Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma


Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of Recombinant SMAD4
2.2. Synthesis and Characterization of Anti-SMAD4 DNA Aptamer
2.3. Aptamer Immobilization on Magnetic Beads
2.4. Pull-Down of rSMAD4 from Blood Plasma
2.5. ‘In Silico’ Selection of Proteotypic Peptides for SMAD4 and Synthesis of Internal Standards
2.6. Tryptic Digestion
2.7. SRM Analysis and Data Processing
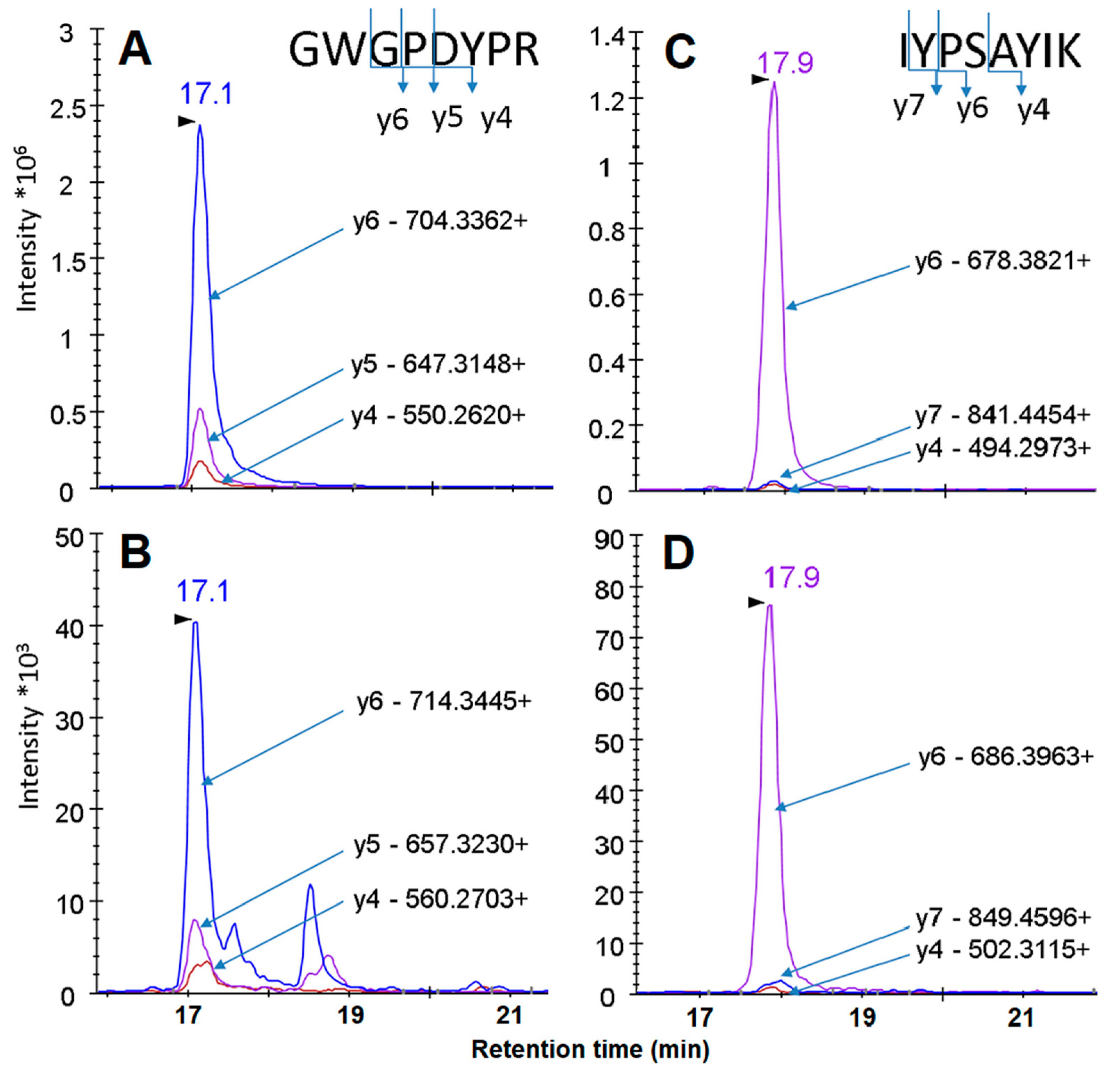
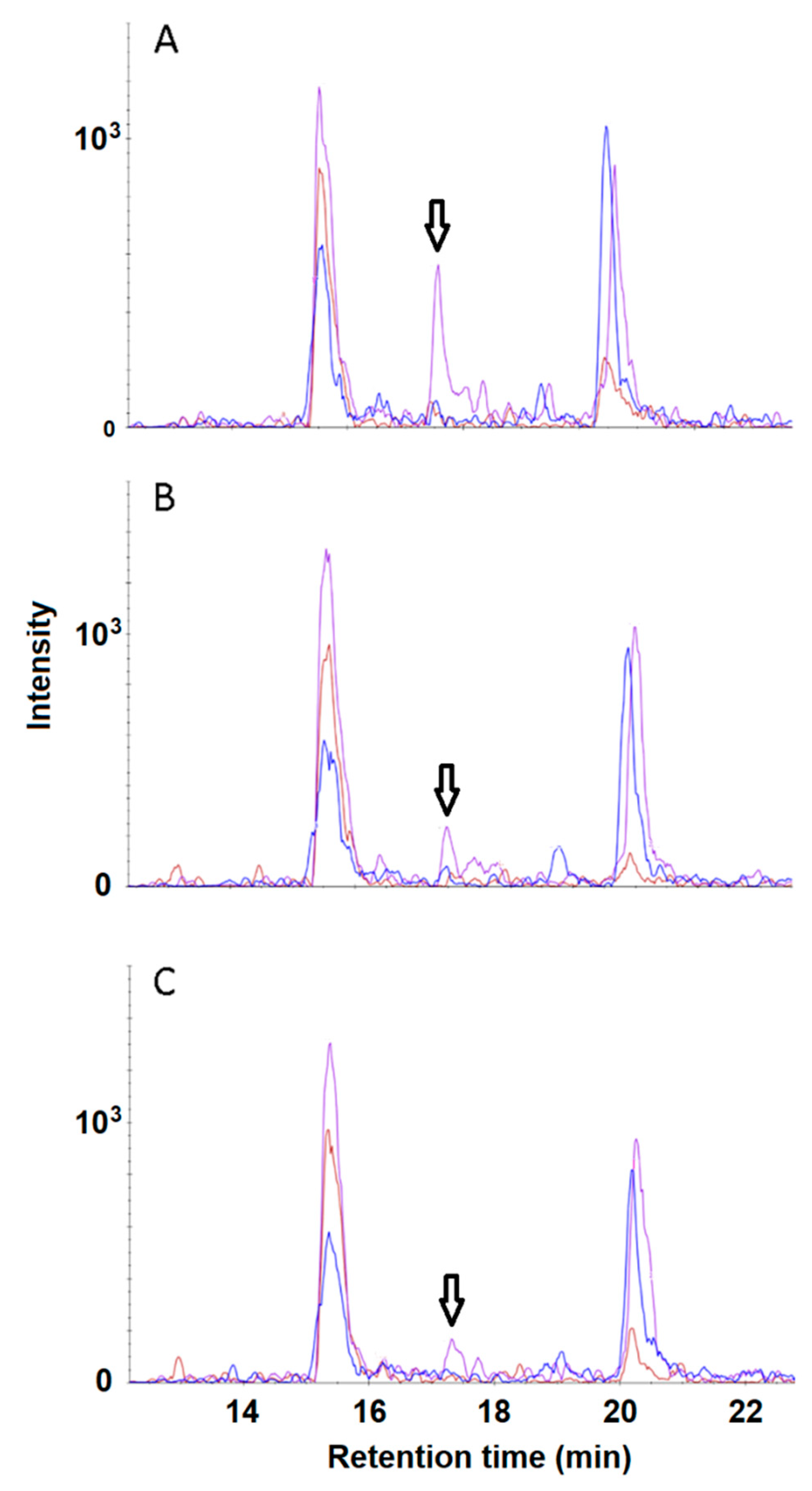
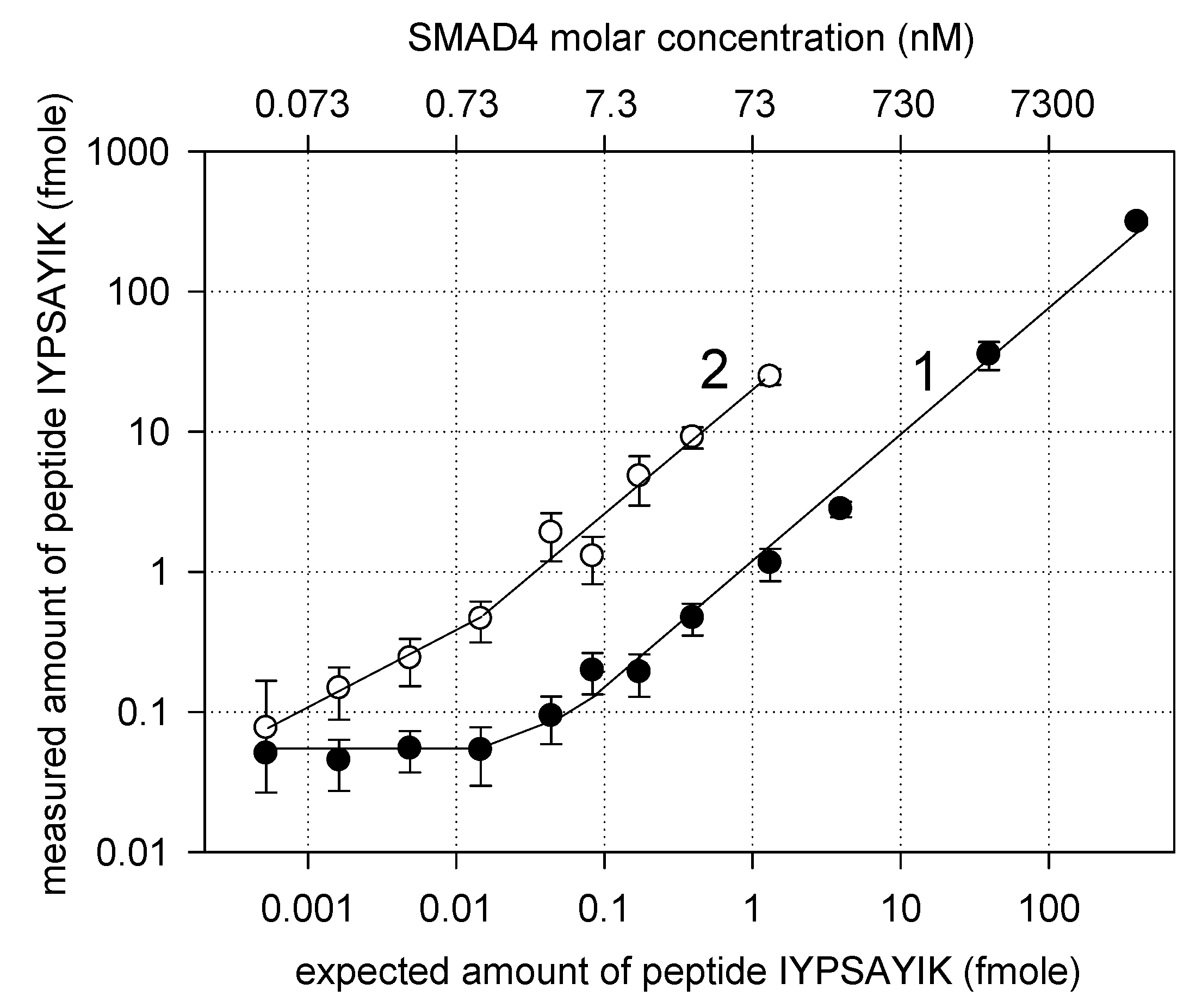
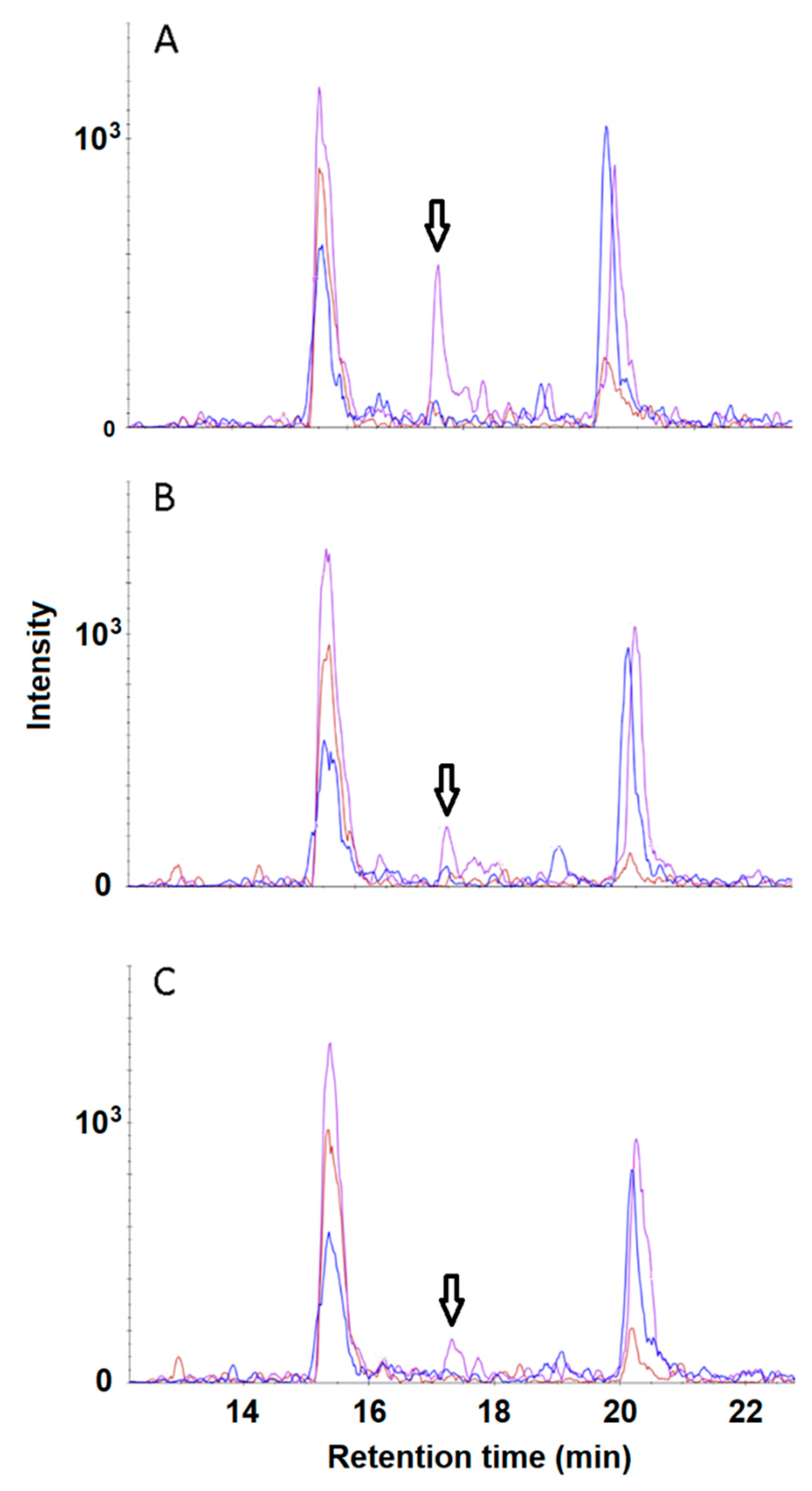
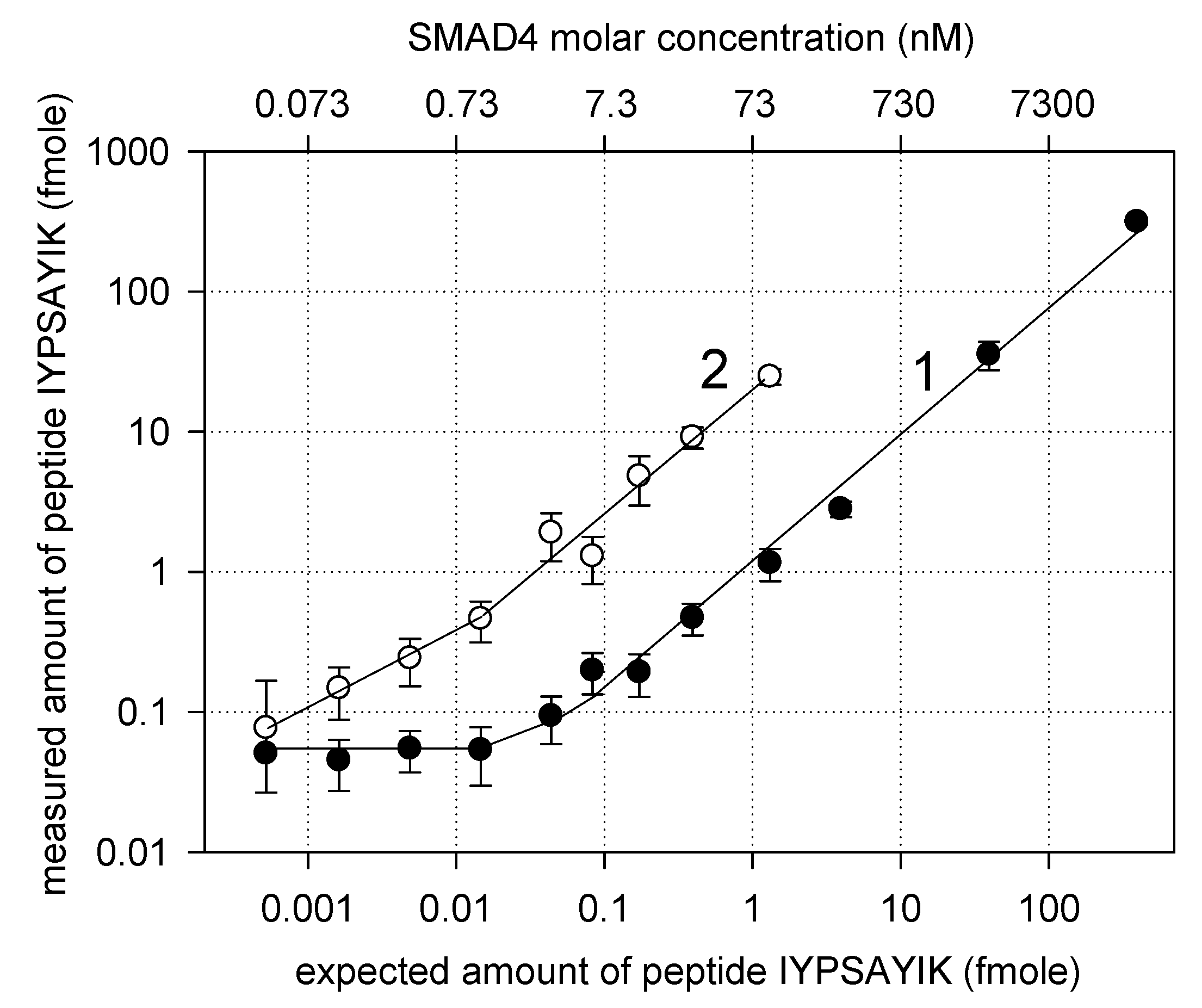
3. Results and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MS | mass spectrometry |
| MALDI-ToF-MS | matrix assisted laser desorption/ionization - time of flight - mass spectrometry |
| SRM | selected reaction monitoring |
| LAPs | low abundant proteins |
| mAbs | monoclonal antibodies |
| SELEX | Systematic Evolution of Ligands by EXponential enrichment |
| SBA | SMAD-binding element |
| LOD | limit of detection |
| LOB | limit of blank |
| SD | standard deviation |
| BCA | bicinchoninic acid |
| RT | room temperature |
| FASP | filter aided sample preparation |
References
- Chen, Y.; Liu, L. Targeted Proteomics. Breast Cancer 2019, 1871, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.G.; Polanski, M.; Pieper, R.; Gatlin, T.; Tirumalai, R.S.; Conrads, T.P.; Veenstra, T.D.; Adkins, J.N.; Pounds, J.G.; Fagan, R.; et al. The Human Plasma Proteome: A nonredundant list developed by combination of four separate sources. Mol. Cell. Proteom. 2004, 3, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, P.; Vondriska, T.M.; Creighton, C.J.; Gandhi, T.; Yang, Z.; Menon, R.; Kwon, M.-S.; Cho, S.Y.; Drwal, G.; Kellmann, M.; et al. A functional annotation of subproteomes in human plasma. Proteomics 2005, 5, 3506–3519. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, A.; Ponomarenko, E.A.; Ilgisonis, E.V.; Pyatnitskiy, M.; Lisitsa, A.V.; Poverennaya, E.V.; Kiseleva, O.I.; Farafonova, T.E.; Tikhonova, O.V.; Zavialova, M.; et al. 200+ Protein Concentrations in Healthy Human Blood Plasma: Targeted Quantitative SRM SIS Screening of Chromosomes 18, 13, Y, and the Mitochondrial Chromosome Encoded Proteome. J. Proteome Res. 2018, 18, 120–129. [Google Scholar] [CrossRef]
- Beck, H.C.; Overgaard, M.; Rasmussen, L.M. Plasma proteomics to identify biomarkers – application to cardiovascular diseases. Transl. Proteom. 2015, 7, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.-J.; Kaleta, D.T.; Petritis, B.O.; Jiang, H.; Liu, T.; Zhang, X.; Mottaz, H.M.; Varnum, S.M.; Camp, D.G.; Huang, L.; et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell. Proteom. 2008, 7, 1963–1973. [Google Scholar] [CrossRef] [Green Version]
- Millioni, R.; Tolin, S.; Puricelli, L.; Sbrignadello, S.; Fadini, G.P.; Tessari, P.; Arrigoni, G. High Abundance Proteins Depletion vs Low Abundance Proteins Enrichment: Comparison of Methods to Reduce the Plasma Proteome Complexity. PLoS ONE 2011, 6, e19603. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Shiota, M.; Nakao, T.; Uemura, R.; Nishi, S.; Ohkawa, Y.; Matsumoto, M.; Yamaguchi, M.; Osada-Oka, M.; Inagaki, A.; et al. Identification of low-abundance proteins in serum via the isolation of HSP72 complexes. J. Proteom. 2016, 136, 214–221. [Google Scholar] [CrossRef]
- Anderson, N.L. The Human Plasma Proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Polanski, M.; Anderson, N.L. A List of Candidate Cancer Biomarkers for Targeted Proteomics. Biomark. Insights 2006, 1, 1–48. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Duan, J.; Liu, T.; Smith, R.D.; Qian, W.-J. Contributions of immunoaffinity chromatography to deep proteome profiling of human biofluids. J. Chromatogr. B 2016, 1021, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Su, D.; Liu, T.; Tang, K.; Camp, D.G.; Qian, W.-J.; Smith, R.D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 12, 1074–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, H.; Wei, D.; Dong, L.; Ghosh, D.; Chen, S.; Qian, M.G. Perspectives on potentiating immunocapture-LC–MS for the bioanalysis of biotherapeutics and biomarkers. Bioanalysis 2018, 10, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Tombelli, S.; Minunni, M.; Mascini, M. Analytical applications of aptamers. Biosens. Bioelectron. 2005, 20, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Pichon, V.; Brothier, F.; Combes, A. Aptamer-based-sorbents for sample treatment—A review. Anal. Bioanal. Chem. 2014, 407, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.W.; McGown, L.B. Aptamer-Enhanced Laser Desorption/Ionization for Affinity Mass Spectrometry. Anal. Chem. 2004, 76, 3037–3041. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-Y.; Lee, S.W.; Kang, H.S.; Jo, M.; Lee, N.-K.; Laurell, T.; Kim, S. Aptamer Microarray Mediated Capture and Mass Spectrometry Identification of Biomarker in Serum Samples. J. Proteome Res. 2010, 9, 5568–5573. [Google Scholar] [CrossRef]
- Zhao, Y.; Widen, S.G.; Jamaluddin, M.; Tian, B.; Wood, T.G.; Edeh, C.B.; Brasier, A.R. Quantification of activated NF-kB/RelA complexes using ssDNA aptamer affinity-stable isotope dilution-selected reaction monitoring-mass spectrometry. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, S.; Xiong, Y.; Deng, C.; Zhang, X. Development of a MALDI-TOF MS Strategy for the High-Throughput Analysis of Biomarkers: On-Target Aptamer Immobilization and Laser-Accelerated Proteolysis. Angew. Chem. Int. Ed. 2013, 52, 6055–6058. [Google Scholar] [CrossRef]
- Xiong, Y.; Deng, C.; Zhang, X. Development of aptamer-conjugated magnetic graphene/gold nanoparticle hybrid nanocomposites for specific enrichment and rapid analysis of thrombin by MALDI-TOF MS. Talanta 2014, 129, 282–289. [Google Scholar] [CrossRef]
- Du, F.; Alam, N.; Pawliszyn, J. Aptamer-functionalized solid phase microextraction–liquid chromatography/tandem mass spectrometry for selective enrichment and determination of thrombin. Anal. Chim. Acta 2014, 845, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Adler, B.; Ekström, S.; Rezeli, M.; Vegvari, A.; Park, J.-W.; Malm, J.; Laurell, T. Aptamer/ISET-MS: A New Affinity-Based MALDI Technique for Improved Detection of Biomarkers. Anal. Chem. 2014, 86, 7627–7634. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Lassman, M.E.; McAvoy, T.; Lee, A.Y.; Chappell, D.L.; Laterza, O.F. An evaluation of an aptamer for use as an affinity reagent with MS: PCSK9 as an example protein. Bioanal. 2016, 8, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Ptitsyn, K.G.; Novikova, S.E.; Kiseleva, Y.Y.; Moysa, A.A.; Kurbatov, L.K.; Farafonova, T.E.; Radko, S.P.; Zgoda, V.G.; Archakov, A.I. Use of DNA-Aptamers for Enrichment of Low Abundant Proteins in Cellular Extracts for Quantitative Detection by Selected Reaction Monitoring. Biochem. Suppl. Ser. B Biomed. Chem. 2018, 12, 176–180. [Google Scholar] [CrossRef]
- Ge, K.; Peng, Y.; Lu, Z.; Hu, Y.; Li, G. Aptamer-gold nanoparticle doped covalent organic framework followed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for selective enrichment and detection of human insulin. J. Chromatogr. A 2020, 1615, 460741. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, F.; Li, D.; Chen, Y. Improved Detection of HER2 by a Quasi-Targeted Proteomics Approach Using Aptamer–Peptide Probe and Liquid Chromatography–Tandem Mass Spectrometry. Clin. Chem. 2018, 64, 526–535. [Google Scholar] [CrossRef]
- Borrás, E.; Sabidó, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 2017, 17, 1700180. [Google Scholar] [CrossRef]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full Dynamic Range Proteome Analysis of S. cerevisiae by Targeted Proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Chutipongtanate, S.; Chatchen, S.; Svasti, J. Plasma prefractionation methods for proteomic analysis and perspectives in clinical applications. Proteom.—Clin. Appl. 2017, 11, 1600135. [Google Scholar] [CrossRef]
- Radko, S.P.; Lapa, S.A.; Chudinov, A.V.; Khmeleva, S.A.; Mannanova, M.M.; Kurbatov, L.K.; Kiseleva, Y.Y.; Zasedatelev, A.S.; Lisitsa, A.V. Evaluation of the Diversity of Random DNA-Libraries by the Shape of Amplification Curves for Estimation of the Efficiency of Aptamer Selection. Biochem. Suppl. Ser. B Biomed. Chem. 2020, 14, 159–167. [Google Scholar] [CrossRef]
- Kopylov, A.; Ilgisonis, E.V.; Moysa, A.A.; Tikhonova, O.V.; Zavialova, M.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.A.; Moshkovskii, S.A.; Markin, A.A.; et al. Targeted Quantitative Screening of Chromosome 18 Encoded Proteome in Plasma Samples of Astronaut Candidates. J. Proteome Res. 2016, 15, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Overall, C.M.; Van Eyk, J.E.; Baker, M.S.; Paik, Y.-K.; Weintraub, S.T.; Lane, L.; Martens, L.; Vandenbrouck, Y.; Kusebauch, U.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Hood, C.A.; Fuentes, G.; Patel, H.; Page, K.; Menakuru, M.; Park, J.H. Fast conventional Fmoc solid-phase peptide synthesis with HCTU. J. Pept. Sci. 2007, 14, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poverennaya, E.V.; Kopylov, A.; Ponomarenko, E.A.; Ilgisonis, E.V.; Zgoda, V.G.; Tikhonova, O.V.; Novikova, S.E.; Farafonova, T.E.; Kiseleva, Y.Y.; Radko, S.P.; et al. State of the Art of Chromosome 18-Centric HPP in 2016: Transcriptome and Proteome Profiling of Liver Tissue and HepG2 Cells. J. Proteome Res. 2016, 15, 4030–4038. [Google Scholar] [CrossRef]
- Fekkes, D. State-of-the-art of high-performance liquid chromatographic analysis of amino acids in physiological samples. J. Chromatogr. B Biomed. Sci. Appl. 1996, 682, 3–22. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [Green Version]
- Zgoda, V.G.; Kopylov, A.; Tikhonova, O.V.; Moisa, A.A.; Pyndyk, N.V.; Farafonova, T.E.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.A.; Poverennaya, E.V.; et al. Chromosome 18 Transcriptome Profiling and Targeted Proteome Mapping in Depleted Plasma, Liver Tissue and HepG2 Cells. J. Proteome Res. 2012, 12, 123–134. [Google Scholar] [CrossRef]
- Ullah, I.; Sun, W.; Tang, L.; Feng, J. Roles of Smads Family and Alternative Splicing Variants of Smad4 in Different Cancers. J. Cancer 2018, 9, 4018–4028. [Google Scholar] [CrossRef] [Green Version]
- Mallick, P.; Schirle, M.; Chen, S.S.; Flory, M.R.; Lee, H.; Martin, D.; Ranish, J.; Raught, B.; Schmitt, R.; Werner, T.; et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 2006, 25, 125–131. [Google Scholar] [CrossRef]
- Kitteringham, N.R.; Jenkins, R.E.; Lane, C.S.; Elliott, V.L.; Park, B.K. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J. Chromatogr. B 2009, 877, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, D.A.; Pry, T. Limit of Blank, Limit of Detection and Limit of Quantitation. Clin. Biochem. Rev. 2008, 29, S49–S52. [Google Scholar] [PubMed]
- Nicol, G.R.; Han, M.; Kim, J.; Birse, C.E.; Brand, E.; Nguyen, A.; Mesri, M.; Fitzhugh, W.; Kaminker, P.; Moore, P.A.; et al. Use of an Immunoaffinity-Mass Spectrometry-based Approach for the Quantification of Protein Biomarkers from Serum Samples of Lung Cancer Patients. Mol. Cell. Proteom. 2008, 7, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneck, N.A.; Phinney, K.W.; Lee, S.B.; Lowenthal, M.S. Quantification of cardiac troponin I in human plasma by immunoaffinity enrichment and targeted mass spectrometry. Anal. Bioanal. Chem. 2018, 410, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Säll, A.; Corbee, D.; Vikström, S.; Ottosson, F.; Persson, H.; Waldemarson, S. Advancing the immunoaffinity platform AFFIRM to targeted measurements of proteins in serum in the pg/ml range. PLoS ONE 2018, 13, e0189116. [Google Scholar] [CrossRef] [Green Version]
- Ciancio, D.R.; Vargas, M.R.; Thiel, W.; Bruno, M.A.; Giangrande, P.H.; Mestre, M.B. Aptamers as Diagnostic Tools in Cancer. Pharmaceuticals 2018, 11, 86. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef] [Green Version]
- Lapa, S.A.; Chudinov, A.V.; Timofeev, E.N. The Toolbox for Modified Aptamers. Mol. Biotechnol. 2015, 58, 79–92. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Aptamer Name | Aptamer Sequence |
|---|---|
| Anti-SMAD4 | 5'-CGAAGTCTAGACAGCGTTTTCGCTGTCTAGACTTCGAAAAA-3'-M |
| Peptide Sequences | m/z of Precursor Ion | m/z of Fragment Ion | Collision Energy, eV | Ion Type |
|---|---|---|---|---|
| GWGPDYPR | 474.222 +2 | 704.336 + 1 | 17.1 | y6 |
| 647.315 + 1 | y5 | |||
| 550.262 + 1 | y4 | |||
| 479.226 +2 | 714.344 + 1 | y6 (“heavy”) | ||
| 657.323 + 1 | y5 (“heavy”) | |||
| 560.270 + 1 | y4 (“heavy”) | |||
| IYPSAYIK | 477.768 +2 | 841.4454 + 1 | 17.2 | y7 |
| 678.3821 + 1 | y6 | |||
| 494.2973 + 1 | y4 | |||
| 481.775 +2 | 849.4596 + 1 | y7 (“heavy”) | ||
| 686.3963 + 1 | y6 (“heavy”) | |||
| 502.3115 + 1 | y4 (“heavy”) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radko, S.; Ptitsyn, K.; Novikova, S.; Kiseleva, Y.; Moysa, A.; Kurbatov, L.; Mannanova, M.; Zgoda, V.; Ponomarenko, E.; Lisitsa, A.; et al. Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma. Biomedicines 2020, 8, 133. https://doi.org/10.3390/biomedicines8050133
Radko S, Ptitsyn K, Novikova S, Kiseleva Y, Moysa A, Kurbatov L, Mannanova M, Zgoda V, Ponomarenko E, Lisitsa A, et al. Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma. Biomedicines. 2020; 8(5):133. https://doi.org/10.3390/biomedicines8050133
Chicago/Turabian StyleRadko, Sergey, Konstantin Ptitsyn, Svetlana Novikova, Yana Kiseleva, Alexander Moysa, Leonid Kurbatov, Maria Mannanova, Victor Zgoda, Elena Ponomarenko, Andrey Lisitsa, and et al. 2020. "Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma" Biomedicines 8, no. 5: 133. https://doi.org/10.3390/biomedicines8050133
APA StyleRadko, S., Ptitsyn, K., Novikova, S., Kiseleva, Y., Moysa, A., Kurbatov, L., Mannanova, M., Zgoda, V., Ponomarenko, E., Lisitsa, A., & Archakov, A. (2020). Evaluation of Aptamers as Affinity Reagents for an Enhancement of SRM-Based Detection of Low-Abundance Proteins in Blood Plasma. Biomedicines, 8(5), 133. https://doi.org/10.3390/biomedicines8050133