Authentication of Primary Murine Cell Lines by a Microfluidics-Based Lab-On-Chip System

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Reagents, Plasmids and Media
2.3. Surface Marker Detection by FACS
2.4. Virus Production
2.5. Cell Lines
2.6. Differentiation of Immortalized HoxB8-FL Cells
2.7. DNA and RNA Isolation and Transcriptome Analysis
2.8. Short Tandem Repeat (STR)-Based Multiplex PCR
2.9. Fragment Length Analysis
3. Results
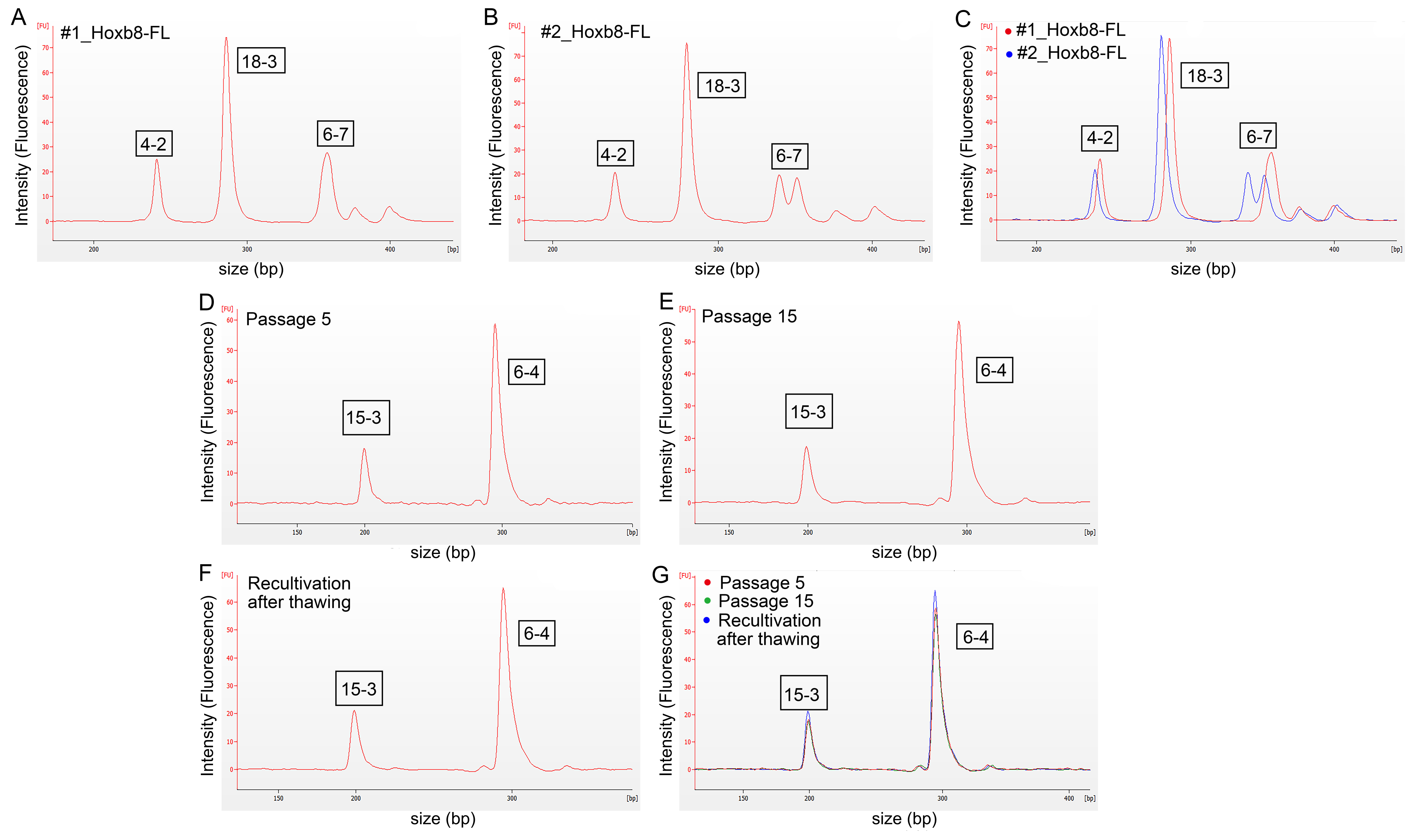
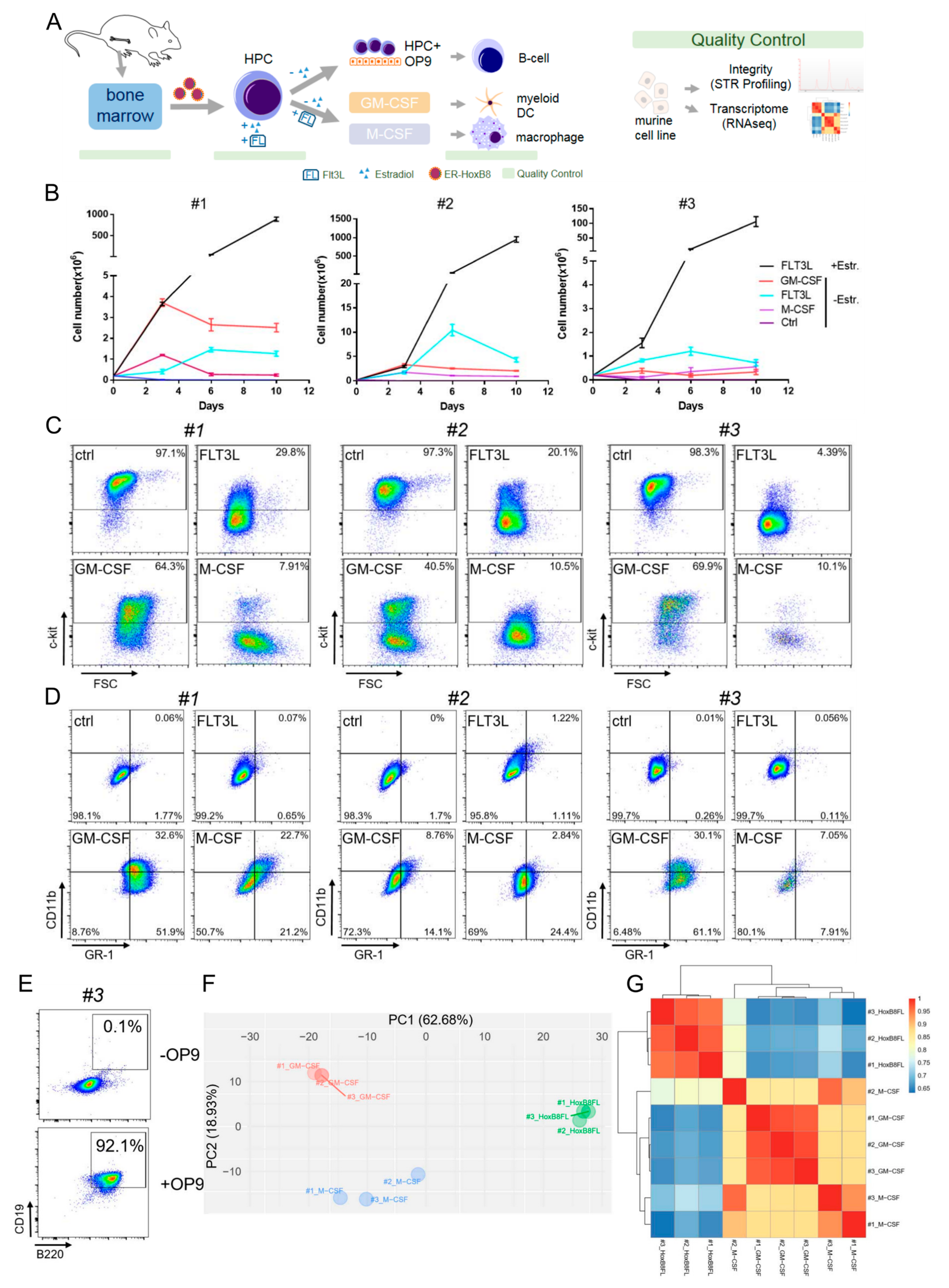
3.1. Differentiation and Characterization of Conditionally Immortalized HoxB8-FL Hematopoietic Progenitors
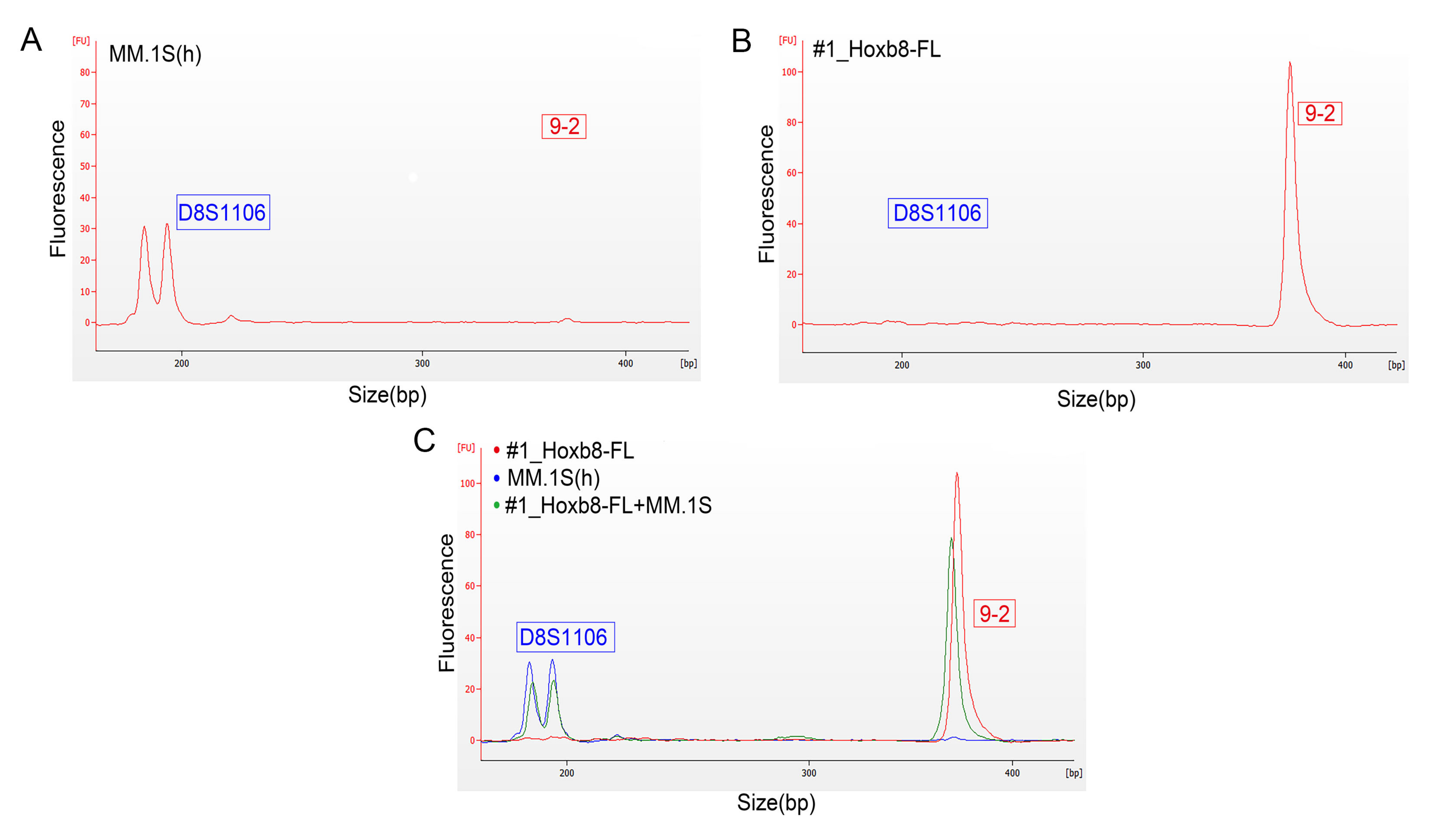
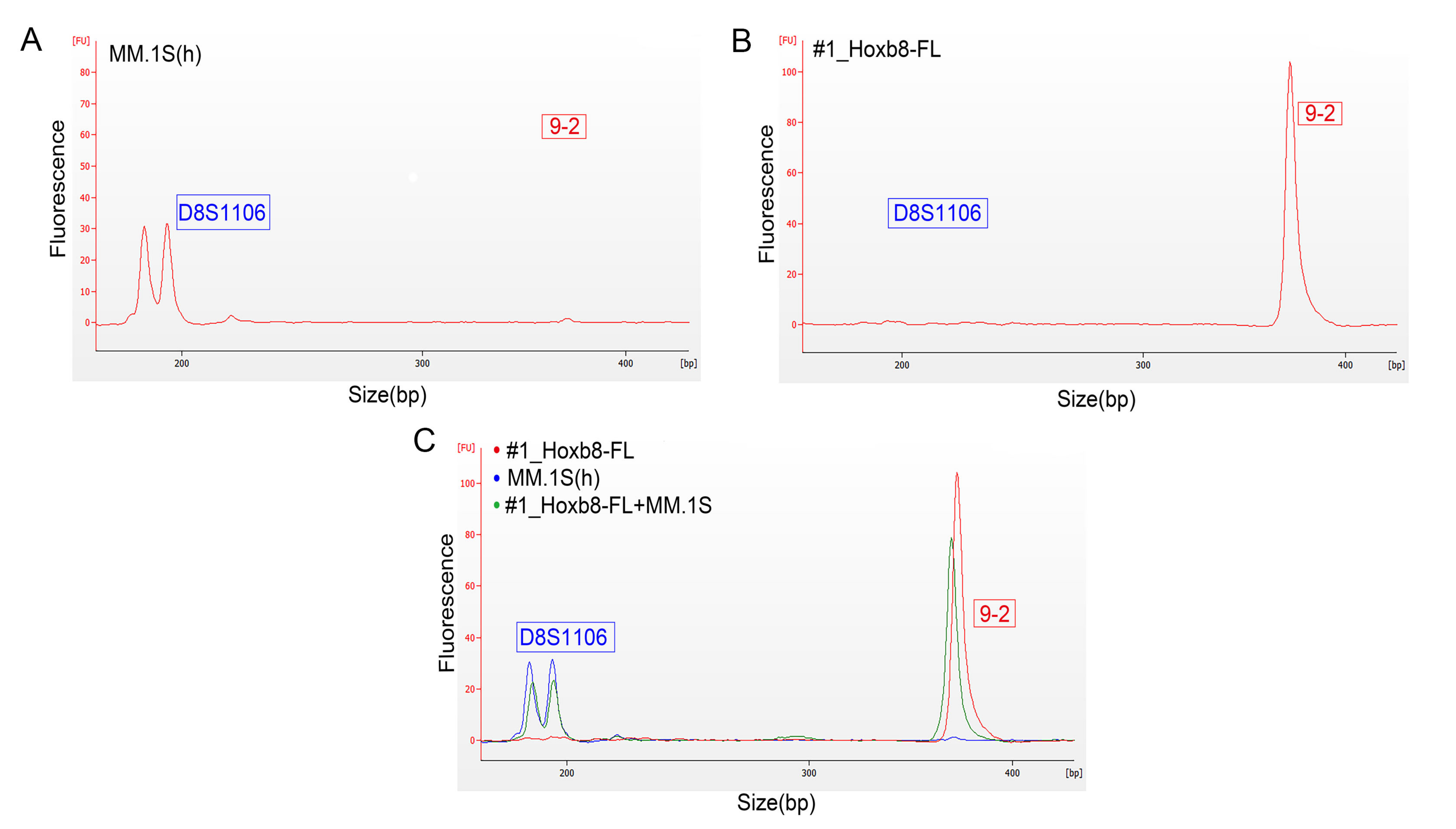
3.2. Authentication of HoxB8-FL Cell Lines by STR Profiling
3.3. Validation of Microfluidics-Based STR Profiling in Primary Murine Pancreatic Cancer Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rieger, M.A.; Schroeder, T. Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int. 2018, 2018, 3569493. [Google Scholar] [CrossRef] [Green Version]
- Redecke, V.; Wu, R.; Zhou, J.; Finkelstein, D.; Chaturvedi, V.; High, A.A.; Hacker, H. Hematopoietic progenitor cell lines with myeloid and lymphoid potential. Nat. Methods 2013, 10, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, S.I.; Werth, K.; Rothe, M.; Galla, M.; Permanyer, M.; Patzer, G.E.; Bubke, A.; Frenk, D.N.; Selich, A.; Lange, L.; et al. CRISPR/Cas9 Immunoengineering of Hoxb8-Immortalized Progenitor Cells for Revealing CCR7-Mediated Dendritic Cell Signaling and Migration Mechanisms in vivo. Front. Immunol. 2018, 9, 1949. [Google Scholar] [CrossRef]
- Zach, F.; Mueller, A.; Gessner, A. Production and Functional Characterization of Murine Osteoclasts Differentiated from ER-Hoxb8-Immortalized Myeloid Progenitor Cells. PLoS ONE 2015, 10, e0142211. [Google Scholar] [CrossRef]
- Wang, G.G.; Calvo, K.R.; Pasillas, M.P.; Sykes, D.B.; Hacker, H.; Kamps, M.P. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 2006, 3, 287–293. [Google Scholar] [CrossRef]
- Graham, M.L.; Prescott, M.J. The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur. J. Pharmacol. 2015, 759, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Freedman, L.P.; Gibson, M.C.; Ethier, S.P.; Soule, H.R.; Neve, R.M.; Reid, Y.A. Reproducibility: Changing the policies and culture of cell line authentication. Nat. Methods 2015, 12, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Marx, V. Cell-line authentication demystified. Nat. Methods 2014, 11, 483–488. [Google Scholar] [CrossRef]
- Masters, J.R.; Thomson, J.A.; Daly-Burns, B.; Reid, Y.A.; Dirks, W.G.; Packer, P.; Toji, L.H.; Ohno, T.; Tanabe, H.; Arlett, C.F.; et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl. Acad. Sci. USA 2001, 98, 8012–8017. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.L.; Hill, C.R.; Cole, K.D. Mouse cell line authentication. Cytotechnology 2014, 66, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.M. Genetics and genomics of core short tandem repeat loci used in human identity testing. J. Forensic Sci. 2006, 51, 253–265. [Google Scholar] [CrossRef]
- Cattoretti, G.; Pasqualucci, L.; Ballon, G.; Tam, W.; Nandula, S.V.; Shen, Q.; Mo, T.; Murty, V.V.; Dalla-Favera, R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005, 7, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, C.; Wittner, M.; Nguyen, J.; Rondeau, V.; Biajoux, V.; Aknin, M.L.; Gaudin, F.; Beaussant-Cohen, S.; Bertrand, Y.; Bellanne-Chantelot, C.; et al. Lymphoid differentiation of hematopoietic stem cells requires efficient Cxcr4 desensitization. J. Exp. Med. 2017, 214, 2023–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amend, S.R.; Valkenburg, K.C.; Pienta, K.J. Murine Hind Limb Long Bone Dissection and Bone Marrow Isolation. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrovolny, P.L.; Bess, D. Optimized PCR-based detection of mycoplasma. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef] [PubMed]
- Bamopoulos, S.A.; Batcha, A.M.N.; Jurinovic, V.; Rothenberg-Thurley, M.; Janke, H.; Ksienzyk, B.; Philippou-Massier, J.; Graf, A.; Krebs, S.; Blum, H.; et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia 2020. [Google Scholar] [CrossRef]
- Agilent DNA 1000; Agilent Technologies, Inc.: Santa Clara, CA, USA, 2016; Volume G2938-90014, p. 24.
- Carlyle, J.R.; Michie, A.M.; Furlonger, C.; Nakano, T.; Lenardo, M.J.; Paige, C.J.; Zuniga-Pflucker, J.C. Identification of a novel developmental stage marking lineage commitment of progenitor thymocytes. J. Exp. Med. 1997, 186, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, T.M.; Zuniga-Pflucker, J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Barallon, R.; Bauer, S.R.; Butler, J.; Capes-Davis, A.; Dirks, W.G.; Elmore, E.; Furtado, M.; Kline, M.C.; Kohara, A.; Los, G.V.; et al. Recommendation of short tandem repeat profiling for authenticating human cell lines, stem cells, and tissues. In Vitro Cell Dev. Biol. Anim. 2010, 46, 727–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Q.; Fillmore, H.L.; Vouri, M.; Pilkington, G.J. Brain tumor cell line authentication, an efficient alternative to capillary electrophoresis by using a microfluidics-based system. Neuro Oncol. 2014, 16, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.L.; Cole, K.D.; Plant, A.L. Standards for Cell Line Authentication and Beyond. PLoS Biol. 2016, 14, e1002476. [Google Scholar] [CrossRef]
- Aboud, M.J.; Gassmann, M.; McCord, B.R. The development of mini pentameric STR loci for rapid analysis of forensic DNA samples on a microfluidic system. Electrophoresis 2010, 31, 2672–2679. [Google Scholar] [CrossRef]
- Cardoso, S.; Valverde, L.; Odriozola, A.; Elcoroaristizabal, X.; de Pancorbo, M.M. Quality standards in Biobanking: Authentication by genetic profiling of blood spots from donor’s original sample. Eur. J. Hum. Genet. 2010, 18, 848–851. [Google Scholar] [CrossRef]
- Yuille, M.; van Ommen, G.J.; Brechot, C.; Cambon-Thomsen, A.; Dagher, G.; Landegren, U.; Litton, J.E.; Pasterk, M.; Peltonen, L.; Taussig, M.; et al. Biobanking for Europe. Brief. Bioinform. 2008, 9, 14–24. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Medium | Concentration | Media/Additive | Catalog No. | Storage |
|---|---|---|---|---|
| RP-10 Medium | RPMI1640 | Gibco: 21875-034 | 4 °C | |
| 10% | FBS (not heat inactivated) | Gibco: 10270-106 | −20 °C | |
| 0.10% | Mercapto Ethanol | Gibco: 31350-010 50 mM | 4 °C | |
| 1% | Antibiotic Antimycotic | Gibco: 15240-062 | −20 °C | |
| Progenitor Outgrowth Medium | RPMI1640 | Gibco: 21875-034 | 4 °C | |
| 10% | FBS (not heat inactivated) | Gibco: 10270-106 | −20 °C | |
| 0.10% | Mercapto Ethanol | Gibco: 31350-010 50 mM | 4 °C | |
| 1% | Antibiotic Antimycotic | Gibco: 15240-062 | −20 °C | |
| 1 µM | β-estradiol | Sigma: E-2758 (stock 100 mM) | −20 °C | |
| 5% | FLT3L-SNT | from B16 melanoma cell line | −20 °C | |
| B-Cell Differentiation Medium | RPMI1640 | Gibco: 21875-034 | 4 °C | |
| 10% | FBS (not heat inactivated) | Gibco: 10270-106 | −20 °C | |
| 0.10% | Mercapto Ethanol | Gibco: 31350-010 50 mM | 4 °C | |
| 1% | Antibiotic Antimycotic | Gibco: 15240-062 | −20 °C | |
| 1 µM | β-estradiol | Sigma: E-2758 (stock 100 mM) | −20 °C | |
| 5 ng/mL | FLT3L | R & D Systems: 427-FL | −20 °C | |
| 25 ng/mL | SCF | R & D Systems: 455-MC | −20 °C | |
| 7 ng/mL | IL-7 | R & D Systems: 407-ML | −20 °C | |
| BBMM (Basal Bone Marrow Medium) | IMDM | Gibco: 12440-053 | 4 °C | |
| 30% | FBS (not heat inactivated) | Gibco: 10270-106 | −20 °C | |
| 0.20% | Mercapto Ethanol | Gibco: 31350-010 50 mM | 4 °C | |
| 0.50% | Antibiotic Antimycotic | Gibco: 15240-062 | −20 °C | |
| 1% | Glutamine | Corning: 25-005-CV | −20 °C | |
| 0.50% | BSA | Sigma-Aldrich: B6917 | 4 °C | |
| HF2 Buffer | ddH2O | Ampuwa: 250166 | RT | |
| 30% | FBS (not heat inactivated) | Gibco: 10270-106 | −20 °C | |
| 0.50% | Antibiotic Antimycotic | Gibco: 15240-062 | −20 °C | |
| 1% | HEPES | Gibco: 15630-049 | 4 °C |
| Cell Line | 18-3 | 4-2 | 6-7 | 9-2 | 15-3 | 6-4 | 12-1 | 5-5 | X-1 |
|---|---|---|---|---|---|---|---|---|---|
| #1_HoxB8FL (wild-type) | 16 | 19 | 19 | 17 | 22 | 19 | 15 | 16 | 23 |
| #2_HoxB8FL (Iμ-HA-Bcl6) | 15 | 18 | 15, 18 | 15 | 20 | 18 | 16 | 17 | 24 |
| #3_HoxB8FL (Cxcr4WHIM) | 16 | 19 | 19 | 15 | 22 | 19 | 16 | 17 | 24 |
| Cell Line | 18-3 | 4-2 | 6-7 | 9-2 | 15-3 | 6-4 | 12-1 | 5-5 | X-1 |
|---|---|---|---|---|---|---|---|---|---|
| mPDAC06 | 17, 18 | 18 | 20 | 16 | 20, 22 | 17 | 16, 20 | 14, 16 | 20, 24 |
| mPDAC09 | 17, 19 | 17, 19 | 20, 23 | 18, 22 | 20 | 18 | 16 | 16, 17 | 18, 25 |
| mPDAC95 | 17 | 19, 19 | 17 | 18, 20 | 21 | 19 | 16 | 16, 17 | 26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Singh, N.; Bamopoulos, S.; Gjerga, E.; Schmalbrock, L.K.; Balabanian, K.; Schick, M.; Keller, U.; Wirth, M. Authentication of Primary Murine Cell Lines by a Microfluidics-Based Lab-On-Chip System. Biomedicines 2020, 8, 590. https://doi.org/10.3390/biomedicines8120590
Hong Y, Singh N, Bamopoulos S, Gjerga E, Schmalbrock LK, Balabanian K, Schick M, Keller U, Wirth M. Authentication of Primary Murine Cell Lines by a Microfluidics-Based Lab-On-Chip System. Biomedicines. 2020; 8(12):590. https://doi.org/10.3390/biomedicines8120590
Chicago/Turabian StyleHong, Yingfen, Nikita Singh, Stefanos Bamopoulos, Enio Gjerga, Laura K. Schmalbrock, Karl Balabanian, Markus Schick, Ulrich Keller, and Matthias Wirth. 2020. "Authentication of Primary Murine Cell Lines by a Microfluidics-Based Lab-On-Chip System" Biomedicines 8, no. 12: 590. https://doi.org/10.3390/biomedicines8120590