Environmental and Nutritional “Stressors” and Oligodendrocyte Dysfunction: Role of Mitochondrial and Endoplasmatic Reticulum Impairment



 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- Interfascicular oligodendrocytes, which myelinate neuronal axons in white matter tracts;
- Perivascular oligodendrocytes which, which metabolically support axons;
- Perineuronal oligodendrocytes, which constitute a cellular pool for remyelination processes, if necessary, and regulate neuronal excitability.
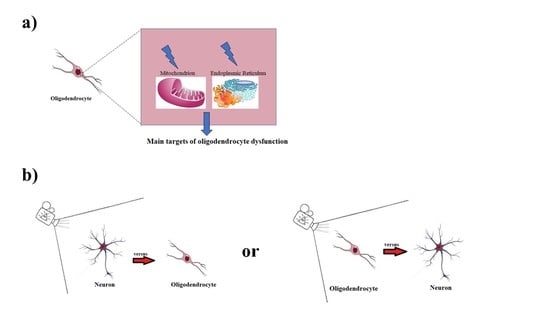
2. Mitochondria and Endoplasmic Reticulum in Oligodendrocytes
- during the formation of myelin, they provide the substrates to generate the necessary and sufficient energy for the synthesis of lipids;
- during the development of brain functions, the mitochondrial production of ATP will be used to contribute to the support of axonal function;
- in the case of pathological changes, the mitochondria are able to buffer excess calcium ion or to induce the activation of apoptosis [27].
3. Environmental and Nutritional “Stressors” and Oligodendrocytes Impairment
3.1. Chronic Consumption of Alcohol and Oligodendrocytes
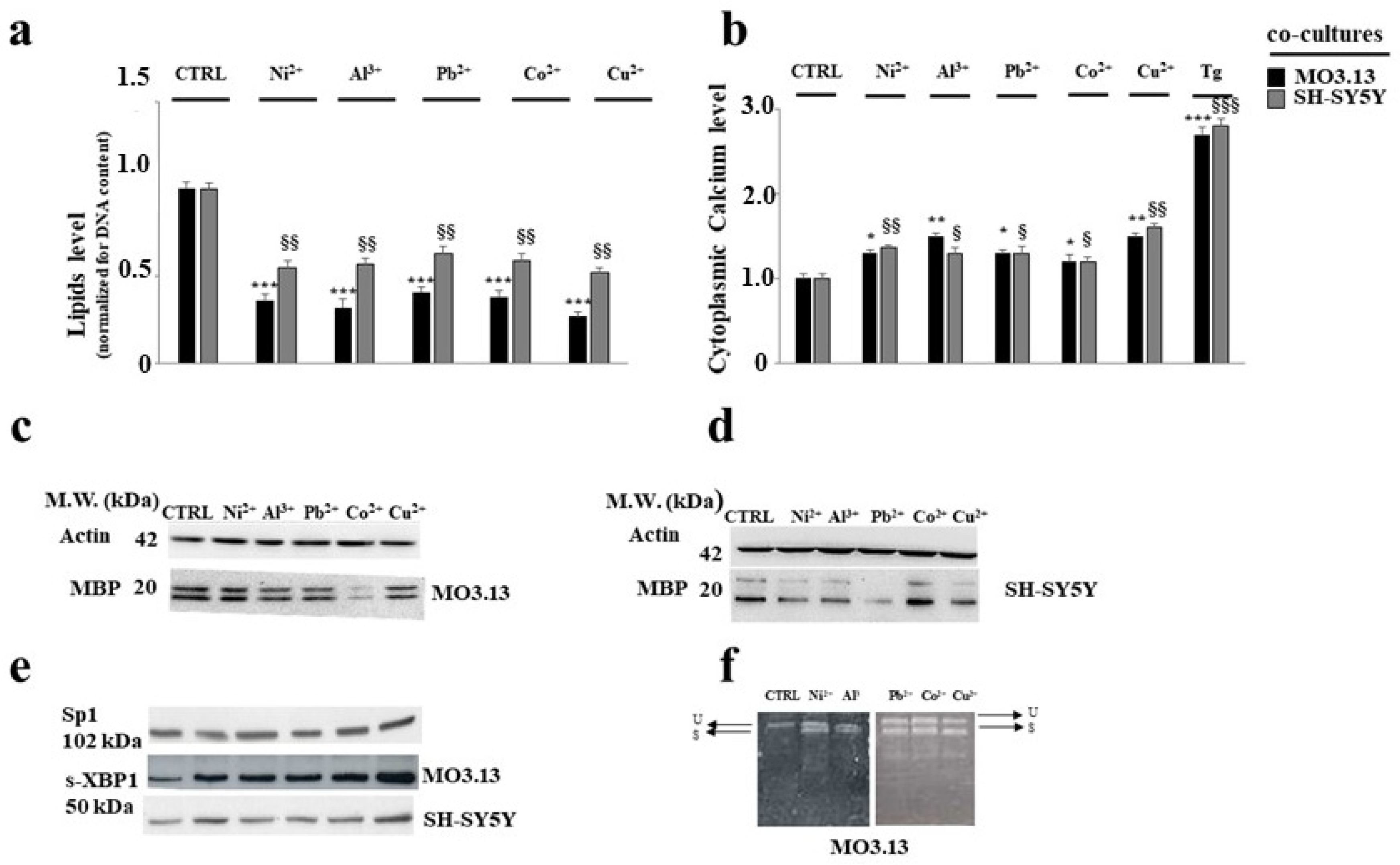
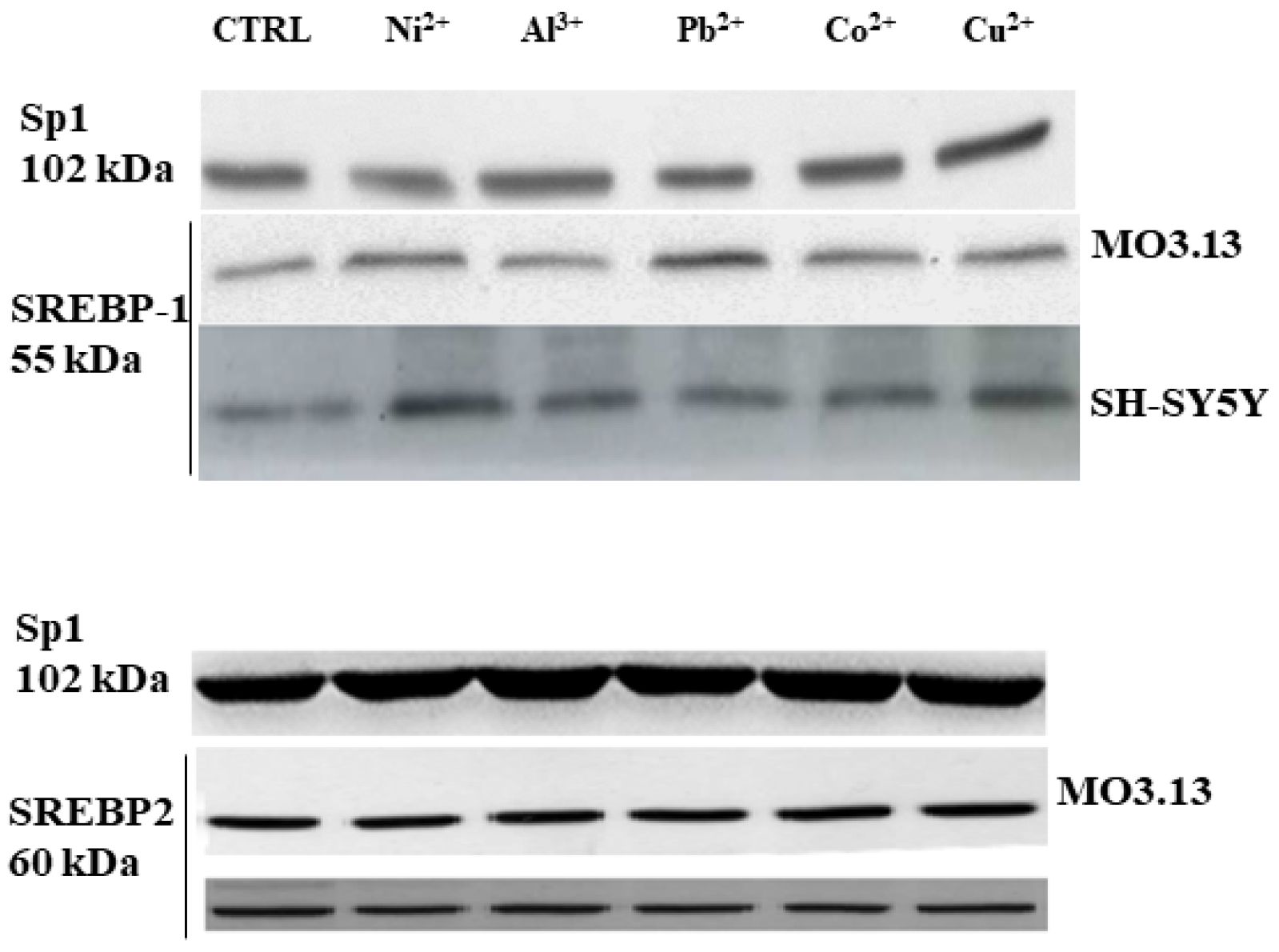
3.2. Intake of Heavy Metals and Oligodendrocytes
4. Discussion and Conclusions
- (1)
- Beginning to consider mitochondria and the endoplasmic reticulum as the main targets of oligodendrocyte dysfunction and trying to restore their conditions in order to evaluate any positive modulations in various neurological pathologies;
- (2)
- Changing the direction in which neurological pathologies are observed—from the oligodendrocyte to the neuron, instead of from the neuron to the oligodendrocyte. In this way, oligodendrocytes, astrocytes, and microglia could be considered as the reference point of the neuron in health and disease conditions.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Huguenard, J.R.; Pașca, S. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Suzuki, N. Heterogeneity of Oligodendrocytes and Their Precursor Cells. Adv. Exp. Med. Biol. 2019, 1190, 53–62. [Google Scholar] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Hasel, P.; Dando, O.; Jiwaj, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef]
- Wellman, S.M.; Cambi, F.; Kozai, T.D.Y. The role of oligodendrocytes and their progenitors on neural interface technology: A novel perspective on tissue regeneration and repair. Biomaterials 2018, 183, 200–217. [Google Scholar] [CrossRef]
- Gouveia, L.; Betsholtz, C.; Andrae, J. Expression analysis of platelet-derived growth factor receptor alpha and its ligands in the developing mouse lung. Physiol. Rep. 2017, 5, e13092. [Google Scholar] [CrossRef]
- Flores-Obando, R.E.; Freidin, M.M.; Abrams, C.K. Rapid and Specific Immunomagnetic Isolation of Mouse Primary Oligodendrocytes. J. Vis. Exp. 2018, 135, 57543. [Google Scholar] [CrossRef]
- Dugas, J.C.; Tai, Y.C.; Speed, T.P.; Ngai, J.; Barres, B.A. Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci. 2006, 26, 10967–10983. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Lu, F.F.; Shao, Q.; Zhu, M.; Cao, L. Progress in metabolism and function of myelin lipids. Sheng Li Xue Bao 2017, 69, 817–829. [Google Scholar] [PubMed]
- Taveggia, C. Schwann cells-axon interaction in myelination. Curr. Opin. Neurobiol. 2016, 39, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Castelvetri, L.C.; Simons, M. Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta 2015, 1851, 999–1005. [Google Scholar] [CrossRef]
- Grigoletto, J.; Pukaß, K.; Gamliel, A.; Davidi, D.; Katz-Brull, R.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Higher levels of myelin phospholipids in brains of neuronal α-Synuclein transgenic mice precede myelin loss. Acta Neuropathol. Commun. 2017, 5, 37. [Google Scholar] [CrossRef]
- Widder, K.; Harauz, G.; Hinderberger, D. Myelin basic protein (MBP) charge variants show different sphingomyelin-mediated interactions with myelin-like lipid monolayers. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183077. [Google Scholar] [CrossRef]
- Fu, M.M.; McAlear, T.S.; Nguyen, H.; Oses-Prieto, J.A.; Valenzuela, A.; Shi, R.D.; Perrino, J.J.; Huang, T.T.; Burlingame, A.L.; Bechstedt, S.; et al. The Golgi Outpost Protein TPPP Nucleates Microtubules and Is Critical for Myelination. Cell 2019, 179, 132–146.e14. [Google Scholar] [CrossRef]
- Ishii, A.; Han, D.; Bansal, R. Proteomics Analysis of Myelin Composition. Methods Mol. Biol. 2018, 1791, 67–77. [Google Scholar]
- Forbes, T.A.; Gallo, V. All wrapped up: Environmental effects on myelination. Trends Neurosci. 2017, 40, 572–587. [Google Scholar] [CrossRef]
- Ishii, A.; Furusho, M.; Macklin, W.; Bansal, R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia 2019, 67, 1277–1295. [Google Scholar] [CrossRef]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2018, 66, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Dulamea, A.O. Role of Oligodendrocyte Dysfunction in Demyelination, Remyelination and Neurodegeneration in Multiple Sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127. [Google Scholar] [PubMed]
- Chamberlain, K.A.; Sheng, Z.H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Maretta, A.; Gliozzi, M.; Musolino, V.; Carresi, C.R.; Macrì, R.; Nuvera, S.; Scicchitano, M.; Scarano, F.; Bosco, F.; et al. The Role of the Endoplasmic Reticulum in Early Damage Generated by Contact of Neurons and Oligodendrocytes with Heavy Metals in a Co-culture. J. Nutr. Food Sci. 2020. Available online: Hdl.handle.net/20.500.12317/62481 (accessed on 28 November 2020).
- Jha, M.K.; Morrison, B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018, 309, 23–31. [Google Scholar] [CrossRef]
- Sheng, Z.H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the MousCorpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Rinholm, J.E.; Vervaeke, K.; Tadross, M.R.; Tkachuk, A.N.; Kopek, B.G.; Bergersen, L.H.; Clayton, D.A. Movement and Structure of Mitochondria in Oligodendrocytes and Their Myelin Sheaths. Glia 2016, 64, 810–825. [Google Scholar] [CrossRef]
- Sghaier, R.; Zarrouk, A.; Nury, T.; Badreddine, I.; O’Brien, N.; Mackrill, J.J.; Vejux, A.; Samadi, M.; Nasser, B.; Caccia, C.; et al. Biotin attenuation of oxidative stress, mitochondrial dysfunction, lipid metabolism alteration and 7β-hydroxycholesterol-induced cell death in 158N murine oligodendrocytes. Free Radic. Res. 2019, 53, 535–561. [Google Scholar] [CrossRef]
- Oppedisano, F.; Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Scicchitano, M.; Scarano, F.; Bosco, F.; Macrì, R.; et al. The Potential for Natural Antioxidant Supplementation in the Early Stages of Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 2618. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Hernández, D.E.; Salvadores, N.A.; Moya-Alvarado, G.; Catalán, R.J.; Bronfman, F.C.; Court, F.A. Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J. Cell Sci. 2018, 131, jcs214684. [Google Scholar] [CrossRef] [PubMed]
- Corasaniti, M.T.; Maiuolo, J.; Maida, S.; Fratto, V.; Navarra, M.; Amantea, D.; Morrone, L.A.; Bagetta, G. Cell signaling pathways in the mechanisms of neuroprotection afforded by bergamot essential oil against NMDA-induced cell death in vitro. Br. J. Pharmacol. 2007, 151, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, M.; O’Neill, N.; Lariccia, V.; Amoroso, S.; Sylantyev, S.; Angelova, P.R.; Abramov, A.Y. Inorganic Polyphosphate Regulates AMPA and NMDA Receptors and Protects Against Glutamate Excitotoxicity via Activation of P2Y Receptors. J. Neurosci. 2019, 39, 6038–6048. [Google Scholar] [CrossRef]
- Sui, Y.; Nguyen, H.B.; Thai, T.Q.; Ikenaka, K.; Ohno, N. Mitochondrial Dynamics in Physiology and Pathology of Myelinated Axons. Adv. Exp. Med. Biol. 2019, 1190, 145–163. [Google Scholar]
- Volpi, V.G.; Touvier, T.; D’Antonio, M. Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front. Mol. Neurosci. 2017, 9, 162. [Google Scholar] [CrossRef]
- Clayton, B.L.L.; Popko, B. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res. 2016, 1648 Pt. B, 594–602. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Herbst, L.; Alexis, M.; Karnell, J.; Davidson, T.; Dutta, R.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelinationand remyelination in the CNS. Exp. Neurol. 2016, 283 Pt B, 501–511. [Google Scholar] [CrossRef]
- Nobuta, H.; Yang, N.; Ng, Y.H.; Marro, S.G.; Sabeur, K.; Chavali, M.; Stockley, J.H.; Killilea, D.W.; Walter, P.B.; Zhao, C.; et al. Oligodendrocyte Death in Pelizaeus-Merzbacher Disease Is Rescued by Iron Chelation. Cell Stem Cell 2019, 25, 531–541.e6. [Google Scholar] [CrossRef]
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 7832–7837. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Chen, L.; Huang, Z.; Wang, W.; Zhang, B.; Xu, Y.; Mao, Z.; Hu, H.; Geng, Q. Autophagy Activation by Rapamycin before Hypoxia-Reoxygenation Reduces Endoplasmic Reticulum Stress in Alveolar Epithelial Cells. Cell. Physiol. Biochem. 2017, 41, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Way, S.W.; Popko, B. Harnessing the integrated stress response for the treatment of multiple sclerosis. Lancet Neurol. 2016, 15, 434–443. [Google Scholar] [CrossRef]
- Kouga, T.; Koizume, S.; Aoki, S.; Jimbo, E.; Yamagata, T.; Inoue, K.; Osaka, H. Drug screening for Pelizaeus-Merzbacher disease by quantifying the total levels and membrane localization of PLP1. Mol. Genet. Metab. Rep. 2019, 20, 100474. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, J.; Jiang, Y.; Wu, Y.; Hao, H.; Ji, T. Different Mutations of Gap Junction Connexin 47 Lead to Discrepant Activation of Unfolded Protein Response Pathway in Pelizaeus-Merzbacher-Like Disease. Neuropediatrics 2017, 48, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Ramchandren, S. Charcot-Marie-Tooth Disease and Other Genetic Polyneuropathies. Continuum (Minneap. Minn.) 2017, 23, 1360–1377. [Google Scholar] [CrossRef] [PubMed]
- Morena, J.; Gupta, A.; Hoyle, J.C. Charcot-Marie-Tooth: From Molecules to Therapy. Int. J. Mol. Sci. 2019, 20, 3419. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Lin, W.; Stone, S. Unfolded protein response in myelin disorders. Neural Regen. Res. 2020, 15, 636–645. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Shai, N.; Schuldiner, M.; Bohnert, M. A Tether Is a Tether Is a Tether: Tethering at Membrane Contact Sites. Dev. Cell 2016, 39, 395–409. [Google Scholar] [CrossRef]
- Gatta, A.T.; Levine, T.P. Piecing Together the Patchwork of Contact Sites. Trends Cell Biol. 2017, 27, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Vance, V.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [PubMed]
- Jeong, H.; Park, J.; Jun, Y.; Lee, C. Crystal structures of Mmm1 and Mdm12-Mmm1 reveal mechanistic insight into phospholipid trafficking at ER-mitochondria contact sites. Proc. Natl. Acad. Sci. USA 2017, 114, E9502–E9511. [Google Scholar] [CrossRef]
- Kawano, S.; Tamura, Y.; Kojima, R.; Bala, S.; Riezman, I.H.; Sakae, Y.; Okamoto, Y.; Endo, T. Structure-function insights into direct lipid transfer between membranes by Mmm1-Mdm12 of ERMES. J. Cell Biol. 2018, 217, 959–974. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.-S.; Huh, S.; Liu, S.; Hong, S.H.; Yu, K.; Lu, B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca(2+) Homeostasis in Neural Stem Cell Development. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef]
- Armon-Omer, A.; Waldman, C.; Simaan, N.; Neuman, H.; Tamir, S.; Shahien, R. New Insights on the Nutrition Status and Antioxidant Capacity in Multiple Sclerosis Patients. Nutrients 2019, 11, 427. [Google Scholar] [CrossRef]
- Balto, J.M.; Ensari, I.; Hubbard, E.A.; Khan, N.; Barnes, J.L.; Motl, R.W. Individual and Co-occurring SNAP Risk Factors: Smoking, Nutrition, Alcohol Consumption, and Physical Activity in People with Multiple Sclerosis. Int. J. MS Care 2016, 18, 298–304. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention’s Office on Smoking and Tobacco Use. National Adult Tobacco Survey (NATS). 2014. Available online: http://www.cdc.gov/tobacco/data_statistics/surveys/nats (accessed on 5 May 2016).
- Friend, K.B.; Mernoff, S.T.; Block, P.; Reeve, G. Smoking rates and smoking cessation among individuals with multiple sclerosis. Disabil. Rehabil. 2006, 28, 1135–1141. [Google Scholar] [CrossRef]
- Biddle, S.J.; Bennie, J.A.; Bauman, A.E.; Chau, J.Y.; Dunstan, D.; Owen, N.; Stamatakis, E.; van Uffelen, J.G.Z. Too much sitting and all-cause mortality: Is there a causal link? BMC Public Health 2016, 16, 635. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Roberts, C.K.; Thyfault, J.P.; Ruegsegger, G.N.; Toedebusch, R.G. Role of Inactivity in Chronic Diseases: Evolutionary Insight and Pathophysiological Mechanisms. Physiol. Rev. 2017, 97, 1351–1402. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. J. 2018, 24, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Rohde, L.E.; Beck-da-Silva, L. Alcohol and the heart: The good, the bad and the worse in heart failure. Heart 2018, 104, 1641–1642. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef]
- Maiuolo, J.; Maretta, A.; Gliozzi, M.; Musolino, V.; Carresi, C.; Bosco, F.; Mollace, R.; Scarano, F.; Palma, E.; Scicchitano, M.; et al. Ethanol-induced cardiomyocyte toxicity implicit autophagy and NFkB transcription factor. Pharmacol. Res. 2018, 133, 141–150. [Google Scholar] [CrossRef]
- Coleman, L.G., Jr.; Crews, F.T. Innate Immune Signaling and Alcohol Use Disorders. Handb. Exp. Pharmacol. 2018, 248, 369–396. [Google Scholar]
- Campollo, O. Alcohol and the Liver: The Return of the Prodigal Son. Ann. Hepatol. 2019, 18, 6–10. [Google Scholar] [CrossRef]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol Res. 2017, 38, 163–171. [Google Scholar]
- Conte, R.; Ladd, F.V.L.; Ladd, A.A.B.L.; Moreira, A.L.; Sueur-Maluf, L.L.; de Barros Viana, M.; Céspedes, I.C. Behavioral and stereological analysis of the prefrontal cortex of rats submitted to chronic alcohol intake. Behav. Brain Res. 2019, 362, 21–27. [Google Scholar] [CrossRef]
- Melbourne, J.K.; Thompson, K.R.; Peng, H.; Nixon, K. It’s complicated: The relationship between alcohol and microglia in the search for novel pharmacotherapeutic targets for alcohol use disorders. Prog. Mol. Biol. Transl. Sci. 2019, 167, 179–221. [Google Scholar] [PubMed]
- de la Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2014, 127, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.J. Cognitive Decline and Recovery in Alcohol Abuse. J. Mol. Neurosci. 2016, 60, 383–389. [Google Scholar] [CrossRef]
- Topiwala, A.; Allan, C.L.; Valkanova, V.; Zsoldos, E.; Filippini, N.; Sexton, C.; Mahmood, A.; Fooks, P.; Singh-Manoux, A.; Mackay, C.E.; et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: Longitudinal cohort study. BMJ 2017, 357, j2353. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Kay, J.; Yalcin, E.B.; Kril, J.J.; Sheedy, D.; Sutherland, G.T. Imaging mass spectrometry of frontal white matter lipid changes in human alcoholics. Alcohol 2018, 67, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Gu, C. Function and Mechanism of Myelin Regulation in Alcohol Abuse and Alcoholism. Bioessays 2019, 41, e1800255. [Google Scholar] [CrossRef]
- Newville, J.; Valenzuela, C.F.; Li, L.; Jantzie, L.L.; Cunningham, L.A. Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of fetal alcohol spectrum disorder. Glia 2017, 65, 1317–1332. [Google Scholar] [CrossRef]
- Vangipuram, S.D.; Lyman, W.D. Ethanol affects differentiation-related pathways and suppresses Wnt signaling protein expression in human neural stem cells. Alcohol. Clin. Exp. Res. 2012, 36, 788–797. [Google Scholar] [CrossRef]
- Xu, H.; Liu, D.; Chen, J.; Li, H.; Xu, M. Effects of Chronic Voluntary Alcohol Drinking on Thiamine Concentrations, Endoplasmic Reticulum Stress, and Oxidative Stress in the Brain of Crossed High Alcohol Preferring Mice. Neurotox. Res. 2019, 36, 777–787. [Google Scholar] [CrossRef]
- Navarro, A.I.; Mandyam, C.D. Protracted abstinence from chronic ethanol exposure alters the structure of neurons and expression of oligodendrocytes and myelin in the medial prefrontal cortex. Neuroscience 2015, 293, 35–44. [Google Scholar] [CrossRef]
- Cahill, A.; Cunningham, C.C. Effects of chronic ethanol feeding on the protein composition of mitochondrial ribosomes. Electrophoresis 2000, 21, 3420–3426. [Google Scholar] [CrossRef]
- George, A.K.; Behera, J.; Kimberly, K.E.; Zhai, Y.; Neetu, T. Hydrogen sulfide, endoplasmic reticulum stress and alcohol mediated neurotoxicity. Brain Res. Bull. 2017, 130, 251–256. [Google Scholar]
- Placido, A.I.; Pereira, C.M.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. Modulation of endoplasmic reticulum stress: An opportunity to prevent neurodegeneration? CNS Neurol. Disord. Drug Targets 2015, 14, 518–533. [Google Scholar] [CrossRef]
- Alahabadi, A.; Malvandi, H. Contamination and ecological risk assessment of heavy metals and metalloids in surface sediments of the Tajan River, Iran. Mar. Pollut. Bull. 2018, 133, 741–749. [Google Scholar] [CrossRef]
- Kyakuwaire, M.; Olupot, G.; Amoding, A.; Nkedi-Kizza, P.; Basamba, T.A. How Safe is Chicken Litter for Land Application as an Organic Fertilizer? A Review. Int. J. Environ. Res. Public Health 2019, 16, 3521. [Google Scholar] [CrossRef]
- Ayangbenro, A.S.; Babalola, O. A New Strategy for Heavy Metal Polluted Environments. Int. J. Environ. Res. Public Health 2017, 14, 94. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Khan, M.A.; Qamar, Z.; Waqas, M. The uptake and bioaccumulation of heavy metals by food plants, their effects on plants nutrients, and associated health risk: A review. Environ. Sci. Pollut. Res. Int. 2015, 22, 13772–13799. [Google Scholar] [CrossRef]
- Green, A.J.; Planchart, A. The neurological toxicity of heavy metals: A fish perspective. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2018, 208, 12–19. [Google Scholar] [CrossRef]
- Soares, F.A.; Fagundez, D.A.; Avila, D.S. Neurodegeneration Induced by Metals in Caenorhabditis elegans. Adv. Neurobiol. 2017, 18, 355–383. [Google Scholar]
- Karri, V.; Ramos, D.; Martinez, J.B.; Odena, A.; Oliveira, E.; Coort, S.L.; Evelo, C.T.; Mariman, E.C.M.; Marta Schuhmacher, M.; Kumar, V. Differential protein expression of hippocampal cells associated with heavy metals (Pb, As, and MeHg) neurotoxicity: Deepening into the molecular mechanism of neurodegenerative diseases. J. Proteomics 2018, 187, 106–125. [Google Scholar] [CrossRef]
- Flores, K.P.; Blohowiak, S.E.; Winzerling, J.J.; Georgieff, M.K.; Kling, P.J. The impact of erythropoietin and iron status on brain myelination in the newborn rat. J. Neurosci. Res. 2018, 96, 1586–1599. [Google Scholar] [CrossRef] [PubMed]
- Rosato-Siri, M.V.; Marziali, L.; Guitart, M.E.; Badaracco, M.E.; Puntel, M.; Pitossi, F.; Correale, J.; Pasquini, J.M. Iron Availability Compromises Not Only Oligodendrocytes but Also Astrocytes and Microglial Cells. Mol. Neurobiol. 2018, 55, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Macrì, R.; Bava, I.; Gliozzi, M.; Musolino, V.; Nucera, S.; Carresi, C.; Scicchitano, M.; Bosco, F.; Scarano, F.; et al. Myelin Disturbances Produced by Sub-Toxic Concentration of Heavy Metals: The Role of Oligodendrocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 4554. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Mitra, M.; Agarwal, P.; Mahapatra, K.; De, S.; Sett, U.; Roy, S. Oxidative and genotoxic damages in plants in response to heavy metal stress and maintenance of genome stability. Plant. Signal. Behav. 2018, 13, e1460048. [Google Scholar] [CrossRef]
- Hossain, S.; Liu, H.N.; Nguyen, M.; Shore, G.; Almazan, G. Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology 2019, 30, 544–554. [Google Scholar] [CrossRef]
- Kahrizi, F.; Salimi, A.; Noorbakhsh, F.; Faizi, M.; Mehri, F.; Naserzadeh, P.; Naderi, N.; Pourahmad, J. Repeated Administration of Mercury Intensifies Brain Damage in Multiple Sclerosis through Mitochondrial Dysfunction. Iran. J. Pharm. Res. 2016, 15, 834–841. [Google Scholar]
- Thomason, M.E.; Hect, J.L.; Rauh, V.A.; Trentacosta, C.; Wheelock, A.T.; Eggebrecht, A.T.; Espinoza-Heredia, C.; Burt, A. Prenatal lead exposure impacts cross-hemispheric and long-range connectivity in the human fetal brain. NeuroImage 2019, 191, 186–192. [Google Scholar] [CrossRef]
- Kim, J.I.; Kim, J.W.; Lee, J.M.; Yun, H.J.; Sohn, C.H.; Shin, M.-S.; Kim, B.; Chae, J.; Roh, J.; Kim, B.-N. Interaction between DRD2 and lead exposure on the cortical thickness of the frontal lobe in youth with attention-deficit/hyperactivity disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 169–176. [Google Scholar] [CrossRef]
- Ohno, N.; Ikenaka, K. Axonal and neuronal degeneration in myelin diseases. Neurosci. Res. 2019, 139, 48–57. [Google Scholar] [CrossRef]
- Barateiro, A.; Brites, D.; Fernandes, A. Oligodendrocyte Development and Myelination in Neurodevelopment: Molecular Mechanisms in Health and Disease. Curr. Pharm. Des. 2016, 22, 656–679. [Google Scholar] [CrossRef]
- Seixas, A.I.; Azevedo, M.M.; Paes de Faria, J.; Fernandes, D.; Mendes Pinto, I.; Relvas, J.B. Evolvability of the actin cytoskeleton in oligodendrocytes during central nervous system development and aging. Cell. Mol. Life Sci. 2019, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; French-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef]
- Sun, L.O.; Mulinyawe, S.B.; Collins, H.Y.; Ibrahim, A.; Li, Q.; Simon, D.J.; Tessier-Lavigne, M.; Barres, B.A. Spatiotemporal Control of CNS Myelination by Oligodendrocyte Programmed Cell Death through the TFEB-PUMA Axis. Cell 2018, 175, 1811–1826.e21. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed]
- Ntranos, A.; Casaccia, P. The Microbiome-Gut-Behavior Axis: Crosstalk Between the Gut Microbiome and Oligodendrocytes Modulates Behavioral Responses. Neurotherapeutics 2018, 15, 31–35. [Google Scholar] [CrossRef]
- Lu, J.; Lu, L.; Yu, Y.; Cluette-Brown, J.; Martin, C.R.; Claud, E.C. Effects of Intestinal Microbiota on Brain Development in Humanized Gnotobiotic Mice. Sci. Rep. 2018, 8, 5443. [Google Scholar] [CrossRef]
- Goldman, S.A.; Kuypers, N.J. How to make an oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.M.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef]
- Langley, M.R.; Yoon, H.; Kim, H.N.; Choi, C.I.; Simon, W.; Kleppe, L.; Kleppe, L.; Lanza, I.R.; LeBrasseur, N.K.; Matveyenko, A.; et al. High fat diet consumption results in mitochondrial dysfunction, oxidative stress, and oligodendrocyte loss in the central nervous system. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165630. [Google Scholar] [CrossRef]
- Langley, M.R.; Triplet, E.M.; Scarisbrick, I.A. Dietary influence on central nervous system myelin production, injury, and regeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165779. [Google Scholar] [CrossRef]
- Bennet, L.; Dhillon, S.; Lear, C.A.; van den Heuij, L.; King, V.; Wassink, G.; Davidson, J.O.; Gunn, A.J. Chronic inflammation and impaired development of the preterm brain. J. Reprod. Immunol. 2018, 125, 45–55. [Google Scholar] [CrossRef]
- Caritis, S.N.; Panigrahy, A. Opioids affect the fetal brain: Reframing the detoxification debate. Am. J. Obstet. Gynecol. 2019, 221, 602–608. [Google Scholar] [CrossRef]
- Dillenburg, A.; Ireland, G.; Holloway, R.K.; Davies, C.L.; Evans, F.L.; Swire, M.; Bechler, M.E.; Soong, D.; Yuen, T.J.; Su, G.H.; et al. Activin receptors regulate the oligodendrocyte lineage in health and disease. Acta Neuropathol. 2018, 135, 887–906. [Google Scholar] [CrossRef]
- Ammitzbøll, C.; von Essen, M.R.; Börnsen, L.; Petersen, E.R.; McWilliam, O.; Ratzer, R.; Romme Christensen, J.; Oturai, A.B.; Søndergaard, H.B.; Sellebjerg, F. GPR15+ T cells are Th17 like, increased in smokers and associated with multiple sclerosis. J. Autoimmun. 2019, 97, 114–121. [Google Scholar] [CrossRef]
- Tong, M.; Andreani, T.; Krotow, A.; Gundogan, F.; de la Monte, S.M. Potential Contributions of the Tobacco Nicotine-Derived Nitrosamine Ketone to White Matter Molecular Pathology in Fetal Alcohol Spectrum Disorder. Int. J. Neurol. Brain Disord. 2016, 3. [Google Scholar] [CrossRef]
- Yu, R.; Deochand, C.; Krotow, A.; Leão, R.; Tong, M.; Agarwal, A.R.; Cadenas, E.; de la Monte, S.M. Tobacco Smoke-Induced Brain White Matter Myelin Dysfunction: Potential Co-Factor Role of Smoking in Neurodegeneration. J. Alzheimers Dis. 2016, 50, 133–148. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Scicchitano, M.; Scarano, F.; Bosco, F.; Oppedisano, F.; Macrì, R.; et al. Environmental and Nutritional “Stressors” and Oligodendrocyte Dysfunction: Role of Mitochondrial and Endoplasmatic Reticulum Impairment. Biomedicines 2020, 8, 553. https://doi.org/10.3390/biomedicines8120553
Maiuolo J, Gliozzi M, Musolino V, Carresi C, Nucera S, Scicchitano M, Scarano F, Bosco F, Oppedisano F, Macrì R, et al. Environmental and Nutritional “Stressors” and Oligodendrocyte Dysfunction: Role of Mitochondrial and Endoplasmatic Reticulum Impairment. Biomedicines. 2020; 8(12):553. https://doi.org/10.3390/biomedicines8120553
Chicago/Turabian StyleMaiuolo, Jessica, Micaela Gliozzi, Vincenzo Musolino, Cristina Carresi, Saverio Nucera, Miriam Scicchitano, Federica Scarano, Francesca Bosco, Francesca Oppedisano, Roberta Macrì, and et al. 2020. "Environmental and Nutritional “Stressors” and Oligodendrocyte Dysfunction: Role of Mitochondrial and Endoplasmatic Reticulum Impairment" Biomedicines 8, no. 12: 553. https://doi.org/10.3390/biomedicines8120553
APA StyleMaiuolo, J., Gliozzi, M., Musolino, V., Carresi, C., Nucera, S., Scicchitano, M., Scarano, F., Bosco, F., Oppedisano, F., Macrì, R., & Mollace, V. (2020). Environmental and Nutritional “Stressors” and Oligodendrocyte Dysfunction: Role of Mitochondrial and Endoplasmatic Reticulum Impairment. Biomedicines, 8(12), 553. https://doi.org/10.3390/biomedicines8120553