Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding

 ,
,  , and
, and
Abstract

1. Introduction
2. Materials and Methods
3. Results and Discussion
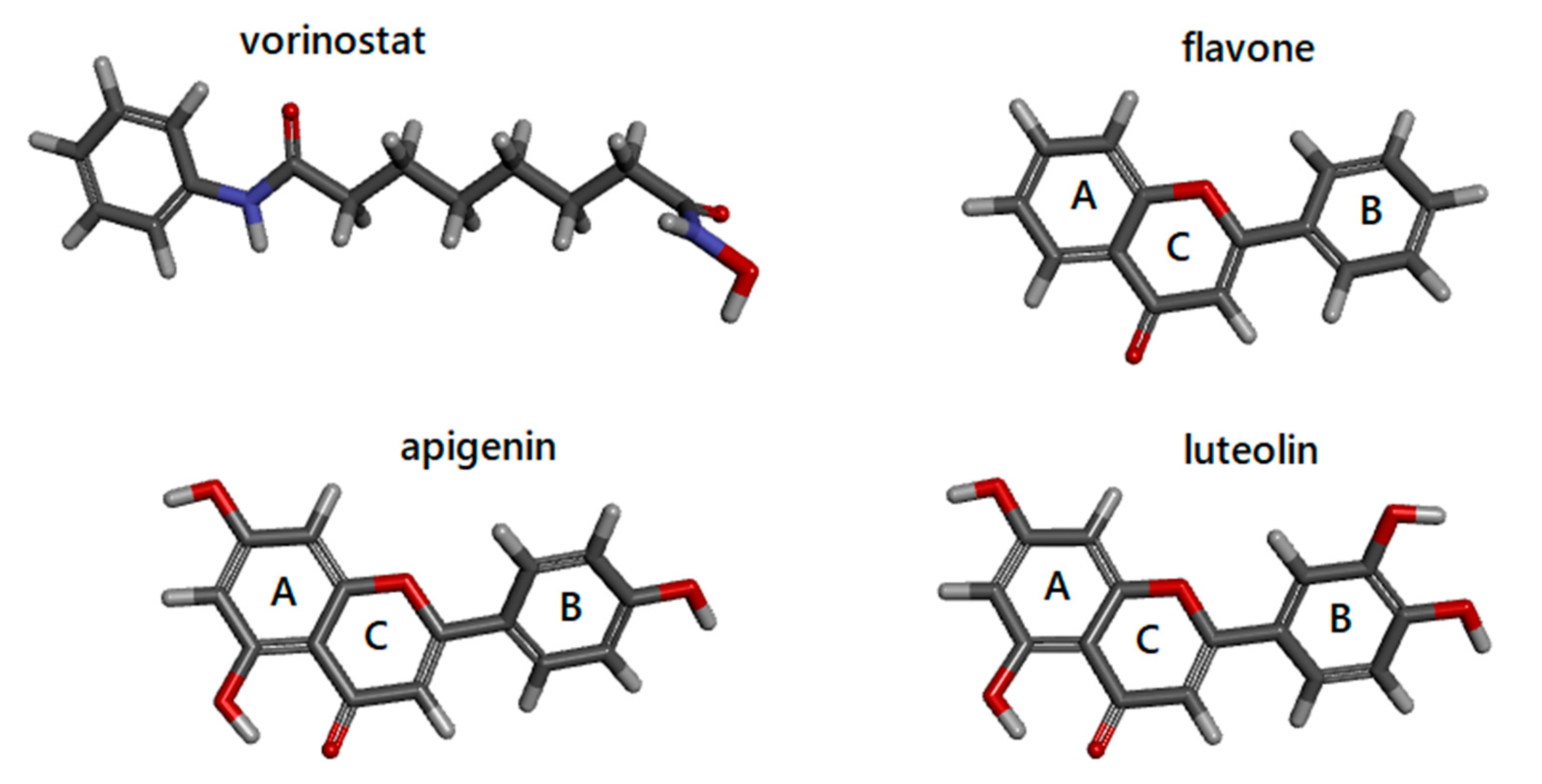
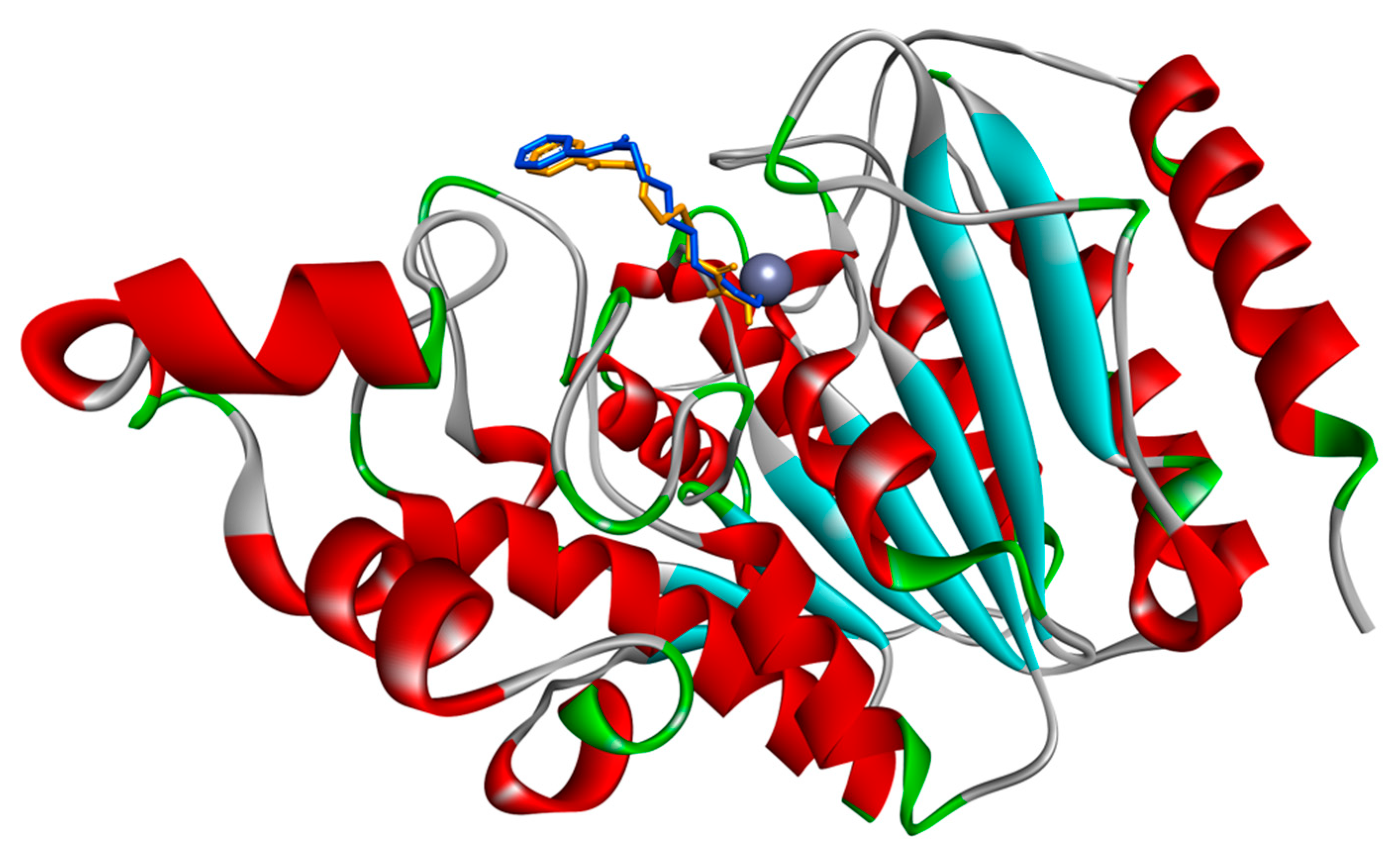
3.1. Docking Simulations of Inhibitor Vorinostat Interaction with HDAC1 and HDAC2
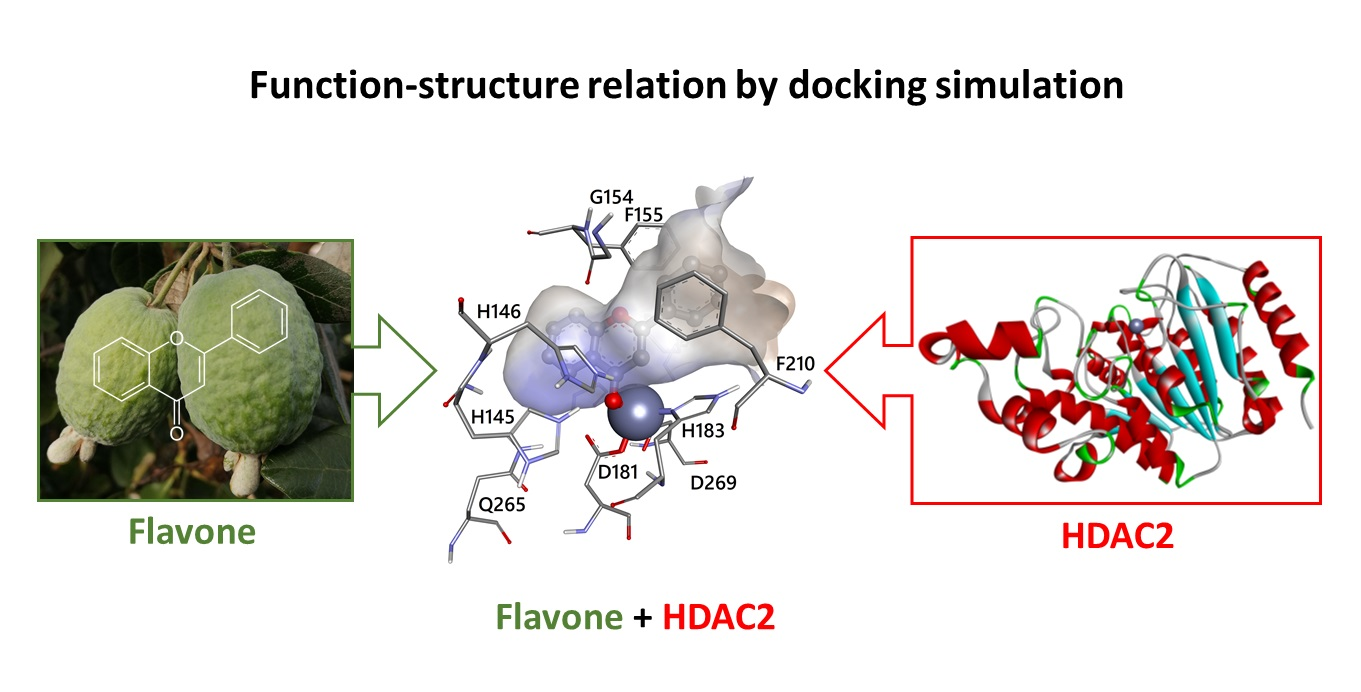
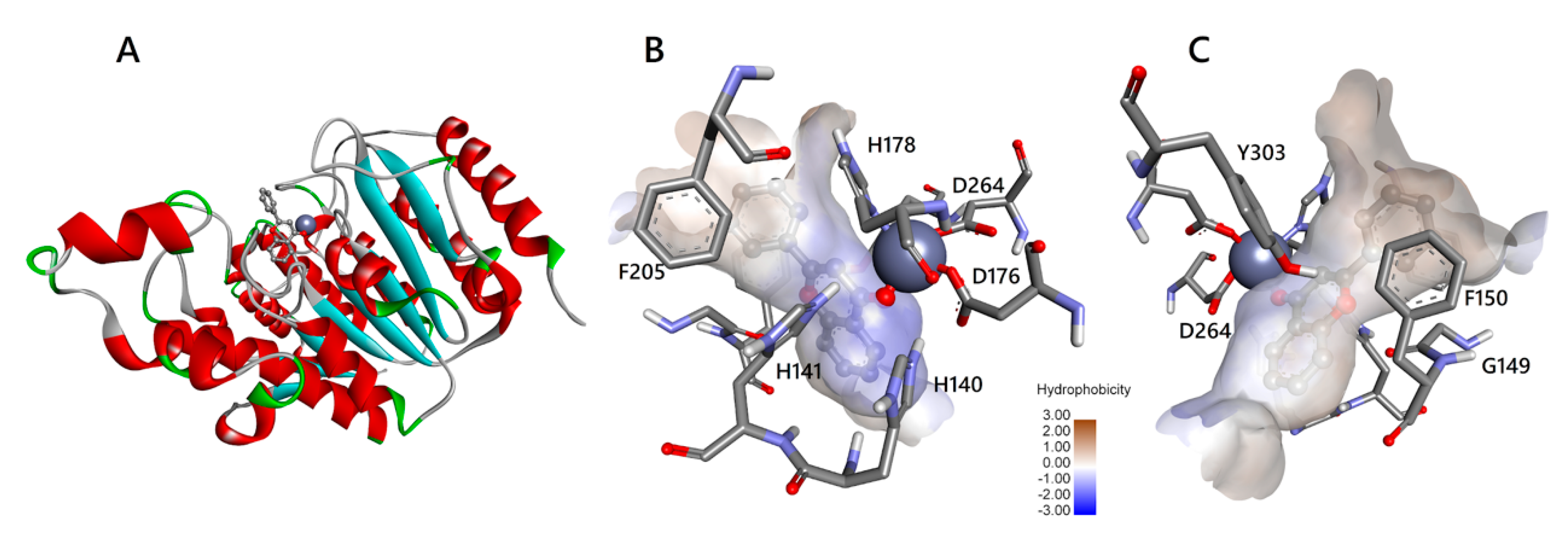
3.2. Docking Simulations of Flavones Interaction with HDAC1 and HDAC2
3.3. Molecular Interactions of Flavones with HDAC1 and HDAC2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.; Bontempo, P.; Nebbioso, A.; Altucci, L. Natural compounds in epigenetics: A current view. Food Chem. Toxicol. 2014, 73, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold. Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, R.; Srikanta, T.; Rath, K. Anticancer potential of the histone deacetylase inhibitor-like effects of flavones, a subclass of polyphenolic compounds: A review. Mol. Biol. Rep. 2015, 42, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2–S6. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Bontempo, P.; Mita, L.; Miceli, M.; Doto, A.; Nebbioso, A.; De Bellis, F.; Conte, M.; Minichiello, A.; Manzo, F.; Carafa, V.; et al. Feijoa sellowiana derived natural Flavone exerts anti-cancer action displaying HDAC inhibitory activities. Int. J. Biochem. Cell Biol. 2007, 39, 1902–1914. [Google Scholar] [CrossRef]
- Bontempo, P.; Rigano, D.; Doto, A.; Formisano, C.; Conte, M.; Nebbioso, A.; Carafa, V.; Caserta, G.; Sica, V.; Molinari, A.M.; et al. Genista sessilifolia DC. extracts induce apoptosis across a range of cancer cell lines. Cell Proliferat. 2013, 46, 183–192. [Google Scholar] [CrossRef]
- Magri, A.; Adiletta, G.; Petriccione, M. Evaluation of antioxidant systems and ascorbate-glutathione cycle in Feijoa edible flowers at different flowering stages. Foods 2020, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F. Chemical and biological properties of feijoa (Acca sellowiana). Trends Food Sci. Technol. 2018, 81, 121–131. [Google Scholar] [CrossRef]
- Pandey, M.; Kaur, P.; Shukla, S.; Abbas, A.; Fu, P.; Gupta, S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: In vitro and in vivo study. Mol. Carcinog. 2012, 51, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Attoub, S.; Hassan, A.H.; Vanhoecke, B.; Iratni, R.; Takahashi, T.; Gaben, A.M.; Bracke, M.; Awad, S.; John, A.; Kamalboor, H.A.; et al. Inhibition of cell survival, invasion, tumor growth and histone deacetylase activity by the dietary flavonoid luteolin in human epithelioid cancer cells. Eur. J. Pharmacol. 2011, 651, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Scafuri, B.; Marabotti, A.; Carbone, V.; Minasi, P.; Dotolo, S.; Facchiano, A. A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Sci. Rep. 2016, 6, 32516. [Google Scholar] [CrossRef]
- Scafuri, B.; Varriale, A.; Facchiano, A.; D’Auria, S.; Raggi, M.E.; Marabotti, A. Binding of mycotoxins to proteins involved in neuronal plasticity: A combined in silico/wet investigation. Sci. Rep. 2017, 7, 15156. [Google Scholar] [CrossRef]
- Ganai, S.A.; Farooq, Z.; Banday, S.; Altaf, M. In silico approaches for investigating the binding propensity of apigenin and luteolin against class I HDAC isoforms. Future Med. Chem. 2018, 10, 1925–1945. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Burkard, M.; Leischner, C.; Lauer, U.M.; Frank, J.; Venturelli, S. Epigenetic activities of flavonoids in the prevention and treatment of cancer. Clin. Epigenet. 2015, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Wiest, O.; Helquist, P.; Lan-Hargest, H.Y.; Wiech, N.L. On the function of the 14 Å long internal cavity of histone deacetylase-like protein: Implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Wascholowski, V.; Giannis, A. Epigenetics—An epicenter of gene regulation: Histones and histone-modifying enzymes. Angew. Chem. Int. Ed. Engl. 2005, 44, 3186–3216. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Volpari, C.; Filocamo, G.; Caroli Casavoli, E.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, C.; Wang, J.; Zou, Y.; Chen, X.; Liu, T.; Li, Y.; Zhao, Y.; Li, Y.; He, B. Nε-acetyl lysine derivatives with zinc binding groups as novel HDAC inhibitors. R. Soc. Open Sci. 2019, 6, 190338. [Google Scholar] [CrossRef]
- Wu, R.; Lu, Z.; Cao, Z.; Zhang, Y. Zinc Chelation with Hydroxamate in Histone Deacetylases Modulated by Water Access to the Linker Binding Channel. J. Am. Chem. Soc. 2011, 133, 6110–6113. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein–Ligand Complex | Free Energy of Interaction (Kcal/mol) | Explicit Interaction with Zn2+ Ion | Rigid/Flexible Docking | Notes |
|---|---|---|---|---|
| Blind docking | ||||
| HDAC1-flavone | −8.50 | yes | Rigid | |
| HDAC1-luteolin | −7.09 | yes | Rigid | |
| HDAC1-apigenin | −7.61 | yes | Rigid | |
| HDAC1-vorinostat | −7.23 | yes | Rigid | |
| HDAC2-flavone | −7.94 | yes | Rigid | |
| HDAC2-luteolin | −6.84 | no | Rigid | |
| HDAC2-apigenin | −8.98 | yes | flexible | hydrated with 3 water molecules |
| HDAC2-vorinostat | −7.45 | yes | Rigid | Hydrated |
| Focused docking | ||||
| HDAC1-flavone | −10.25 | Yes | flexible | |
| HDAC1-luteolin | −9.41 | Yes | flexible | |
| HDAC1-apigenin | −9.25 | Yes | flexible | |
| HDAC1-vorinostat | −8.46 | Yes | rigid | hydrated |
| HDAC2-flavone | −8.90 | Yes | rigid | |
| HDAC2-luteolin | −9.26 | Yes | flexible | |
| HDAC2-apigenin | −9.32 | Yes | flexible | |
| HDAC2-vorinostat | −8.45 | Yes | rigid | 2 water molecules involved |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scafuri, B.; Bontempo, P.; Altucci, L.; De Masi, L.; Facchiano, A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines 2020, 8, 568. https://doi.org/10.3390/biomedicines8120568
Scafuri B, Bontempo P, Altucci L, De Masi L, Facchiano A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines. 2020; 8(12):568. https://doi.org/10.3390/biomedicines8120568
Chicago/Turabian StyleScafuri, Bernardina, Paola Bontempo, Lucia Altucci, Luigi De Masi, and Angelo Facchiano. 2020. "Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding" Biomedicines 8, no. 12: 568. https://doi.org/10.3390/biomedicines8120568
APA StyleScafuri, B., Bontempo, P., Altucci, L., De Masi, L., & Facchiano, A. (2020). Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines, 8(12), 568. https://doi.org/10.3390/biomedicines8120568