NAFLD Preclinical Models: More than a Handful, Less of a Concern?


Abstract
1. Introduction
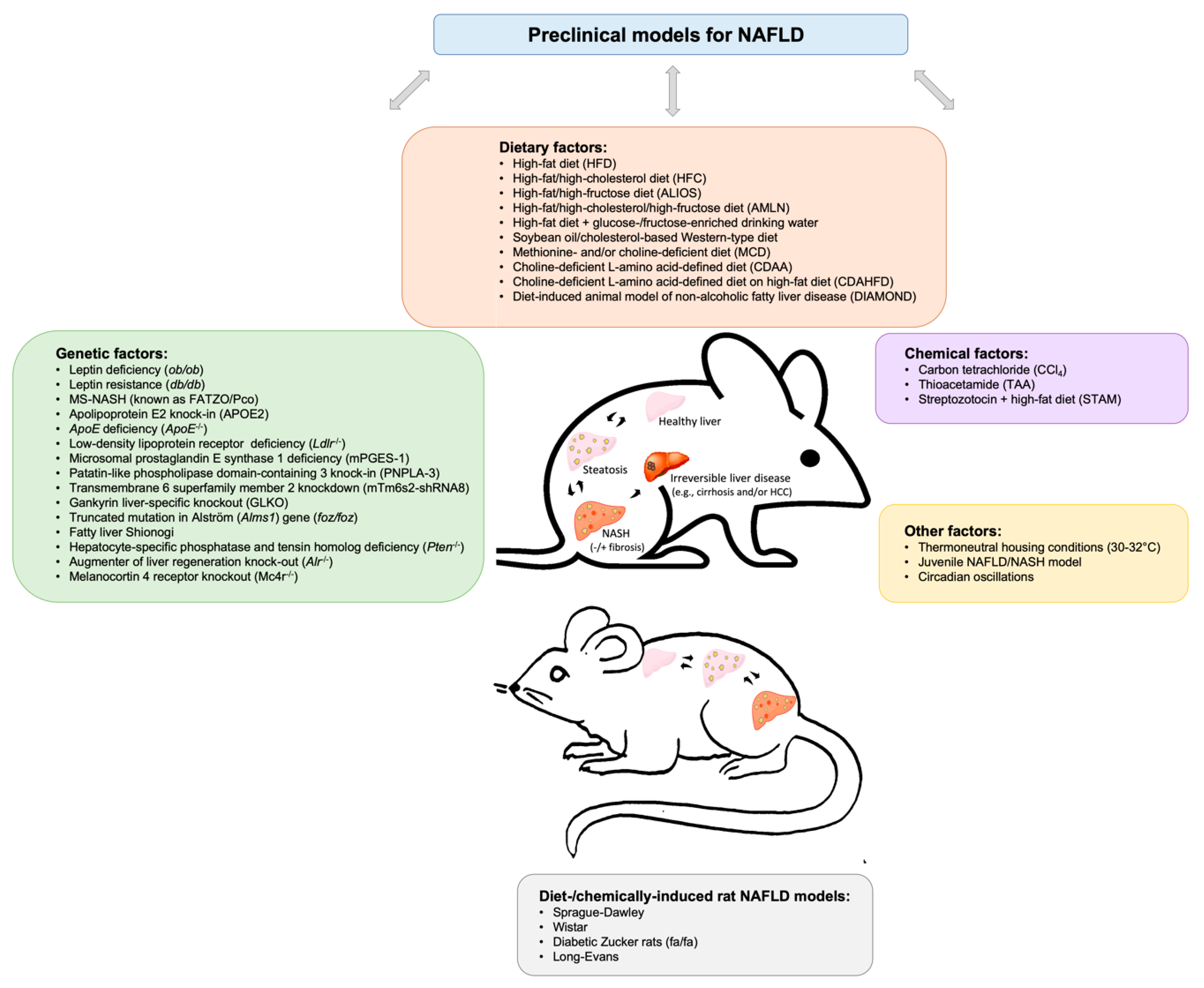
2. Insights into Available Preclinical Models for Non-alcoholic Fatty Liver Disease
2.1. Dietary Murine Models
2.2. Genetic Murine Models
2.3. Chemically-induced Murine Models
2.4. Other Murine Models
2.5. Rat NAFLD Models
3. Therapeutic Approaches in Preclinical NAFLD Models
4. Clinical Relevance: Comparisons with Clinical Data
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALIOS | American Lifestyle Induced Obesity Syndrome |
| ALMS1 | Alström |
| ALR | Augmenter of liver regeneration |
| APOE2ki | Apolipoprotein E2 knock-in |
| CCl4 | Carbon tetrachloride |
| CD | Diet deficient for/low in choline |
| CDAA | Choline-deficient L-amino acid-defined diet |
| DIAMOND | Diet-induced animal model of non-alcoholic fatty liver disease |
| FLS | Fatty liver Shionogi |
| FXR | Farnesoid X receptor |
| Gank | Gankyrin |
| GFER | Growth Factor ERV1 homolog of Saccharomyces cerevisiae |
| HCC | Hepatocellular carcinoma |
| HFC | High-fat/high-cholesterol diet |
| HFD | High-fat diet |
| Ldlr | Low-density lipoprotein receptor |
| MAT1a | methionine adenosyltransferase 1A |
| MC4R | Melanocortin 4 receptor |
| MetS | Metabolic Syndrome |
| MS-NASH | Mice formerly known as FATZO/Pco |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NEMO | NF-κB essential modulator |
| PNPLA3 | Patatin-like phospholipase domain-containing 3 |
| PPAR | Peroxisome proliferator-activated receptor |
| PTEN | Phosphatase and tensin homolog |
| T2D | Type 2 Diabetes |
| TAA | Thioacetamide |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| TNFα | Tumor necrosis factor α |
| TSOD | Tsumura-Suzuki Obese Diabetes |
References
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Jensen, V.S.; Tveden-Nyborg, P.; Zacho-Rasmussen, C.; Quaade, M.L.; Ipsen, D.H.; Hvid, H.; Fledelius, C.; Wulff, E.M.; Lykkesfeldt, J. Variation in diagnostic NAFLD/NASH read-outs in paired liver samples from rodent models. J. Pharmacol. Toxicol. Methods 2019, 101, 106651. [Google Scholar] [CrossRef] [PubMed]
- Ganbold, M.; Owada, Y.; Ozawa, Y.; Shimamoto, Y.; Ferdousi, F.; Tominaga, K.; Zheng, Y.W.; Ohkohchi, N.; Isoda, H. Isorhamnetin Alleviates Steatosis and Fibrosis in Mice with Nonalcoholic Steatohepatitis. Sci. Rep. 2019, 9, 16210. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, C.; Zhan, Y.T. Nonalcoholic Fatty Liver Disease Cirrhosis: A Review of Its Epidemiology, Risk Factors, Clinical Presentation, Diagnosis, Management, and Prognosis. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2784537. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Loomba, R.; Anstee, Q.M.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology 2018, 68, 349–360. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017. [Google Scholar] [CrossRef]
- Haczeyni, F.; Yeh, M.M.; Ioannou, G.N.; Leclercq, I.A.; Goldin, R.; Dan, Y.Y.; Yu, J.; Teoh, N.C.; Farrell, G.C. Mouse models of non-alcoholic steatohepatitis: A reflection on recent literature. J. Gastroenterol. Hepatol. 2018, 33, 1312–1320. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal models of NAFLD from a hepatologist’s point of view. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wu, N.; Chen, X.; Wang, W.; Chu, Y.; Liu, H.; Li, W.; Chen, D.; Li, X.; Xu, B. Pathogenesis of and major animal models used for nonalcoholic fatty liver disease. J. Int. Med. Res. 2019, 47, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Di Pasqua, L.G.; Berardo, C.; Siciliano, V.; Richelmi, P.; Perlini, S.; Ferrigno, A.; Vairetti, M. Animal Models of Steatosis (NAFLD) and Steatohepatitis (NASH) Exhibit Hepatic Lobe-Specific Gelatinases Activity and Oxidative Stress. Can. J. Gastroenterol. Hepatol. 2019, 2019, 5413461. [Google Scholar] [CrossRef]
- Zhong, F.; Zhou, X.; Xu, J.; Gao, L. Rodent Models of Nonalcoholic Fatty Liver Disease. Digestion 2019, 1–14. [Google Scholar] [CrossRef]
- Velazquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Jacobs, A.; Warda, A.S.; Verbeek, J.; Cassiman, D.; Spincemaille, P. An Overview of Mouse Models of Nonalcoholic Steatohepatitis: From Past to Present. Curr. Protoc. Mouse Biol. 2016, 6, 185–200. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Chen, K.; Ma, J.; Jia, X.; Ai, W.; Ma, Z.; Pan, Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 117–125. [Google Scholar] [CrossRef]
- Vonghia, L.; Ruyssers, N.; Schrijvers, D.; Pelckmans, P.; Michielsen, P.; De Clerck, L.; Ramon, A.; Jirillo, E.; Ebo, D.; De Winter, B.; et al. CD4+ROR gamma t++ and Tregs in a Mouse Model of Diet-Induced Nonalcoholic Steatohepatitis. Mediators Inflamm. 2015, 2015, 239623. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Chiu, C.C.; Hung, S.W.; Huang, W.C.; Lee, Y.P.; Liu, J.Y.; Huang, Y.T.; Chen, T.H.; Chuang, H.L. Gnotobiotic mice inoculated with Firmicutes, but not Bacteroidetes, deteriorate nonalcoholic fatty liver disease severity by modulating hepatic lipid metabolism. Nutr. Res. 2019, 69, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875 e861-863. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lutjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Bilsen, M.; van Gorp, P.J.; Bieghs, V.; Lutjohann, D.; Kerksiek, A.; Staels, B.; Hofker, M.H.; Shiri-Sverdlov, R. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010, 584, 1001–1005. [Google Scholar] [CrossRef]
- Zheng, S.; Hoos, L.; Cook, J.; Tetzloff, G.; Davis, H., Jr.; van Heek, M.; Hwa, J.J. Ezetimibe improves high fat and cholesterol diet-induced non-alcoholic fatty liver disease in mice. Eur. J. Pharmacol. 2008, 584, 118–124. [Google Scholar] [CrossRef]
- Clapper, J.R.; Hendricks, M.D.; Gu, G.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 305, G483–G495. [Google Scholar] [CrossRef]
- Kristiansen, M.N.; Veidal, S.S.; Rigbolt, K.T.; Tolbol, K.S.; Roth, J.D.; Jelsing, J.; Vrang, N.; Feigh, M. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J. Hepatol. 2016, 8, 673–684. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- US. Food Drug Administration. Final Determination Regarding Partially Hydrogenated Oils (Removing Trans Fat). Available online: https://www.federalregister.gov/documents/2018/05/21/2018-10714/final-determination-regarding-partially-hydrogenated-oils (accessed on 21 May 2018).
- Boland, M.L.; Oro, D.; Tolbol, K.S.; Thrane, S.T.; Nielsen, J.C.; Cohen, T.S.; Tabor, D.E.; Fernandes, F.; Tovchigrechko, A.; Veidal, S.S.; et al. Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: Impact of dietary fat source. World J. Gastroenterol. 2019, 25, 4904–4920. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Kato, S.; Tsuchida, T.; Sugimoto, K.; Saito, R.; Verschuren, L.; Kleemann, R.; Oka, K. Longitudinal characterization of diet-induced genetic murine models of non-alcoholic steatohepatitis with metabolic, histological, and transcriptomic hallmarks of human patients. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Ellacott, K.L.; King, V.L.; Hasty, A.H. Mouse models of the metabolic syndrome. Dis. Model. Mech. 2010, 3, 156–166. [Google Scholar] [CrossRef]
- Suto, J.; Matsuura, S.; Imamura, K.; Yamanaka, H.; Sekikawa, K. Genetic analysis of non-insulin-dependent diabetes mellitus in KK and KK-Ay mice. Eur. J. Endocrinol. 1998, 139, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jhrens, K.; Weber, D.; Castro, J.P.; Hugo, M.; Schulz, T.J.; Kramer, S.; Schurmann, A.; et al. Induction of steatohepatitis (NASH) with insulin resistance in wildtype B6 mice by a western-type diet containing soybean oil and cholesterol. Mol. Med. 2017, 23, 70–82. [Google Scholar] [CrossRef]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Haigh, W.G.; Williams, J.; Brown, S.; Bell-Anderson, K.S.; Lee, S.P.; Farrell, G.C. Hepatic free fatty acids accumulate in experimental steatohepatitis: Role of adaptive pathways. J. Hepatol. 2008, 48, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, W.; Wei, H.; Sun, R.; Tian, Z.; Chen, Y. Hepatic NK cells attenuate fibrosis progression of non-alcoholic steatohepatitis in dependent of CXCL10-mediated recruitment. Liver Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am. J. Physiol. Gastrointest Liver Physiol. 2004, 287, G264–G273. [Google Scholar] [CrossRef]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar]
- Raubenheimer, P.J.; Nyirenda, M.J.; Walker, B.R. A choline-deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high-fat diet. Diabetes 2006, 55, 2015–2020. [Google Scholar] [CrossRef]
- Ikejima, K.; Okumura, K.; Lang, T.; Honda, H.; Abe, W.; Yamashina, S.; Enomoto, N.; Takei, Y.; Sato, N. The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol. Res. 2005, 33, 151–154. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Al Rajabi, A.; Castro, G.S.; da Silva, R.P.; Nelson, R.C.; Thiesen, A.; Vannucchi, H.; Vine, D.F.; Proctor, S.D.; Field, C.J.; Curtis, J.M.; et al. Choline supplementation protects against liver damage by normalizing cholesterol metabolism in Pemt/Ldlr knockout mice fed a high-fat diet. J. Nutr. 2014, 144, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e327. [Google Scholar] [CrossRef] [PubMed]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef]
- Neff, E.P. Farewell, FATZO: A NASH mouse update. Lab. Anim. (NY) 2019, 48, 151. [Google Scholar] [CrossRef]
- Alexander, J.; Chang, G.Q.; Dourmashkin, J.T.; Leibowitz, S.F. Distinct phenotypes of obesity-prone AKR/J, DBA2J and C57BL/6J mice compared to control strains. Int. J. Obes. (Lond.) 2006, 30, 50–59. [Google Scholar] [CrossRef]
- Peterson, R.G.; Jackson, C.V.; Zimmerman, K.M.; Alsina-Fernandez, J.; Michael, M.D.; Emmerson, P.J.; Coskun, T. Glucose dysregulation and response to common anti-diabetic agents in the FATZO/Pco mouse. PLoS ONE 2017, 12, e0179856. [Google Scholar] [CrossRef]
- Sun, G.; Jackson, C.V.; Zimmerman, K.; Zhang, L.K.; Finnearty, C.M.; Sandusky, G.E.; Zhang, G.; Peterson, R.G.; Wang, Y.J. The FATZO mouse, a next generation model of type 2 diabetes, develops NAFLD and NASH when fed a Western diet supplemented with fructose. BMC Gastroenterol. 2019, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Van Gorp, P.J.; Wouters, K.; Hendrikx, T.; Gijbels, M.J.; van Bilsen, M.; Bakker, J.; Binder, C.J.; Lutjohann, D.; Staels, B.; et al. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e30668. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Maybuchen, L.; Zimmer, S.; Hittatiya, K.; Back, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Johrens, K.; Weiss, T.S.; Jonas, W.; Schurmann, A.; Puschel, G.P. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci. Rep. 2018, 8, 16127. [Google Scholar] [CrossRef]
- Horrillo, R.; Planaguma, A.; Gonzalez-Periz, A.; Ferre, N.; Titos, E.; Miquel, R.; Lopez-Parra, M.; Masferrer, J.L.; Arroyo, V.; Claria, J. Comparative protection against liver inflammation and fibrosis by a selective cyclooxygenase-2 inhibitor and a nonredox-type 5-lipoxygenase inhibitor. J. Pharmacol. Exp. Ther. 2007, 323, 778–786. [Google Scholar] [CrossRef]
- Karck, U.; Peters, T.; Decker, K. The release of tumor necrosis factor from endotoxin-stimulated rat Kupffer cells is regulated by prostaglandin E2 and dexamethasone. J. Hepatol. 1988, 7, 352–361. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver Dis. 2015, 35, 270–290. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Pingitore, P.; Romeo, S. The role of PNPLA3 in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 900–906. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, P.; De, L.; Li, B.; Su, S. The roles of transmembrane 6 superfamily member 2 rs58542926 polymorphism in chronic liver disease: A meta-analysis of 24,147 subjects. Mol. Genet. Genomic Med. 2019, 7, e824. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Cast, A.; Kumbaji, M.; D’Souza, A.; Rodriguez, K.; Gupta, A.; Karns, R.; Timchenko, L.; Timchenko, N. Liver Proliferation Is an Essential Driver of Fibrosis in Mouse Models of Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2019, 3, 1036–1049. [Google Scholar] [CrossRef]
- Qian, Y.W.; Chen, Y.; Yang, W.; Fu, J.; Cao, J.; Ren, Y.B.; Zhu, J.J.; Su, B.; Luo, T.; Zhao, X.F.; et al. p28(GANK) prevents degradation of Oct4 and promotes expansion of tumor-initiating cells in hepatocarcinogenesis. Gastroenterology 2012, 142, 1547–1558.e1514. [Google Scholar] [CrossRef]
- Arsov, T.; Silva, D.G.; O’Bryan, M.K.; Sainsbury, A.; Lee, N.J.; Kennedy, C.; Manji, S.S.; Nelms, K.; Liu, C.; Vinuesa, C.G.; et al. Fat aussie—A new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol. Endocrinol. 2006, 20, 1610–1622. [Google Scholar] [CrossRef]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat. Genet. 2002, 31, 74–78. [Google Scholar] [CrossRef]
- Soga, M.; Kishimoto, Y.; kawaguchi, J.; Nakai, Y.; Kawamura, Y.; Inagaki, S.; Katoh, K.; Oohara, T.; Makino, S.; Oshima, I. The FLS mouse: A new inbred strain with spontaneous fatty liver. Lab. Anim. Sci. 1999, 49, 269–275. [Google Scholar]
- Semba, T.; Nishimura, M.; Nishimura, S.; Ohara, O.; Ishige, T.; Ohno, S.; Nonaka, K.; Sogawa, K.; Satoh, M.; Sawai, S.; et al. The FLS (fatty liver Shionogi) mouse reveals local expressions of lipocalin-2, CXCL1 and CXCL9 in the liver with non-alcoholic steatohepatitis. BMC Gastroenterol. 2013, 13, 120. [Google Scholar] [CrossRef]
- Sugihara, T.; Koda, M.; Kishina, M.; Kato, J.; Tokunaga, S.; Matono, T.; Ueki, M.; Murawaki, Y. Fatty liver Shionogi-ob/ob mouse: A new candidate for a non-alcoholic steatohepatitis model. Hepatol. Res. 2013, 43, 547–556. [Google Scholar] [CrossRef]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Horie, Y.; Kataoka, E.; Sato, W.; Dohmen, T.; Ohshima, S.; Goto, T.; Suzuki, A. Non-alcoholic steatohepatitis and hepatocellular carcinoma: Lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S96–S100. [Google Scholar] [CrossRef]
- Takakura, K.; Oikawa, T.; Tomita, Y.; Mizuno, Y.; Nakano, M.; Saeki, C.; Torisu, Y.; Saruta, M. Mouse models for investigating the underlying mechanisms of nonalcoholic steatohepatitis-derived hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. (Lausanne) 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R.; Chaillet, J.R.; Nalesnik, M.A.; Kumar, S.; Dangi, A.; Demetris, A.J.; Ferrell, R.; Wu, T.; Divanovic, S.; Stankeiwicz, T.; et al. Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 2015, 148, 379–391.e374. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, A.; Vujanovic, N.L.; Polimeno, L.; Azzarone, A.; Iacobellis, A.; Deleo, A.; Hagiya, M.; Whiteside, T.L.; Starzl, T.E. The in vivo effect of hepatotrophic factors augmenter of liver regeneration, hepatocyte growth factor, and insulin-like growth factor-II on liver natural killer cell functions. Hepatology 1997, 25, 411–415. [Google Scholar] [CrossRef][Green Version]
- Ibrahim, S.; Weiss, T.S. Augmenter of liver regeneration: Essential for growth and beyond. Cytokine Growth Factor Rev. 2019, 45, 65–80. [Google Scholar] [CrossRef]
- Gandhi, C.R.; Murase, N.; Starzl, T.E. Cholera toxin-sensitive GTP-binding protein-coupled activation of augmenter of liver regeneration (ALR) receptor and its function in rat kupffer cells. J. Cell Physiol. 2010, 222, 365–373. [Google Scholar] [CrossRef]
- Itoh, M.; Suganami, T.; Nakagawa, N.; Tanaka, M.; Yamamoto, Y.; Kamei, Y.; Terai, S.; Sakaida, I.; Ogawa, Y. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 2011, 179, 2454–2463. [Google Scholar] [CrossRef]
- Yamada, T.; Kashiwagi, Y.; Rokugawa, T.; Kato, H.; Konishi, H.; Hamada, T.; Nagai, R.; Masago, Y.; Itoh, M.; Suganami, T.; et al. Evaluation of hepatic function using dynamic contrast-enhanced magnetic resonance imaging in melanocortin 4 receptor-deficient mice as a model of nonalcoholic steatohepatitis. Magn. Reson. Imaging 2019, 57, 210–217. [Google Scholar] [CrossRef]
- Denk, H.; Abuja, P.M.; Zatloukal, K. Animal models of NAFLD from the pathologist’s point of view. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 929–942. [Google Scholar] [CrossRef]
- Nishida, T.; Tsuneyama, K.; Fujimoto, M.; Nomoto, K.; Hayashi, S.; Miwa, S.; Nakajima, T.; Nakanishi, Y.; Sasaki, Y.; Suzuki, W.; et al. Spontaneous onset of nonalcoholic steatohepatitis and hepatocellular carcinoma in a mouse model of metabolic syndrome. Lab. Invest. 2013, 93, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Mehta, A.K.; Hofer, E.M.; Wohlrab, C.; Golob-Schwarzl, N.; Svendova, V.; Schimek, M.G.; Stumptner, C.; Thuringer, A.; Speicher, M.R.; et al. Keratin 18-deficiency results in steatohepatitis and liver tumors in old mice: A model of steatohepatitis-associated liver carcinogenesis. Oncotarget 2016, 7, 73309–73322. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Buque, X.; Martinez-Una, M.; Aurrekoetxea, I.; Menor, A.; Garcia-Rodriguez, J.L.; Lu, S.C.; Martinez-Chantar, M.L.; Mato, J.M.; Ochoa, B.; et al. Methionine adenosyltransferase 1A gene deletion disrupts hepatic very low-density lipoprotein assembly in mice. Hepatology 2011, 54, 1975–1986. [Google Scholar] [CrossRef]
- Domitrovic, R.; Jakovac, H.; Tomac, J.; Sain, I. Liver fibrosis in mice induced by carbon tetrachloride and its reversion by luteolin. Toxicol. Appl. Pharmacol. 2009, 241, 311–321. [Google Scholar] [CrossRef]
- Sharma, L.; Gupta, D.; Abdullah, S.T. Thioacetamide potentiates high cholesterol and high fat diet induced steato-hepatitic changes in livers of C57BL/6J mice: A novel eight weeks model of fibrosing NASH. Toxicol. Lett. 2019, 304, 21–29. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Liu, H.X.; Kalanetra, K.M.; Mirsoian, A.; Murphy, W.J.; French, S.W.; Krishnan, V.V.; Mills, D.A.; Wan, Y.Y. Western Diet-Induced Dysbiosis in Farnesoid X Receptor Knockout Mice Causes Persistent Hepatic Inflammation after Antibiotic Treatment. Am. J. Pathol. 2017, 187, 1800–1813. [Google Scholar] [CrossRef]
- Norheim, F.; Hui, S.T.; Kulahcioglu, E.; Mehrabian, M.; Cantor, R.M.; Pan, C.; Parks, B.W.; Lusis, A.J. Genetic and hormonal control of hepatic steatosis in female and male mice. J. Lipid Res. 2017, 58, 178–187. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Henkel, J.; Kanuri, G.; Blank, D.; Puschel, G.P.; Bischoff, S.C.; Bergheim, I. Female mice are more susceptible to nonalcoholic fatty liver disease: Sex-specific regulation of the hepatic AMP-activated protein kinase-plasminogen activator inhibitor 1 cascade, but not the hepatic endotoxin response. Mol. Med. 2012, 18, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Rosso, N.; Dal Ben, M.; Raseni, A.; Boschelle, M.; Degrassi, C.; Nemeckova, I.; Nachtigal, P.; Avellini, C.; Tiribelli, C.; et al. An Animal Model for the Juvenile Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. PLoS ONE 2016, 11, e0158817. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, D.; Wang, Z.; Dong, H.; Xu, X.; Zhou, S. Establishment and Comparison of Juvenile Female Mouse Models of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Gastroenterol. Res. Pract. 2018, 2018, 8929620. [Google Scholar] [CrossRef] [PubMed]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Graspeuntner, S.; Cappelletti, M.; Wu, D.; Mukherjee, R.; Chan, C.C.; Lawson, M.J.; Klarquist, J.; et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat. Med. 2017, 23, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Rousso-Noori, L.; Zwighaft, Z.; Neufeld-Cohen, A.; Golik, M.; Kraut-Cohen, J.; Wang, M.; Han, X.; Asher, G. Circadian clocks and feeding time regulate the oscillations and levels of hepatic triglycerides. Cell Metab. 2014, 19, 319–330. [Google Scholar] [CrossRef]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef]
- Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef]
- Goodman, S.N.; Fanelli, D.; Ioannidis, J.P. What does research reproducibility mean? Sci. Transl. Med. 2016, 8, 341ps312. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, J.; Lu, C.; Wang, J.; Ge, J.; Huang, Y.; Zhang, L.; Wang, Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006, 79, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Kucera, O.; Cervinkova, Z. Experimental models of non-alcoholic fatty liver disease in rats. World J. Gastroenterol. 2014, 20, 8364–8376. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.A.; Andrade, R.S.B.; Vasconcelos, D.F.P. Relationship between Experimental Diet in Rats and Nonalcoholic Hepatic Disease: Review of Literature. Int. J. Hepatol. 2018, 2018, 9023027. [Google Scholar] [CrossRef]
- Maciejewska, D.; Lukomska, A.; Dec, K.; Skonieczna-Zydecka, K.; Gutowska, I.; Skorka-Majewicz, M.; Styburski, D.; Misiakiewicz-Has, K.; Pilutin, A.; Palma, J.; et al. Diet-Induced Rat Model of Gradual Development of Non-Alcoholic Fatty Liver Disease (NAFLD) with Lipopolysaccharides (LPS) Secretion. Diagnostics (Basel) 2019, 9, 205. [Google Scholar] [CrossRef]
- Khurana, P.; Yadati, T.; Goyal, S.; Dolas, A.; Houben, T.; Oligschlaeger, Y.; Agarwal, A.K.; Kulkarni, A.; Shiri-Sverdlov, R. Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague(-)Dawley Rats (dagger). Biomolecules 2019, 9, 171. [Google Scholar] [CrossRef]
- Maeso-Diaz, R.; Boyer-Diaz, Z.; Lozano, J.J.; Ortega-Ribera, M.; Peralta, C.; Bosch, J.; Gracia-Sancho, J. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of The Human Disease. Cells 2019, 8, 1062. [Google Scholar] [CrossRef]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef]
- Kirsch, R.; Clarkson, V.; Shephard, E.G.; Marais, D.A.; Jaffer, M.A.; Woodburne, V.E.; Kirsch, R.E.; Hall Pde, L. Rodent nutritional model of non-alcoholic steatohepatitis: Species, strain and sex difference studies. J. Gastroenterol. Hepatol. 2003, 18, 1272–1282. [Google Scholar] [CrossRef]
- Domitrovic, R.; Jakovac, H.; Milin, C.; Radosevic-Stasic, B. Dose- and time-dependent effects of luteolin on carbon tetrachloride-induced hepatotoxicity in mice. Exp. Toxicol. Pathol. 2009, 61, 581–589. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Houben, T.; Rensen, S.S.; Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Oligschlaeger, Y.; Jeurissen, M.L.; Gijbels, M.J.; Buurman, W.A.; et al. Plasma cathepsin D correlates with histological classifications of fatty liver disease in adults and responds to intervention. Sci. Rep. 2016, 6, 38278. [Google Scholar] [CrossRef]
- Houben, T.; Oligschlaeger, Y.; Hendrikx, T.; Bitorina, A.V.; Walenbergh, S.M.A.; van Gorp, P.J.; Gijbels, M.J.J.; Friedrichs, S.; Plat, J.; Schaap, F.G.; et al. Cathepsin D regulates lipid metabolism in murine steatohepatitis. Sci. Rep. 2017, 7, 3494. [Google Scholar] [CrossRef]
- Gehrke, N.; Biedenbach, J.; Huber, Y.; Straub, B.K.; Galle, P.R.; Simon, P.; Schattenberg, J.M. Voluntary exercise in mice fed an obesogenic diet alters the hepatic immune phenotype and improves metabolic parameters—An animal model of life style intervention in NAFLD. Sci. Rep. 2019, 9, 4007. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Niihara, H.; Mizokami, T.; Yada, K.; Suzuki, K. Exercise training attenuates neutrophil infiltration and elastase expression in adipose tissue of high-fat-diet-induced obese mice. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; R, M.R. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Shipman, K.E.; Strange, R.C.; Ramachandran, S. Use of fibrates in the metabolic syndrome: A review. World J. Diabetes 2016, 7, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S.; Group, K.S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor alpha modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Choung, S.; Joung, K.H.; You, B.R.; Park, S.K.; Kim, H.J.; Ku, B.J. Treatment with Lobeglitazone Attenuates Hepatic Steatosis in Diet-Induced Obese Mice. PPAR Res. 2018, 2018, 4292509. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e1145. [Google Scholar] [CrossRef] [PubMed]
- Tolbol, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Alukal, J.J.; Thuluvath, P.J. Reversal of NASH fibrosis with pharmacotherapy. Hepatol. Int. 2019, 13, 534–545. [Google Scholar] [CrossRef]
- Roth, J.D.; Veidal, S.S.; Fensholdt, L.K.D.; Rigbolt, K.T.G.; Papazyan, R.; Nielsen, J.C.; Feigh, M.; Vrang, N.; Young, M.; Jelsing, J.; et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci. Rep. 2019, 9, 9046. [Google Scholar] [CrossRef]
- Hui, S.T.; Kurt, Z.; Tuominen, I.; Norheim, F.; R, C.D.; Pan, C.; Dirks, D.L.; Magyar, C.E.; French, S.W.; Chella Krishnan, K.; et al. The Genetic Architecture of Diet-Induced Hepatic Fibrosis in Mice. Hepatology 2018, 68, 2182–2196. [Google Scholar] [CrossRef]


{kind=link}
| Type of Mouse Model | Treatment or Intervention | Phenotypical Outcome and Relevance to Human Disease | Author(s) |
|---|---|---|---|
| Dietary Models | |||
| High-fat diet (HFD) | HFD containing 60% fat administered to C57BL6/J | After 10-12w, induction of obesity, insulin resistance and hyperlipidemia. After long-term exposure (36w), no or only minimal signs of inflammation and fibrosis. After chronic feeding (80w), hepatic steatosis, cell injury, portal & lobular inflammation and fibrosis. | Velázquez et al. [17], Ito et al. [19], Chen et al. [20], Vonghia et al. [21] |
| Atherogenic diet | Diet containing 1.25% cholesterol and 0.5% cholate | Increased plasma and liver lipid levels. From 6–24 weeks, induction NASH with hepatocellular ballooning in a time-dependent manner. | Matsuzawa et al. [24] |
| High-fat atherogenic diet | HFD containing 1.25% cholesterol and 0.5% cholate | Exacerbated NASH features including hepatic insulin resistance, oxidative stress, activation of hepatic stellate cells. | Matsuzawa et al. [24], Montandon et al. [41], Larter et al. [42] |
| High-fat/high-cholesterol diet (HFC) | HFC containing 21% milk butter, 0.2% cholesterol | After short-term HFC diet, only steatosis in C57BL/6 mice. Steatosis with severe inflammation in female Ldlr-/- and APOE2ki hyperlipidemic mice. After seven days, severe hepatic inflammation but no steatosis in male hyperlipidemic mice. After seven months, development of obesity, hepatomegaly, hepatic steatosis and varying degrees of steatohepatitis in C57BL/6 mice. | Wouters et al. [25,26] Zheng et al. [27] |
| High-fat/high-cholesterol/ high-fructose diet (AMLN) | Diet containing 40% high-fat and 22% fructose, supplemented with ~18% trans-fat and 2% cholesterol | After 26–30w, marked steatosis, moderate lobular inflammation and hepatocellular ballooning in C57BL/6 and ob/ob mice. | Clapper et al. [28], Kristiansen et al. [29] |
| Gubra amylin diet (GAN) | High-fat (40 kcal-%, of which 0% trans-fat and 46% saturated fatty acids by weight), fructose (22%), sucrose (10%), cholesterol (2%) | After 8-16w, more pronounced weight gain and a highly similar phenotype of biopsy-confirmed fibrotic NASH in C57BL/6 and ob/ob mice. | Boland et al. [33] |
| High-fat/high-fructose/high-cholesterol | Composed of 41% fat, 30% fructose, 2% cholesterol | Induction NASH in various models. | Abe et al. [34], Kennedy et al. [35], Suto et al. [36] |
| Soybean-oil-based Western-type diet | Western-type diet containing 25g/100 g n-6-PUFA-rich soybean oil +/- 0.75% cholesterol | After long-term exposure (20w), hepatic steatosis, inflammation and fibrosis, weight gain, insulin resistance, hepatic lipid peroxidation and oxidative stress in C57BL/6 mice. | Henkel et al. [37] |
| High-caloric cholesterol-free HFD | Composed of lard (21g/100g)/soy-bean oil (3g/100g)/5% fructose in drinking water | Only mild steatosis. No signs of hepatic inflammation and fibrosis. | Wouters et al. [25,26], Henkel et al. [37], Subramanian et al. [38], Mari et al. [39], Savard et al. [40] |
| Choline-deficient diet | C57BL/6 mice were fed HFD (45% of calories) for 8 weeks. During the final 4 weeks, diets were choline-deficient (or choline-supplemented) | Amplified liver fat accumulation, while improved glucose tolerance. | Raubenheimer et al. [48] |
| Methionine/choline-deficient diet (MCD) | Diet lacking methionine and choline, but containing high sucrose (40%) and moderate fat (10%) | After 2w, severe steatohepatitis with elevated serum AST and ALT levels. After 10w, additional Kupffer cell infiltration and irreversible fibrosis. After 1.5-4w, no signs of insulin resistance. | Santhekadur et al. [12], Montandon et al. [41], Itagaki et al. [47], Rinella et al. [52], Al Rajabi et al. [53] |
| Choline-deficient L-amino acid-defined diet (CDAA) | Choline-deficient L-amino acid-defined diet containing carbohydrates (68,5%), proteins (17,4%) and fats (14%) | Within a few weeks, fatty liver followed by mild features of NASH in C57BL/6J mice. After >20w, mild-to-moderate fibrosis and insulin resistance. | Van Herck et al. [11], Matsumoto et al. [54], Miura et al. [55] |
| Choline-deficient L-amino acid-defined diet on high-fat diet (CDAHFD) | Choline-deficient, L-amino acid-defined, HFD consisting of 60 kcal% fat and 0.1% methionine by weight | Excessive liver fat accumulation, increased circulating liver enzymes and progressive hepatic fibrosis. | Matsumoto et al. [54] |
| High-fat/high-fructose diet (ALIOS) | HFD with fructose-containing drinking water. Additional administration of a low weekly dose of intraperitoneal carbon tetrachloride (CCl4) | After 16w, substantial steatosis with necro-inflammatory changes and increased ALT levels. No difference in steatosis degree or ALT levels if compared to without additional fructose. Development of progressive stages of human-like fatty liver disease. | Tetri et al. [56], Tsuchida et al. [57] |
| Diet-induced animal model of non-alcoholic fatty liver disease (DIAMOND) | High fat/carbohydrate diet (Western diet) with 42% kcal from fat, containing cholesterol (0.1%), with a high fructose/glucose solution (23.1 g/L d-fructose +18.9 g/L d-glucose) | After 16w, obesity, liver injury, dyslipidemia and insulin resistance, sustained up to 52w. Parent strains 129S1/SvImJ or C57BL/6 lacked insulin resistance and steatohepatitis or developed delayed insulin resistance. | Santhekadur et al. [12], Asgharpour et al. [58] |
| High-fat diet + glucose/ fructose-enriched drinking water | Obesogenic diet containing ((35.5% w/w) crude fat (58 kJ%), 22.8 MJ/kg = 5.45 kcal/g) and fructose (55% w/v) and glucose (45% w/v) enriched drinking water. After 8 weeks of dietary feeding, mice were randomly assigned to a voluntary wheel running group or a sedentary group. | Voluntary wheel running prevented HFD-induced pro-inflammatory / fibrogenic states in C57BL/6 mice. Hepatic steatosis was prevented by alterations in key liver metabolic processes. | Gehrke et al. [125] |
| Genetic Models | |||
| Leptin deficiency (ob/ob) | Leptin-deficient (ob/ob) mice are predisposed to develop NASH and fibrosis, whereas not when maintained on regular chow diet. Treatment with high-fat/high-fructose/high-cholesterol diet. | Lack the ability to spontaneously develop hepatic inflammation. After 12-26w, increased adiposity, total cholesterol and elevated plasma liver enzymes upon diet high in trans-fat (40%), fructose (22%) and cholesterol (2%). After treatment with high-fat/high-fructose/high-cholesterol diet, development of metabolic, histologic and transcriptomic features similar to human NASH. | Kristiansen et al. [29], Abe et al. [34], Trevaskis et al. [51] |
| Leptin resistance (db/db) | db/db mice are deficient in the leptin receptor, with dramatic elevations in circulating leptin concentrations. Dietary intervention with an MCD diet for 4 weeks. | Lack the ability to spontaneously develop hepatic inflammation and thus needs to be combined with a nutritional model for NASH. After 4w MCD diet, mice displayed marked hepatic inflammation and fibrosis. | Kennedy et al. [35], Sahai et al. [45], Hummel et al. [50] |
| MS-NASH (FATZO/Pco) | Mice spontaneous development of obesity | After 20w of fructose-supplemented diet, hepatic steatosis, lobular inflammation, ballooning and fibrosis. | Sun et al. [62] |
| Apolipoprotein E2 knock-in (APOE2) | Murine ApoE replaced by the human APOE2 gene | After 12w of HFC, steatosis in conjunction with early but not sustained hepatic inflammation. | Wouters et al. [25,26], Bieghs et al. [63] |
| ApoE deficiency (ApoE-/-) | Complete deficiency in the murine ApoE gene | After 7w of Western diet, abnormal glucose tolerance, hepatomegaly, weight gain and full spectrum of NASH, while lacking humanized lipoprotein profiles. | Schierwagen et al. [64] |
| Low-density lipoprotein receptor deficiency (Ldlr-/-) | Complete deficiency of the murine Ldl receptor, an important gene regulating the transport of non-modified lipids into macrophages | After 3-12w of HFC diet, resemblance to lifestyle-induced early-onset hepatic inflammation. High and low levels of circulating LDL and HDL, respectively, closely mimicked the human lipoprotein profile. Development of mild fibrosis. | Wouters et al. [25,26], Bieghs et al. [63] |
| Microsomal prostaglandin E synthase 1 (mPGES1) deficiency | Mice with global deletion of mPGES-133 were backcrossed on C57BL/6J | TNFα-dependent inflammatory response in murine liver. Increased severity of diet-induced murine NASH. | Henkel et al. [65] |
| Patatin-like phospholipase domain-containing 3 (PNPLA-3) knock-in | Mice carried I148M mutation in the Pnpla3 gene and were fed a high-sucrose or HFD diet for 4 weeks | Accumulation of PNPLA3 on lipid droplets. Development of hepatic steatosis. | Smagris et al. [72] |
| Transmembrane 6 superfamily member 2 knockdown (mTm6s2-shRNA8) | Adeno-associated virus-mediated short hairpin RNA knockdown of Tm6sf2 in liver of C57BL/6J mice | Increased hepatic fat content and decreased VLDL secretion, recapitulating the effects observed in humans carrying the TM6SF2-167Lys mutation | Kozlitina et al. [74] |
| Gankyrin liver-specific knockout (GLKO) | Cre-Alb mice were backcrossed with LoxP-Gank mice | Gankyrin generally drives liver proliferation. After 6-7 months of HFD, higher degree of hepatic steatosis but prevention of fibrosis development in GLKO mice compared to wild-type mice. | Cast et al. [75] |
| Truncated mutation in Alström (Alms1) gene (foz/foz) | 11-base pair truncating mutation in the Alström gene ALMS1. Lack of knowledge regarding the exact role of Alms1 | After 6 months of HFD, MetS features, including obesity, hyperglycemia / lipidemia and insulin resistance. Mice spontaneously develop steatosis, hepatic inflammation and fibrosis. | Santhekadur et al. [12], Jiang et al. [14], Arsov et al. [77] |
| Fatty liver Shionogi | Spontaneous development of hepatic inflammation with rather a mild degree of fibrosis. Uncontrollable heterogeneity in disease onset | Backcrossing with ob/ob mice resulted in severe liver steatosis, inflammation, advanced fibrosis and spontaneous HCC | He et al. [82] |
| Hepatocyte-specific phosphatase and tensin homolog deficiency (Pten-/-) | PTEN deficiency specific in the liver | After 40w of age, steatosis, inflammation and fibrosis in the liver. After 74-78w of age, HCC was present in 83% of males and 50% of female mice. | Watanabe et al. [83], Takakura et al. [84] |
| Augmenter of liver regeneration knock-out (Alr-/-) | Liver-specific deletion of augmenter of liver regeneration | 4-8w after birth, steatohepatitis with hepatocellular necrosis, ductular proliferation and fibrosis. 1y after birth, HCC in nearly 60% of the mice | Van Herck et al. [11], Gandhi et al. [89] |
| Melanocortin 4 receptor knockout (Mc4r-/-) | Mice with targeted disruption of melanocortin 4 receptor, which is a seven-transmembrane G protein–coupled receptor that is expressed in the hypothalamic nuclei | Development of simple steatosis. Upon feeding HFD, development of human-like NASH, including obesity, insulin resistance and dyslipidemia. After 20w HFD, obesity and NASH with clear signs of moderate fibrosis, functionally mimicking the human NASH disease state. | Itoh et al. [90], Yamada et al. [91] |
| Chemically-induced Models | |||
| Carbon tetrachloride (CCL4) | Biweekly injections of CCl4 | After 6w, increased circulating liver enzymes and dose-dependent progression of liver fibrosis in Balb/C mice | Domitrovic et al. [97] |
| Thioacetamide (TAA) | Three times/week IP injection thioacetamide (75mg/kg) in combination with western-type diet | After 8w, hepatic inflammation, severe diffuse fibrosis and collagen deposition in C57BL/6 mice | Hansen et al. [8] Van Herck et al. [11] Santhekadur et al. [12] |
| Streptozotocin + high-fat diet (STAM) | 200 μg streptozotocin at 2 days after birth and feeding ad libitum with high-fat diet at 4 weeks of age | Between 6-20w of age, hepatic inflammation, hepatocellular ballooning, progressive fibrosis and HCC. Reduced body weight and insulin levels compared to HFD-fed mice | Fujii et al. [99] |
| Other models | |||
| C57BL/6 background | Mice were housed in separate specific pathogen-free units maintained at either 22°C (standard) or 30-33°C (thermoneutral) | After 24w of thermoneutral housing, exacerbated HFD-driven NAFLD pathogenesis. Increased intestinal permeability and alterations in gut microbiome, mimicking the human situation. | Giles et al. [106] |
| C57BL/6 background | Juvenile NASH model: immediately after weaning, mice were fed HFC diet for a total of 16 weeks (4, 8, 12 and 16 weeks of diet) | Hepatic oxidative stress in female juvenile NAFLD/NASH models, whereas hepatic inflammation in males | Marin et al. [104] |
| C57BL/6 background | Juvenile NAFLD/NASH model: HFD were administered 2 weeks before conception and during gestation and lactation | Offspring HFC intake resulted in NAFLD, maternal-offspring fat intake contributed to NASH in juvenile female mice | Zhou et al. [105] |
| Models with circadian oscillations (e.g. Per1/2-/- or liver-specific Bmal1 knockout mice) | The effects of feeding time and circadian clocks on murine liver | Circadian rhythm drives oscillations in hepatic triglyceride levels, inflammation, oxidative stress, mitochondrial dysfunction and hepatic insulin resistance. Chronic disruption of circadian rhythm may spontaneously induce the progression from NAFLD to NASH, fibrosis and HCC | Adamovich et al. [107], Jacobi et al. [108], Kettner et al. [109] |
| C57BL/6 background | Mice (and other rodent models) were fed a high-fat/high-fructose/high-cholesterol for 16 weeks | Significant intraindividual differences in fibrosis score and hepatic biomarkers pointed towards the importance of standardizing sampling site location during preclinical liver biopsy procedures | Jensen et al. [2] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oligschlaeger, Y.; Shiri-Sverdlov, R. NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines 2020, 8, 28. https://doi.org/10.3390/biomedicines8020028
Oligschlaeger Y, Shiri-Sverdlov R. NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines. 2020; 8(2):28. https://doi.org/10.3390/biomedicines8020028
Chicago/Turabian StyleOligschlaeger, Yvonne, and Ronit Shiri-Sverdlov. 2020. "NAFLD Preclinical Models: More than a Handful, Less of a Concern?" Biomedicines 8, no. 2: 28. https://doi.org/10.3390/biomedicines8020028
APA StyleOligschlaeger, Y., & Shiri-Sverdlov, R. (2020). NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines, 8(2), 28. https://doi.org/10.3390/biomedicines8020028