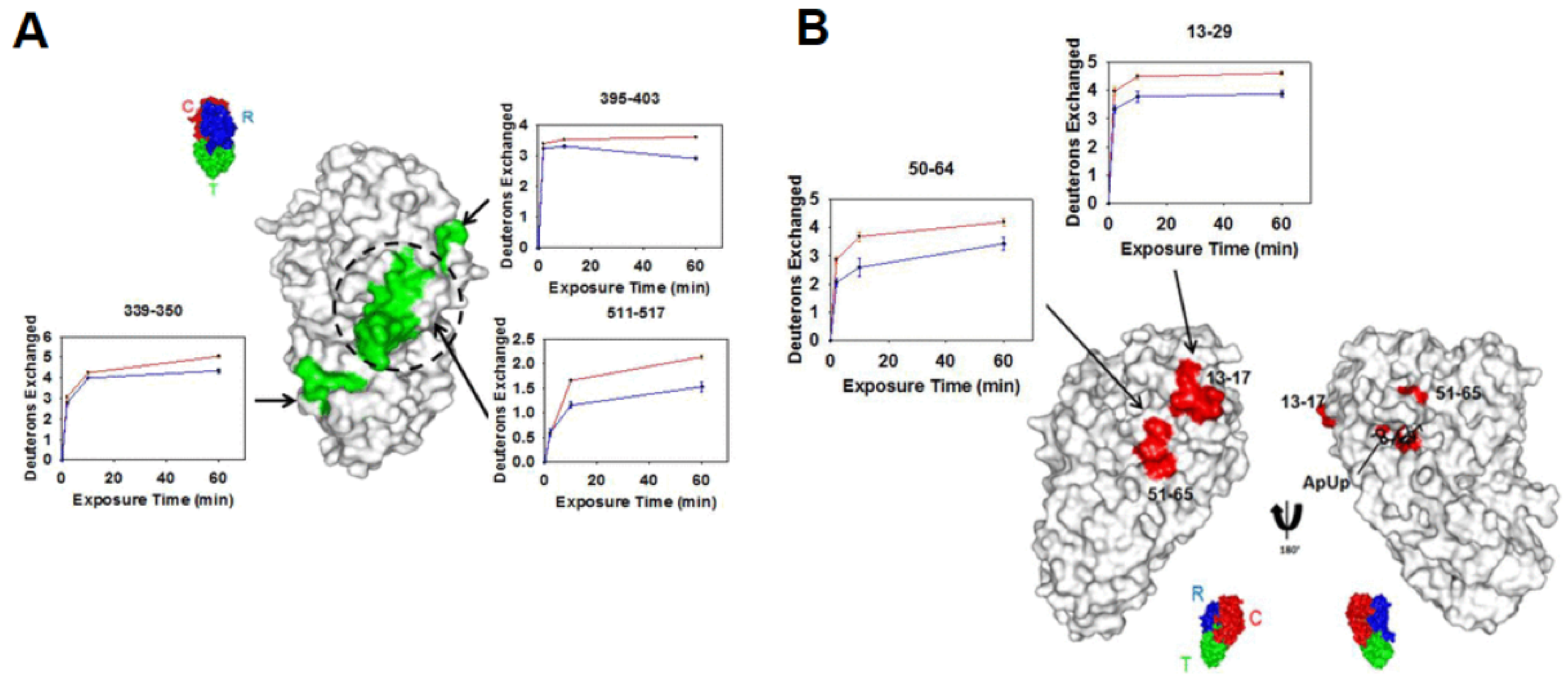
3.3. Epitope Mapping
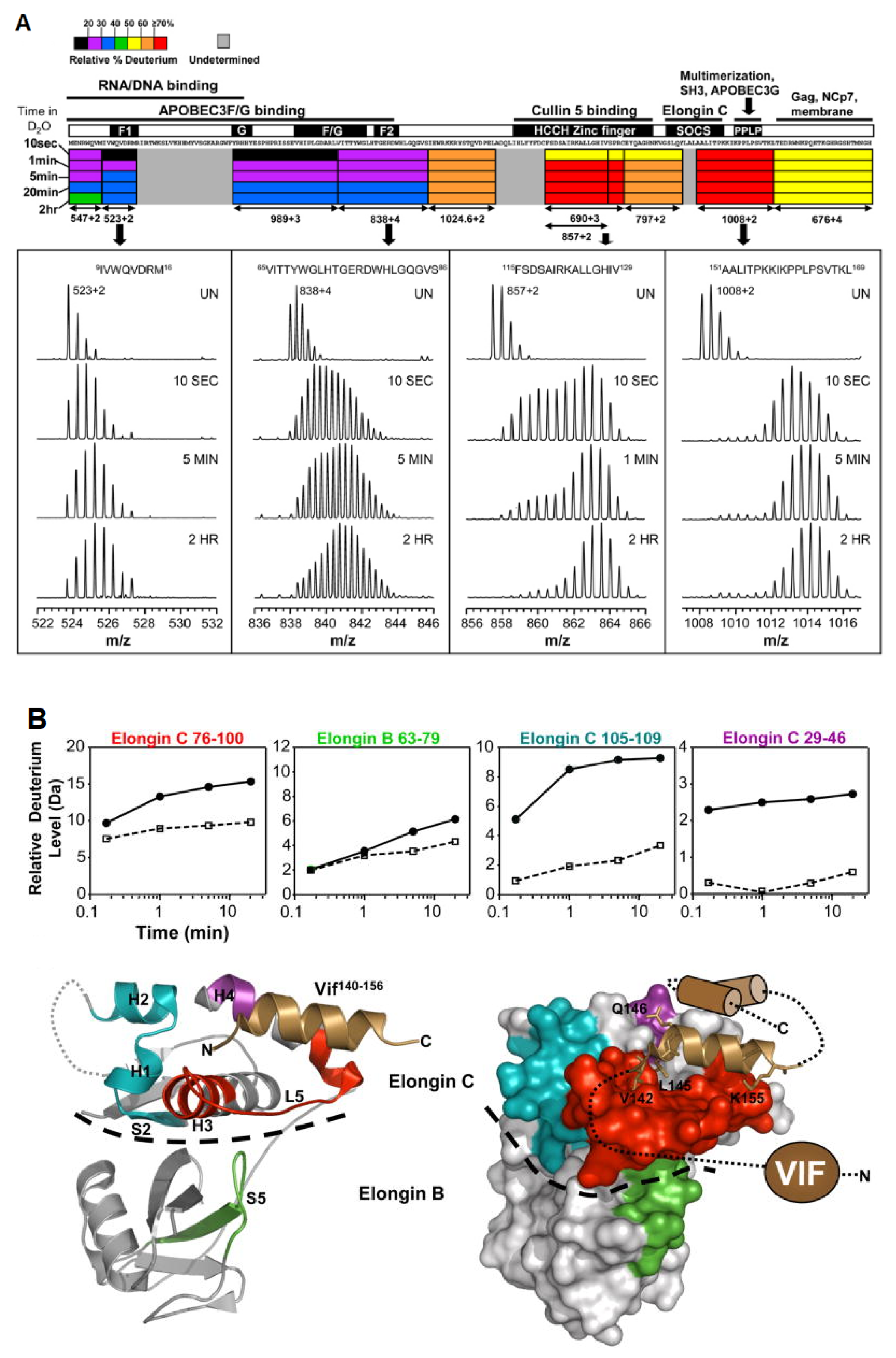
3.4. Multi-Protein Complexes
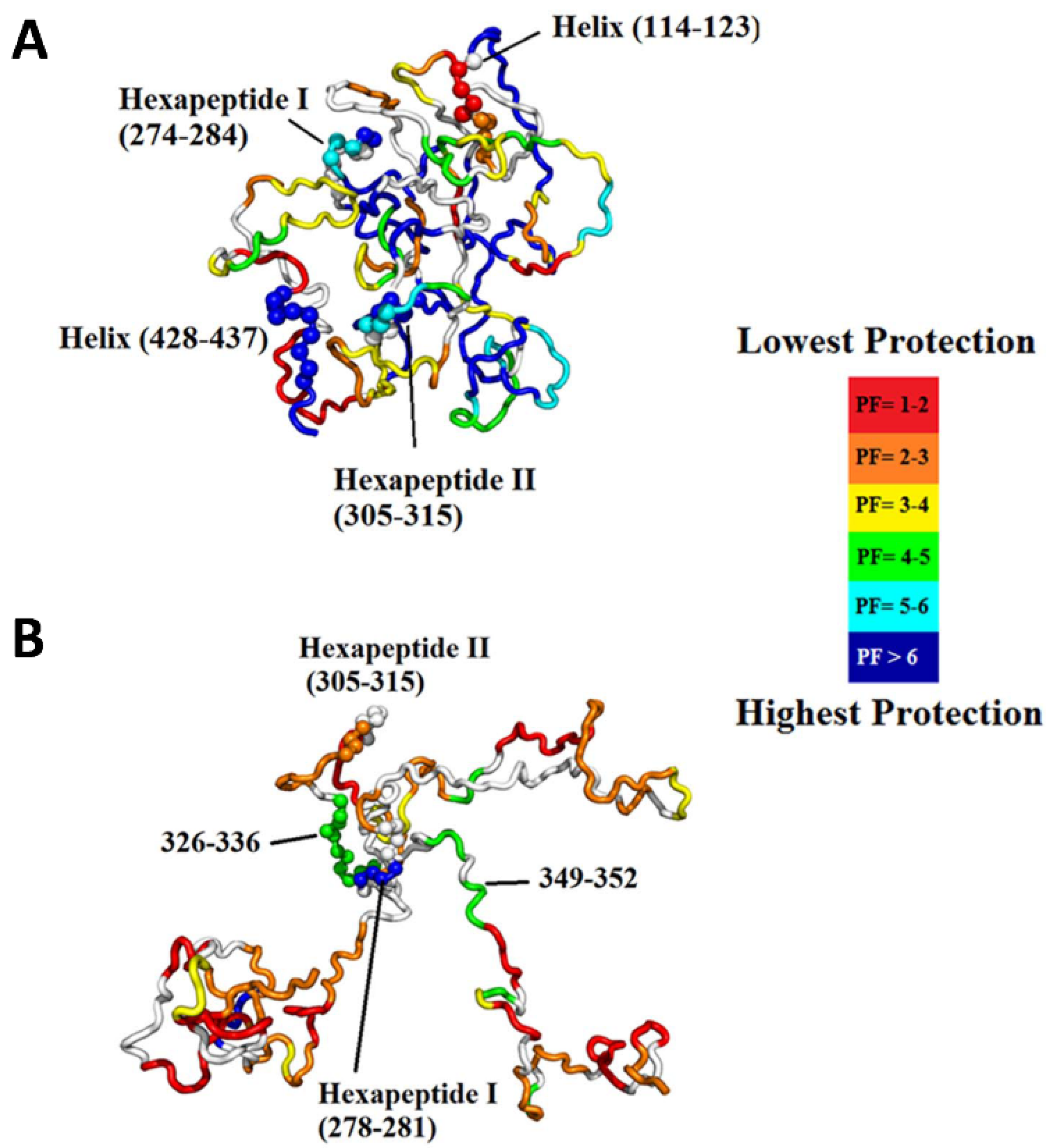
3.5. Intrinsically Disordered Proteins
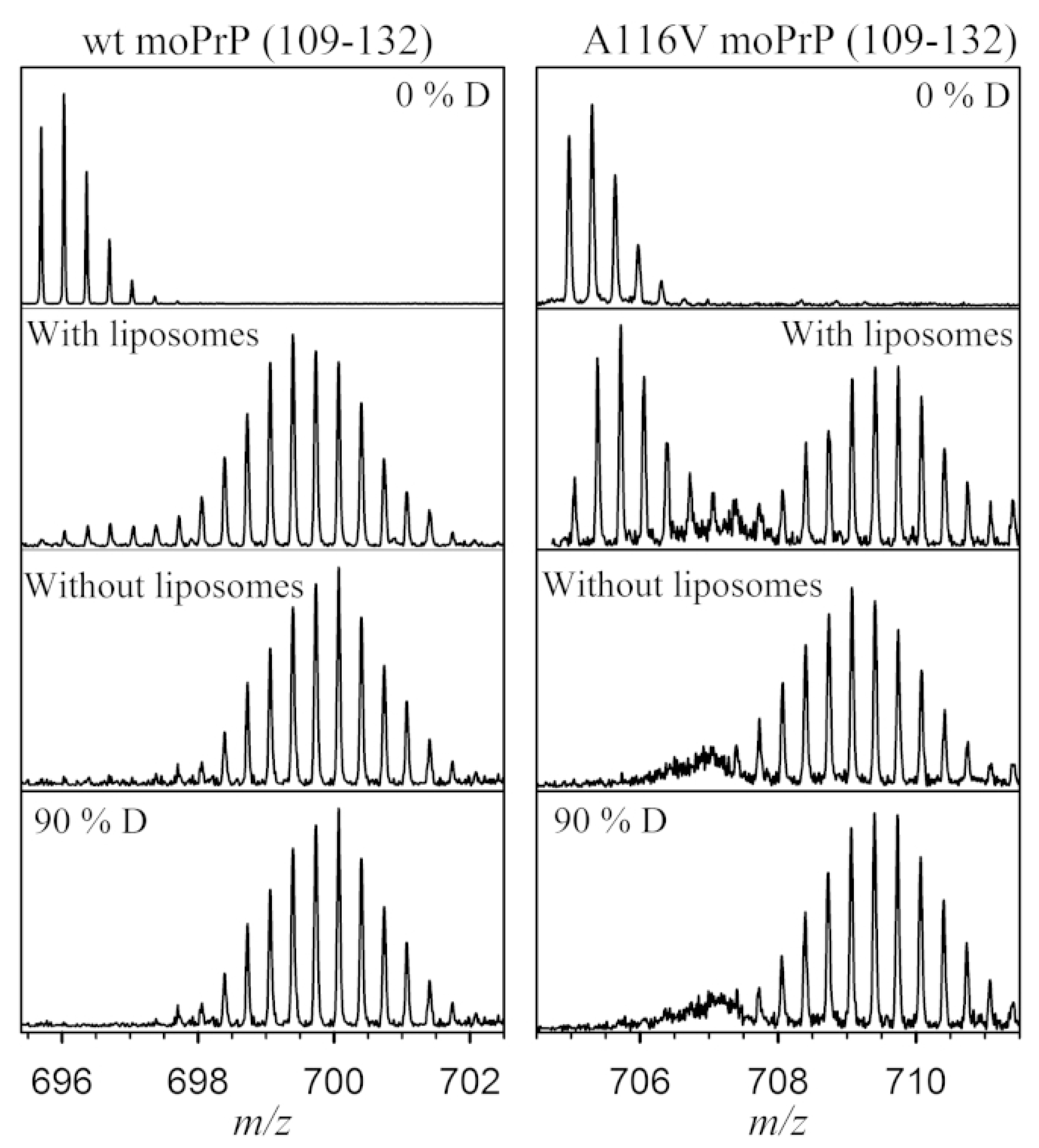
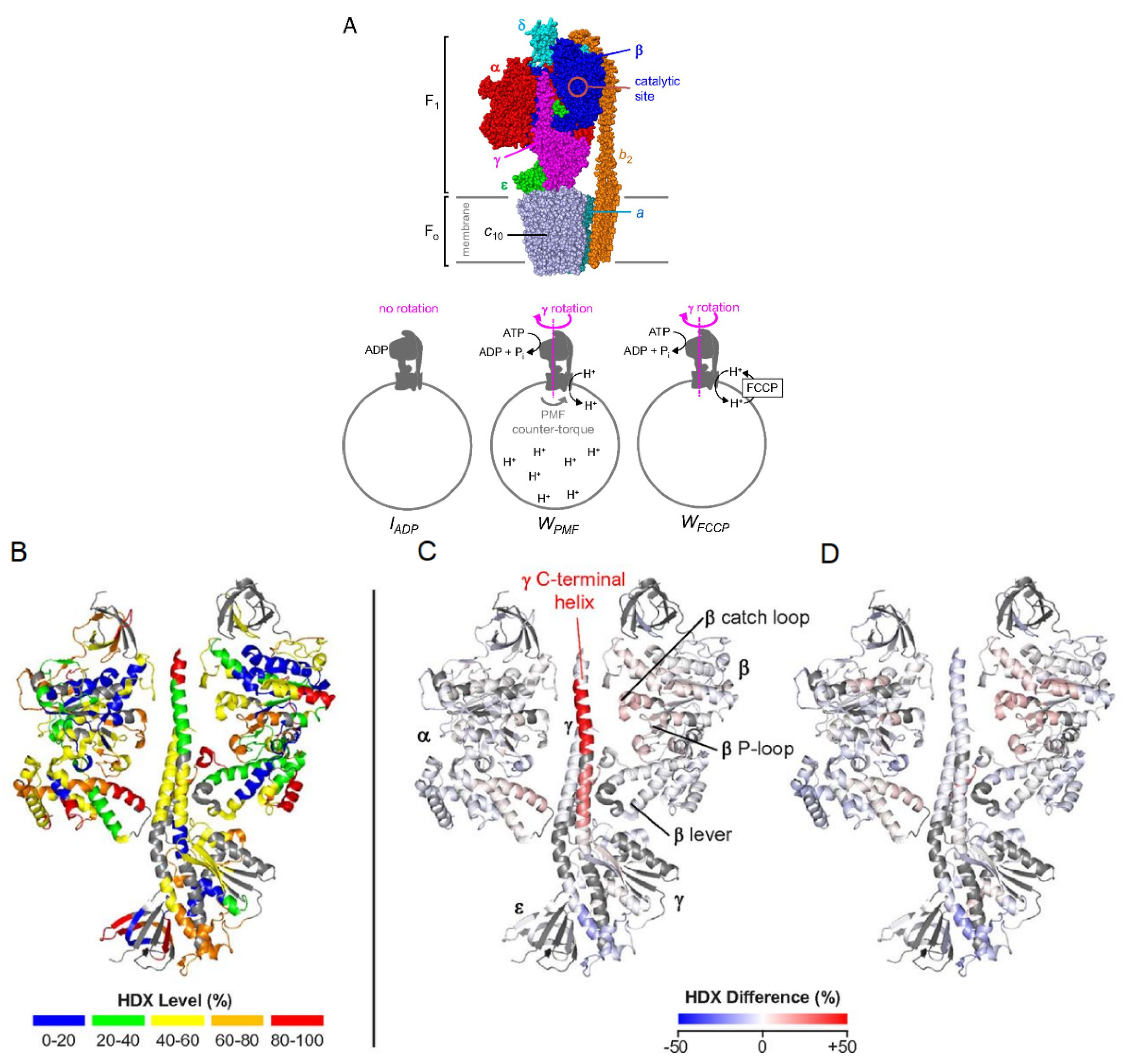
3.6. Membrane Proteins
3.7. Glycosylated Proteins
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, B.; Stevens, R.C. Advancing Chemokine GPCR Structure Based Drug Discovery. Structure 2019, 27, 405–408. [Google Scholar] [CrossRef]
- Deng, B.; Lento, C.; Wilson, D.J. Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development — A review. Anal Chim. Acta. 2016, 940, 8–20. [Google Scholar] [CrossRef]
- Berkowitz, S.A.; Engen, J.R.; Mazzeo, J.R.; Jones, G.B. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat. Rev. Drug Discov. 2012, 11, 527–540. [Google Scholar] [CrossRef]
- Beck, A.; Wagner-Rousset, E.; Ayoub, D.; Dorsselaer, V.A.; Sanglier-Cianférani, S. Characterization of Therapeutic Antibodies and Related Products. Anal Chem. 2013, 85, 715–736. [Google Scholar] [CrossRef]
- Skinner, J.J.; Lim, W.K.; Bédard, S.; Black, B.E.; Englander, S.W. Protein dynamics viewed by hydrogen exchange. Protein Science 2012, 21, 996–1005. [Google Scholar] [CrossRef]
- Hydrogen Exchange Mass Spectrometry of Proteins, Wiley Online Books n.d. Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/9781118703748 (accessed on 6 March 2020).
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins: Struct. Funct. Bioinform. 1993, 17, 75–86. [Google Scholar] [CrossRef]
- Nguyen, D.; Mayne, L.; Phillips, M.C.; Englander, W.S. Reference Parameters for Protein Hydrogen Exchange Rates. J Am Soc. Mass Spectrom. 2018, 29, 1936–1939. [Google Scholar] [CrossRef]
- Oganesyan, I.; Lento, C.; Wilson, D.J. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 2018, 144, 27–42. [Google Scholar] [CrossRef]
- Hvidt, A.; Nielsen, S.O. Hydrogen Exchange in Proteins. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 1966; Volume 21, pp. 287–386. [Google Scholar] [CrossRef]
- Smith, D.L.; Deng, Y.; Zhang, Z. Probing the Non-covalent Structure of Proteins by Amide Hydrogen Exchange and Mass Spectrometry. J. Mass Spectrom. 1997, 32, 135–146. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. of Biophys. 1983, 16, 521–655. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.J.; Wand, A.J. Local Dynamics and Stability of Apocytochrome b562 Examined by Hydrogen Exchange. Biochemistry 1998, 37, 3687–3698. [Google Scholar] [CrossRef] [PubMed]
- Cieplak-Rotowska, M.K.; Tarnowski, K.; Rubin, M.; Fabian, M.R.; Sonenberg, N.; Dadlez, M.; Niedzwiecka, A. Structural Dynamics of the GW182 Silencing Domain Including its RNA Recognition motif (RRM) Revealed by Hydrogen-Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Wildes, D.; Marqusee, S. Hydrogen exchange and ligand binding: Ligand-dependent and ligand-independent protection in the Src SH3 domain. Protein Sci. 2005, 14, 81–88. [Google Scholar] [CrossRef]
- Engen, J.R.; Smithgall, T.E.; Gmeiner, W.H.; Smith, D.L. Identification and Localization of Slow, Natural, Cooperative Unfolding in the Hematopoietic Cell Kinase SH3 Domain by Amide Hydrogen Exchange and Mass Spectrometry. Biochemistry 1997, 36, 14384–14391. [Google Scholar] [CrossRef]
- Slysz, G.W.; Percy, A.J.; Schriemer, D.C. Restraining Expansion of the Peak Envelope in H/D Exchange-MS and Its Application in Detecting Perturbations of Protein Structure/Dynamics. Anal. Chem. 2008, 80, 7004–7011. [Google Scholar] [CrossRef] [PubMed]
- Arora, J.; Hickey, J.M.; Majumdar, R.; Esfandiary, R.; Bishop, S.M.; Samra, H.S.; Middaugh, C.R.; Weis, D.D.; Volkin, D.B. Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. MAbs 2015, 7, 525–539. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, L.; Ruotolo, B.T.; Wang, N. Hydrogen/deuterium exchange-mass spectrometry analysis of high concentration biotherapeutics: Application to phase-separated antibody formulations. MAbs 2019, 11, 779–788. [Google Scholar] [CrossRef]
- Hudgens, J.W.; Gallagher, E.S.; Karageorgos, I.; Anderson, K.W.; Filliben, J.J.; Huang, R.Y.-C.; Chen, G.D.; Bou-Assaf, G.M.; Espada, A.; Chalmers, M.J.; et al. Interlaboratory Comparison of Hydrogen-Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Anal. Chem. 2019, 91, 7336–7345. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nature Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; McLoughlin, S.M.; Frausto, S.D.; Tang, H.; Emmett, MR.; Marshall, A.G. Simultaneous Reduction and Digestion of Proteins with Disulfide Bonds for Hydrogen/Deuterium Exchange Monitored by Mass Spectrometry. Anal. Chem. 2010, 82, 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Cravello, L.; Lascoux, D.; Forest, E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 2003, 17, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Tsiatsiani, L.; Akeroyd, M.; Olsthoorn, M.; Heck, A.J.R. Aspergillus niger Prolyl Endoprotease for Hydrogen–Deuterium Exchange Mass Spectrometry and Protein Structural Studies. Anal. Chem. 2017, 89, 7966–7973. [Google Scholar] [CrossRef]
- Espada, A.; Haro, R.; Castañon, J.; Sayago, C.; Perez-Cozar, F.; Cano, L.; Redero, P.; Molina-Martin, M.; Broughton, H.; Stites, R.E.; et al. A Decoupled Automation Platform for Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. J. Am. Soc. Mass Spectrom. 2019, 30, 2580–2583. [Google Scholar] [CrossRef]
- McAllister, R.G.; Konermann, L. Challenges in the Interpretation of Protein H/D Exchange Data: A Molecular Dynamics Simulation Perspective. Biochemistry 2015, 54, 2683–2692. [Google Scholar] [CrossRef]
- Brown, K.A.; Wilson, D.J. Bottom-up hydrogen deuterium exchange mass spectrometry: Data analysis and interpretation. Analyst 2017, 142, 2874–2886. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, A.; Xiao, G. Improved Protein Hydrogen/Deuterium Exchange Mass Spectrometry Platform with Fully Automated Data Processing. Anal Chem. 2012, 84, 4942–4949. [Google Scholar] [CrossRef]
- Pascal, B.D.; Willis, S.; Lauer, J.L.; Landgraf, R.R.; West, G.M.; Marciano, D.; Novick, S.; Goswami, D.; Chalmers, M.J.; Griffin, P.R. HDX Workbench: Software for the Analysis of H/D Exchange MS Data. J Am Soc. Mass Spectrom. 2012, 23. [Google Scholar] [CrossRef]
- Lumpkin, R.; Komives, E.A. DECA, a comprehensive, automatic post-processing program for HDX-MS data. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef]
- Pascal, B.D.; Chalmers, M.J.; Busby, S.A.; Griffin, P.R. HD Desktop: An integrated platform for the analysis and visualization of H/D exchange data. J. Am. Soc Mass Spectrom. 2009, 20, 601–610. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slysz, G.W.; Baker, C.A.; Bozsa, B.M.; Dang, A.; Percy, A.J.; Bennett, M.; Schriemer, D.C. Hydra: Software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinform. 2009, 10, 162. [Google Scholar] [CrossRef]
- Kan, Z.-Y.; Mayne, L.; Sevugan Chetty, P.; Englander, S.W. ExMS: Data Analysis for HX-MS Experiments. J. Am. Soc. Mass Spectrom. 2011, 22. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Dunn, S.D.; Konermann, L. Conformational Dynamics of Partially Denatured Myoglobin Studied by Time-Resolved Electrospray Mass Spectrometry with Online Hydrogen−Deuterium Exchange. Biochemistry 2003, 42, 9248. [Google Scholar] [CrossRef][Green Version]
- Rob, T.; Liuni, P.; Gill, P.K.; Zhu, S.; Balachandran, N.; Berti, P.J.; Wilson, D.J. Measuring Dynamics in Weakly Structured Regions of Proteins Using Microfluidics-Enabled Subsecond H/D Exchange Mass Spectrometry. Anal. Chem. 2012, 84, 3771–3779. [Google Scholar] [CrossRef]
- Weis, D.D.; Wales, T.E.; Engen, J.R.; Hotchko, M.; Ten Eyck, L.F. Identification and Characterization of EX1 Kinetics in H/D Exchange Mass Spectrometry by Peak Width Analysis. J. Am. Soc. Mass Spectrom. 2006, 17, 1498–1509. [Google Scholar] [CrossRef]
- Rob, T.; Gill, P.K.; Golemi-Kotra, D.; Wilson, D.J. An electrospray ms-coupled microfluidic device for sub-second hydrogen/deuterium exchange pulse-labelling reveals allosteric effects in enzyme inhibition. Lab. Chip 2013, 13, 2528–2532. [Google Scholar] [CrossRef]
- Lento, C.; Zhu, S.; Brown, K.A.; Knox, R.; Liuni, P.; Wilson, D.J. Time-resolved ElectroSpray Ionization Hydrogen-deuterium Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Rob, T.; Wilson, D.J. Time-resolved mass spectrometry for monitoring millisecond time-scale solution-phase processes. Eur..J. Mass Spectrom. 2012, 18, 205–214. [Google Scholar] [CrossRef]
- Wilson, D.J.; Konermann, L. A Capillary Mixer with Adjustable Reaction Chamber Volume for Millisecond Time-Resolved Studies by Electrospray Mass Spectrometry. Anal. Chem. 2003, 75, 6408–6414. [Google Scholar] [CrossRef]
- Knox, R.; Lento, C.; Wilson, DJ. Mapping Conformational Dynamics to Individual Steps in the TEM-1 β-Lactamase Catalytic Mechanism. J. Mol. Biol. 2018, 430, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Zehl, M.; Jensen, O.N.; Jørgensen, T.J.D. Protein Hydrogen Exchange Measured at Single-Residue Resolution by Electron Transfer Dissociation Mass Spectrometry. Anal. Chem. 2009, 81, 5577–5584. [Google Scholar] [CrossRef]
- Rand, K.D.; Zehl, M.; Jørgensen, T.J.D. Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: Overcoming gas-phase hydrogen/deuterium scrambling. Acc. Chem. Res. 2014, 47, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Han, J.; Borchers, C.H.; Konermann, L. Hydrogen/Deuterium Exchange Mass Spectrometry with Top-Down Electron Capture Dissociation for Characterizing Structural Transitions of a 17 kDa Protein. J. Am. Chem. Soc. 2009, 131, 12801–12808. [Google Scholar] [CrossRef] [PubMed]
- Zehl, M.; Rand, K.D.; Jensen, O.N.; Jørgensen, T.J.D. Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc. 2008, 130, 17453–17459. [Google Scholar] [CrossRef]
- Sabareesan, A.T.; Singh, J.; Roy, S.; Udgaonkar, J.B.; Mathew, M.K. The Pathogenic A116V Mutation Enhances Ion-Selective Channel Formation by Prion Protein in Membranes. Biophys. J. 2016, 110, 1766–1776. [Google Scholar] [CrossRef]
- Marcsisin, S.R.; Narute, P.S.; Emert-Sedlak, L.A.; Kloczewiak, M.; Smithgall, T.E.; Engen, J.R. On the Solution Conformation and Dynamics of the HIV-1 Viral Infectivity Factor. J. Mol. Biol. 2011, 410, 1008–1022. [Google Scholar] [CrossRef][Green Version]
- Marcsisin, S.R.; Engen, J.R. Molecular Insight into the Conformational Dynamics of the Elongin BC Complex and Its Interaction with HIV-1 Vif. J. Mol. Biol. 2010, 402, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Walker, D.W. Apolipoprotein D: An overview of its role in aging and age-related diseases. Cell Cycle 2010, 9, 269–273. [Google Scholar] [CrossRef]
- Kielkopf, C.S.; Ghosh, M.; Anand, G.S.; Brown, S.H.J. HDX-MS reveals orthosteric and allosteric changes in apolipoprotein-D structural dynamics upon binding of progesterone. Protein Sci. 2019, 28, 365–374. [Google Scholar] [CrossRef]
- Masson, G.R.; Jenkins, M.L.; Burke, J.E. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin. Drug Discov. 2017, 12, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Baerga-Ortiz, A.; Hughes, C.A.; Mandell, J.G.; Komives, E.A. Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 2002, 11, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Faleri, A.; Lo Surdo, P.; Veggi, D.; Maruggi, G.; Grassi, E.; Cartocci, E.; Bertoldi, I.; Genovese, A.; Santini, L.; et al. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc. Natl. Acad. Sci. USA 2013, 110, 3304–3309. [Google Scholar] [CrossRef]
- Wei, H.; Mo, J.; Tao, L.; Russell, R.J.; Tymiak, A.A.; Chen, G.; Iacob, R.E.; Engen, J.R. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: Methodology and applications. Drug Discov. Today 2014, 19, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Marciano, D.P.; Dharmarajan, V.; Griffin, P.R. HDX-MS guided drug discovery: Small molecules and biopharmaceuticals. Curr. Opin. Struct. Biol. 2014, 28, 105–111. [Google Scholar] [CrossRef]
- Puchades, C.; Kűkrer, B.; Diefenbach, O.; Sneekes-Vriese, E.; Juraszek, J.; Koudstaal, W.; Apetri, A. Epitope mapping of diverse influenza Hemagglutinin drug candidates using HDX-MS. Sci. Rep. 2019, 9, 4735. [Google Scholar] [CrossRef]
- Zhu, S.; Liuni, P.; Ettorre, L.; Chen, T.; Szeto, J.; Carpick, B.; James, D.A.; Wilson, D.J. Hydrogen–Deuterium Exchange Epitope Mapping Reveals Distinct Neutralizing Mechanisms for Two Monoclonal Antibodies against Diphtheria Toxin. Biochemistry 2019, 58, 646–656. [Google Scholar] [CrossRef]
- Vahidi, S.; Bi, Y.; Dunn, S.D.; Konermann, L. Load-dependent destabilization of the γ-rotor shaft in FOF1 ATP synthase revealed by hydrogen/deuterium-exchange mass spectrometry. PNAS 2016, 113, 2412–2417. [Google Scholar] [CrossRef]
- Sheff, J.G.; Hepburn, M.; Yu, Y.; Lees-Miller, S.P.; Schriemer, D.C. Nanospray HX-MS configuration for structural interrogation of large protein systems. Analyst 2017, 142, 904–910. [Google Scholar] [CrossRef]
- Keppel, T.R.; Weis, D.D. Mapping Residual Structure in Intrinsically Disordered Proteins at Residue Resolution Using Millisecond Hydrogen/Deuterium Exchange and Residue Averaging. J. Am. Soc. Mass Spectrom. 2015, 26, 547–554. [Google Scholar] [CrossRef]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D.J. Hyperphosphorylation of Intrinsically Disordered Tau Protein Induces an Amyloidogenic Shift in Its Conformational Ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef]
- Benhaim, M.; Lee, K.K.; Guttman, M. Tracking Higher Order Protein Structure by Hydrogen-Deuterium Exchange Mass Spectrometry. Protein Pept. Lett. 2019, 26, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chien, E.Y.T.; Chalmers, M.J.; Pascal, B.D.; Gatchalian, J.; Stevens, R.C.; Griffin, P.R. Dynamics of the β2-Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen−Deuterium Exchange. Anal. Chem. 2010, 82, 1100–1108. [Google Scholar] [CrossRef]
- Duc, N.M.; Du, Y.; Zhang, C.; Lee, S.Y.; Thorsen, T.S.; Kobilka, B.K.; Chung, K.Y. Effective Application of Bicelles for Conformational Analysis of G Protein-Coupled Receptors by Hydrogen/deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Redhair, M.; Clouser, A.F.; Atkins, W.M. Hydrogen-deuterium exchange mass spectrometry of membrane proteins in lipid nanodiscs. Chem. Phys. Lipids 2019, 220, 14–22. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Hourdel, V.; Chenal, A.; Brier, S. Hydrogen/Deuterium Exchange Mass Spectrometry for the Structural Analysis of Detergent-Solubilized Membrane Proteins. Methods Mol. Biol. 2020, 2127, 339–358. [Google Scholar] [CrossRef]
- Martens, C.; Politis, A. A glimpse into the molecular mechanism of integral membrane proteins through hydrogen-deuterium exchange mass spectrometry. Protein Sci. 2020, 29, 1285–1301. [Google Scholar] [CrossRef]
- Jensen, P.F.; Comamala, G.; Trelle, M.B.; Madsen, J.B.; Jørgensen, T.J.D.; Rand, K.D. Removal of N-Linked Glycosylations at Acidic pH by PNGase A Facilitates Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of N-Linked Glycoproteins. Anal. Chem. 2016, 88, 12479–12488. [Google Scholar] [CrossRef]
- Barton, C.; Li, X.S.; Li, S.P.; Flaherty, B.; Sison, L.; Lu, Q.; Yeung, B.; Wu, S.L. Impact of Glycosylation on the Comparability of the Higher-Order Structures in Idursulfase by Hydrogen-Deuterium Exchange Mass Spectrometry. Anal. Chem. 2020, 92, 8306–8314. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, O.T.; Seneviratne, C.A.; Gallagher, E.S. Applying an Internal Standard to Improve the Repeatability of In-electrospray H/D Exchange of Carbohydrate-Metal Adducts. J. Am. Soc. Mass Spectrom. 2019, 30, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, W.; O’Connor, G. Assessment of the repeatability and reproducibility of hydrogen/deuterium exchange mass spectrometry measurements. Rapid Commun. Mass Spectrom. 2008. [Google Scholar] [CrossRef] [PubMed]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narang, D.; Lento, C.; J. Wilson, D. HDX-MS: An Analytical Tool to Capture Protein Motion in Action. Biomedicines 2020, 8, 224. https://doi.org/10.3390/biomedicines8070224
Narang D, Lento C, J. Wilson D. HDX-MS: An Analytical Tool to Capture Protein Motion in Action. Biomedicines. 2020; 8(7):224. https://doi.org/10.3390/biomedicines8070224
Chicago/Turabian StyleNarang, Dominic, Cristina Lento, and Derek J. Wilson. 2020. "HDX-MS: An Analytical Tool to Capture Protein Motion in Action" Biomedicines 8, no. 7: 224. https://doi.org/10.3390/biomedicines8070224
APA StyleNarang, D., Lento, C., & J. Wilson, D. (2020). HDX-MS: An Analytical Tool to Capture Protein Motion in Action. Biomedicines, 8(7), 224. https://doi.org/10.3390/biomedicines8070224