Chemopreventive Role of Apigenin against the Synergistic Carcinogenesis of Human Papillomavirus and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone

 ,
,  , and
, and 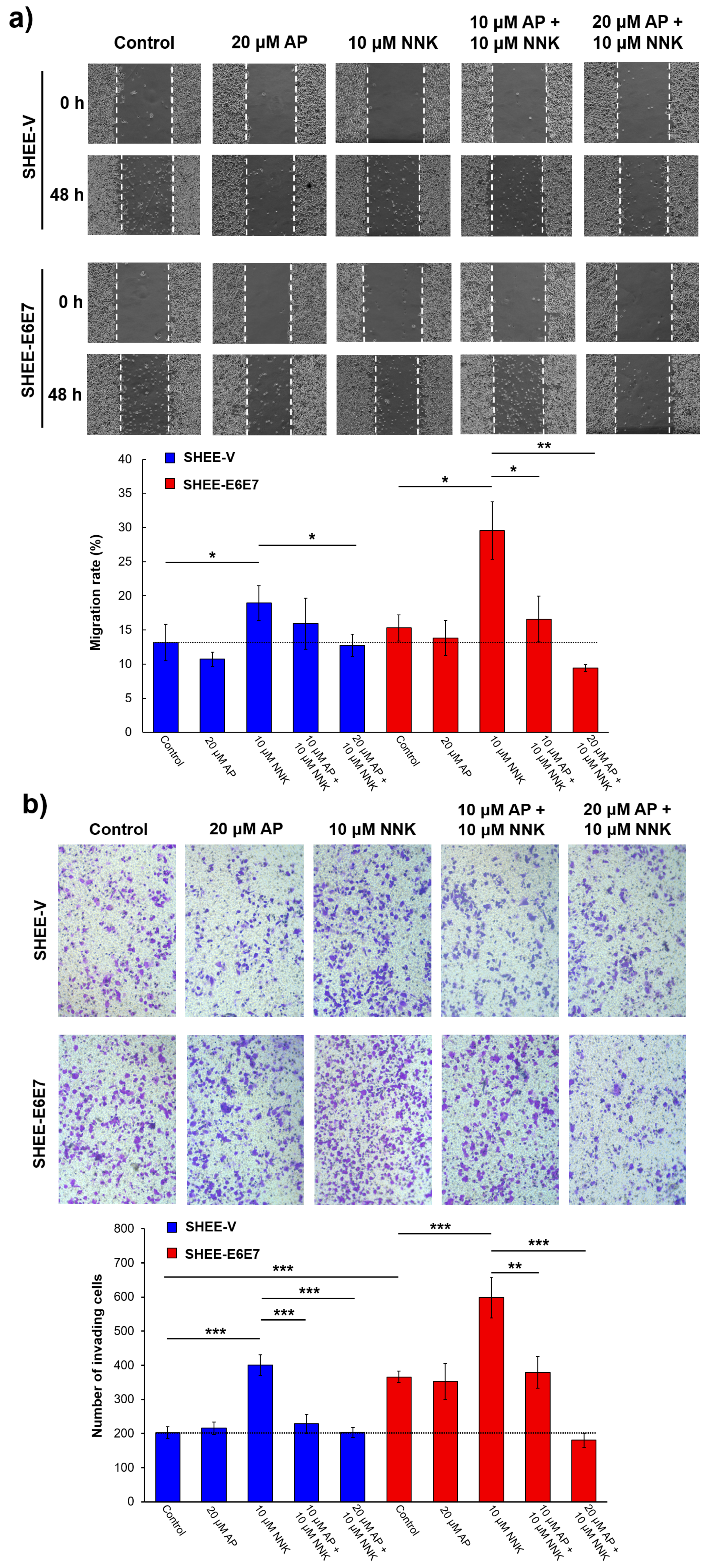{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Wound Healing Assay
2.4. Transwell Assay
2.5. Cell Colony Formation Assay
2.6. Cell Apoptosis and Caspase-3 Activity Assay
2.7. Hochest Staining
2.8. Cell Cycle Analysis
2.9. Western Blot
2.10. HPLC-ESI-MS/MS Quantitation of HPB
2.11. Statistical Analysis
3. Results
3.1. AP Inhibited Literal Migration Ability of SHEE Cells
3.2. AP Suppressed Cell Invasion Promoted by NNK and HPV
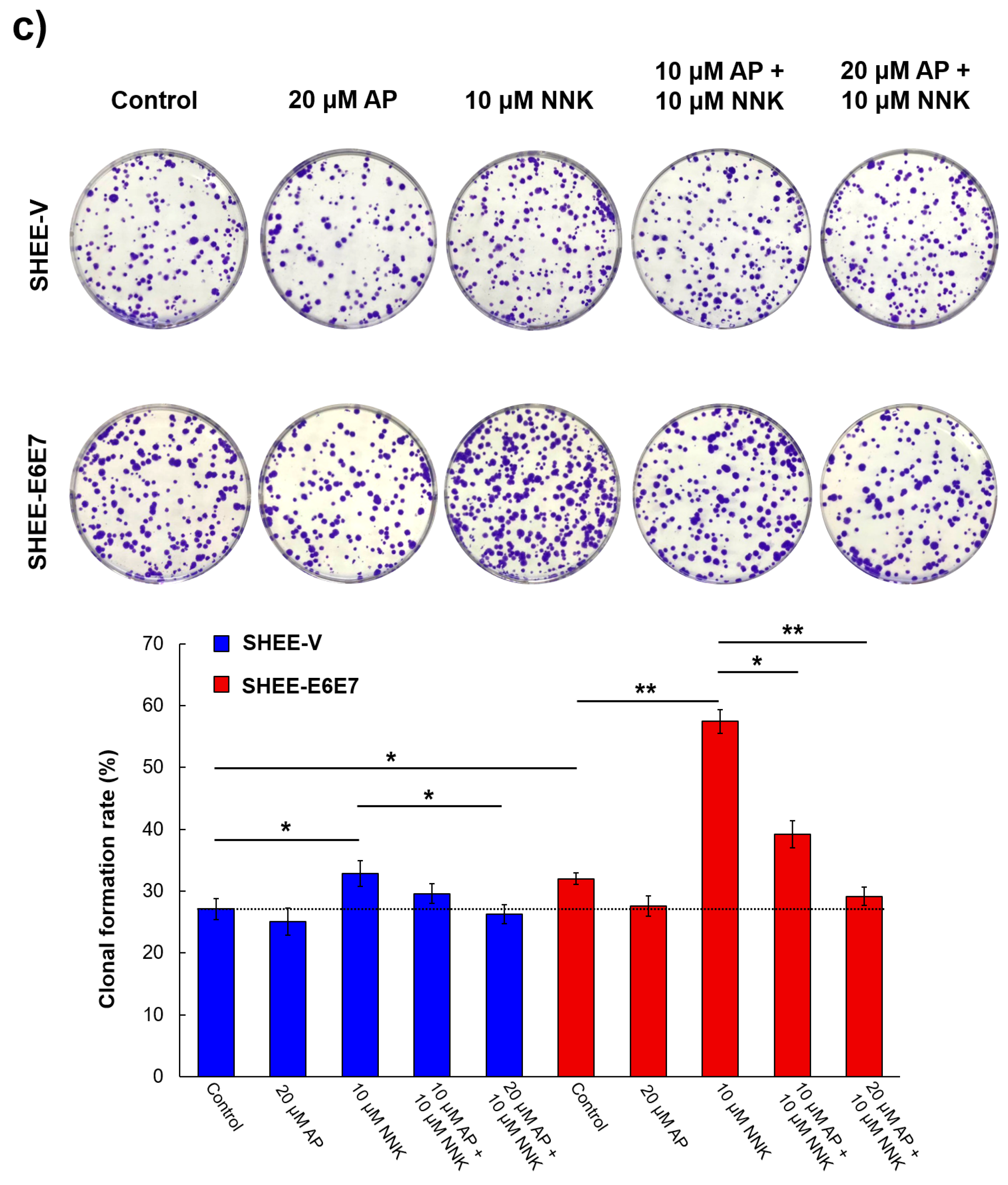
3.3. AP Impeded Cell Colony Formation Induced by NNK and HPV
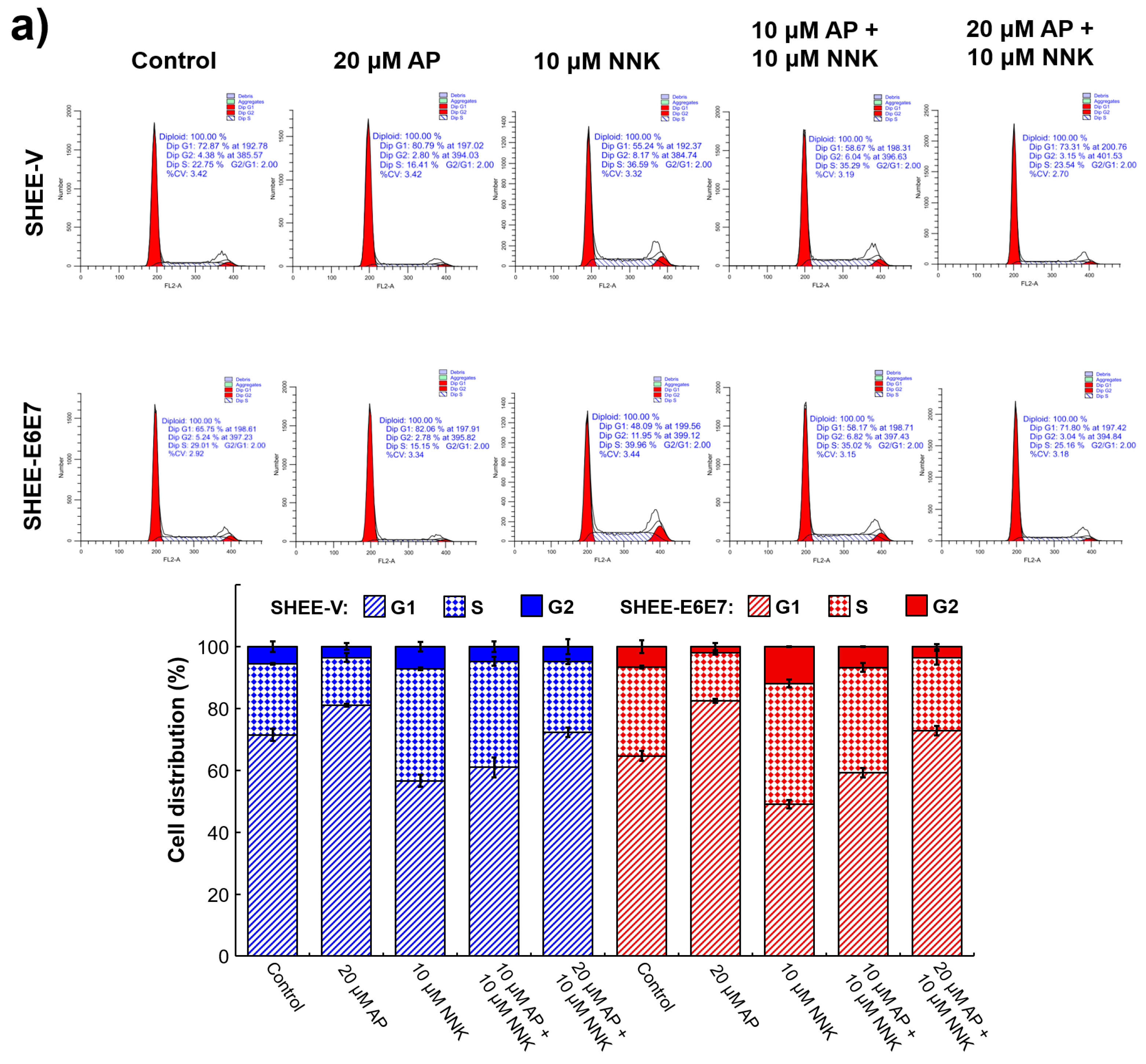
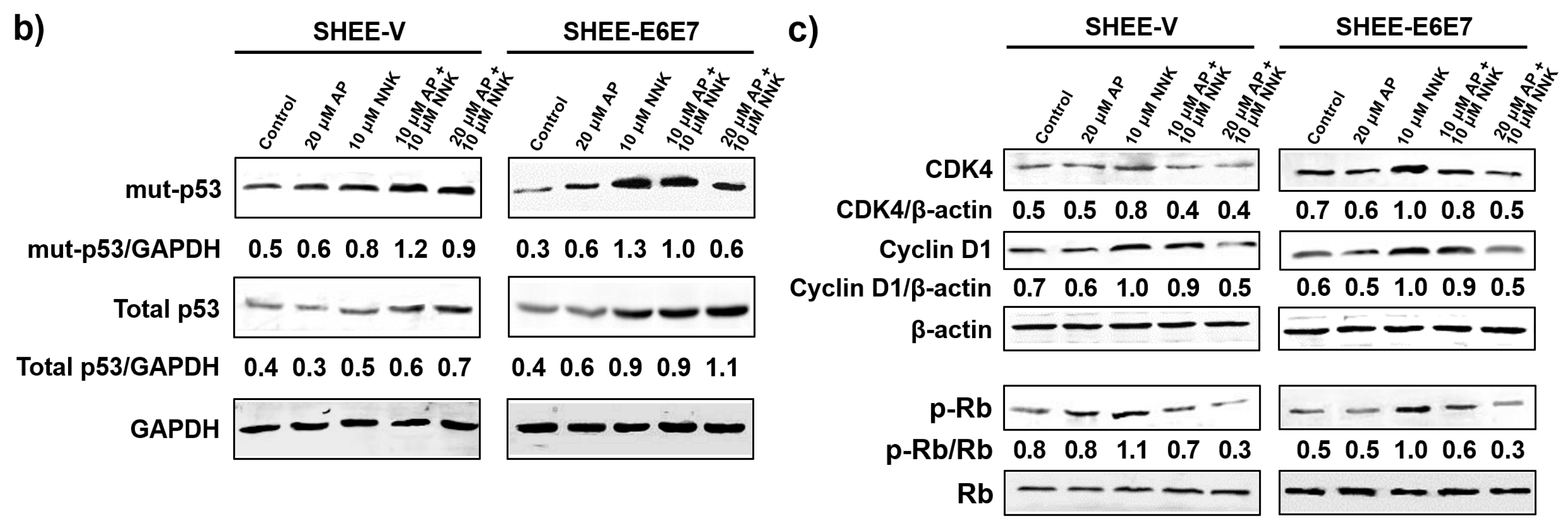
3.4. AP Induced G1 Arrest of SHEE Cells
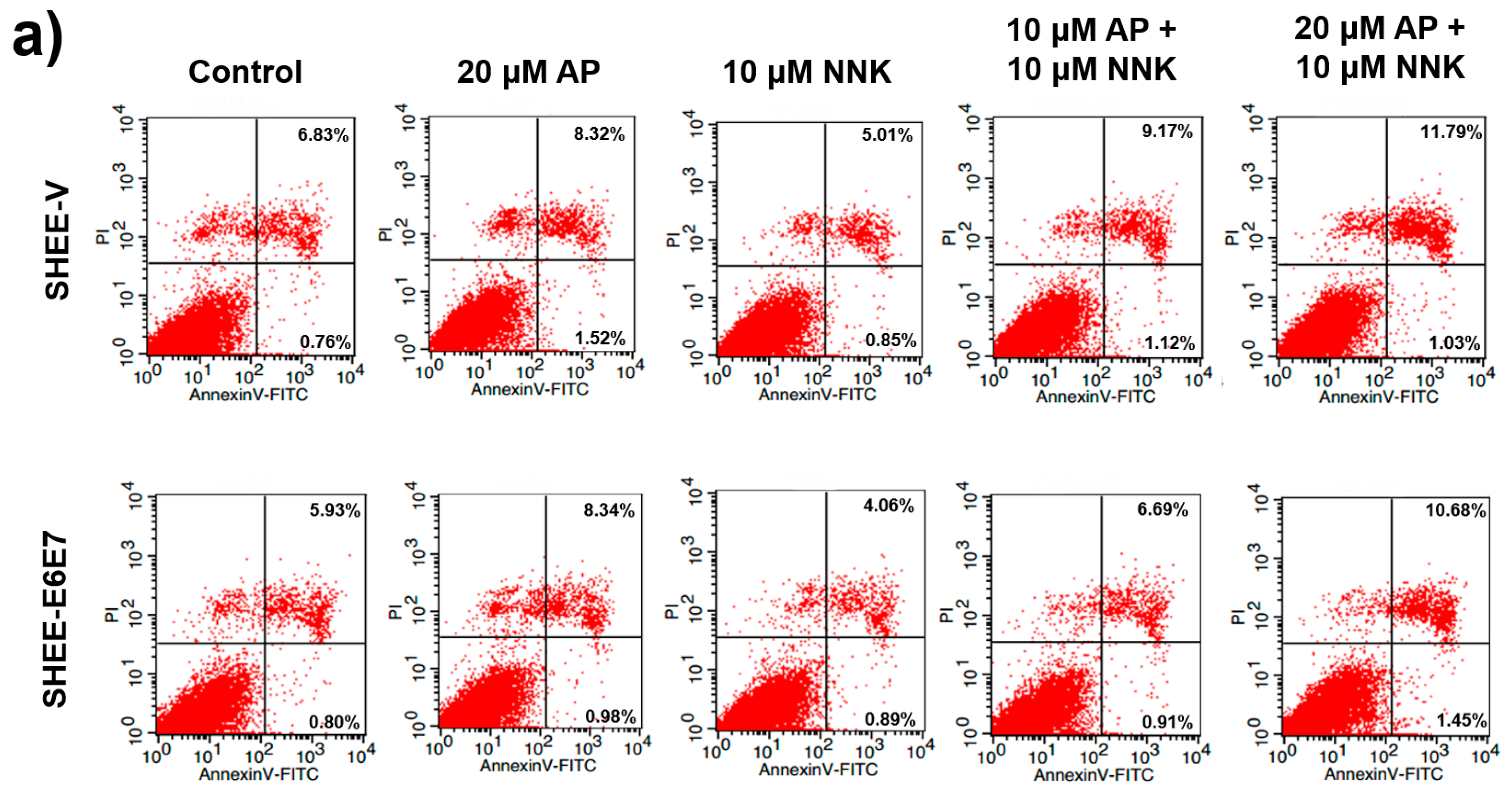
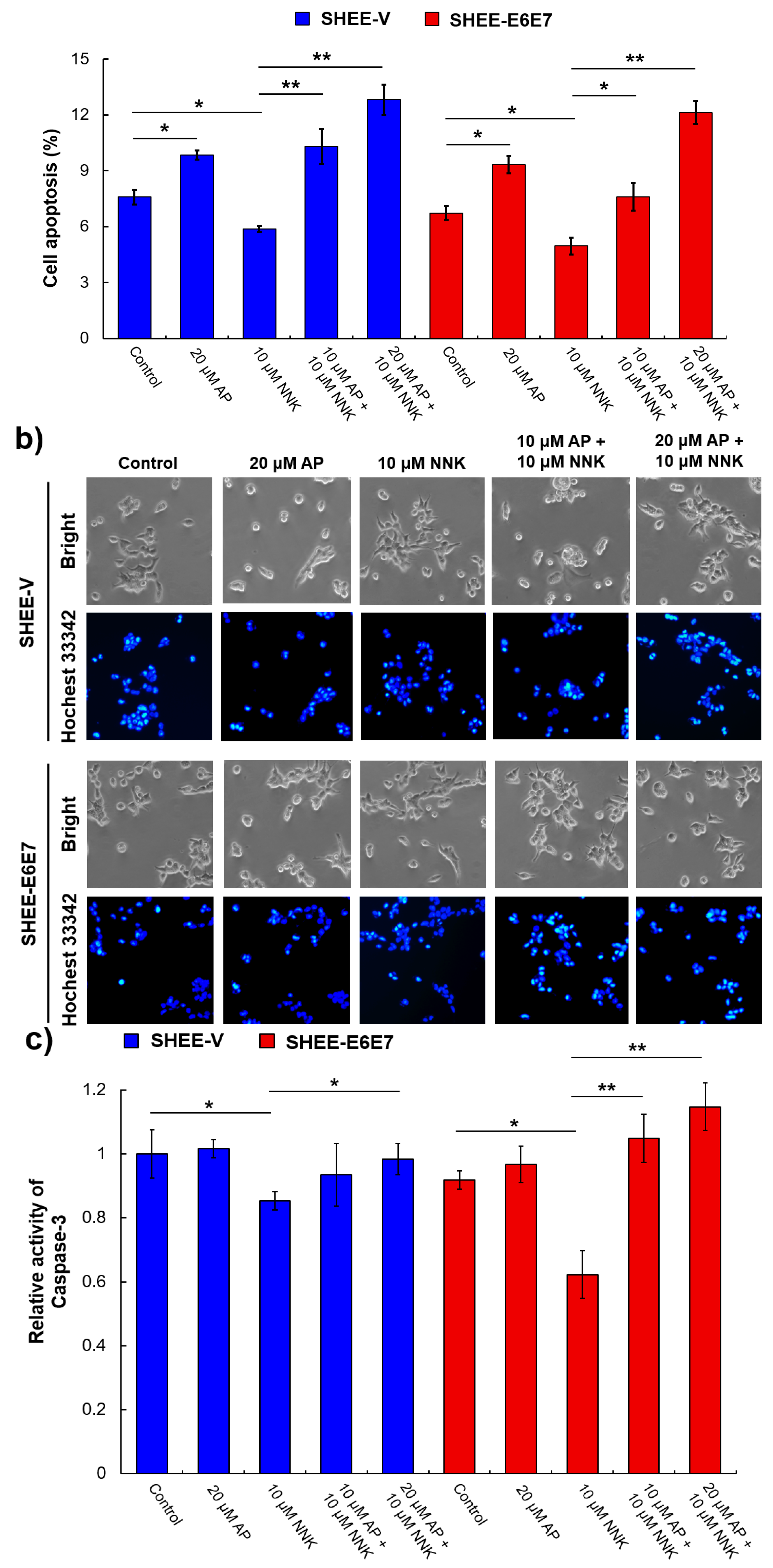
3.5. AP Promoted Apoptosis of SHEE Cells
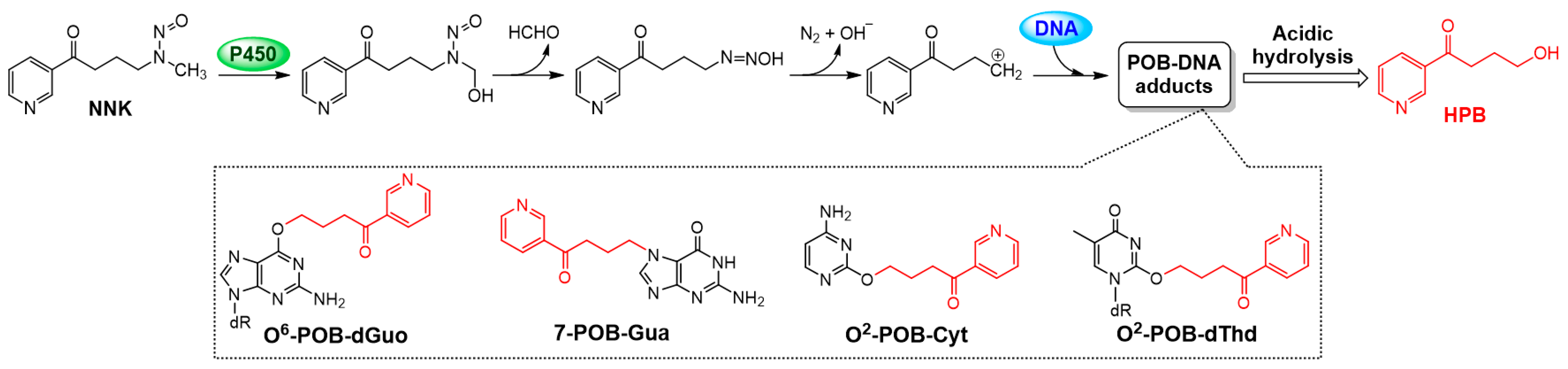
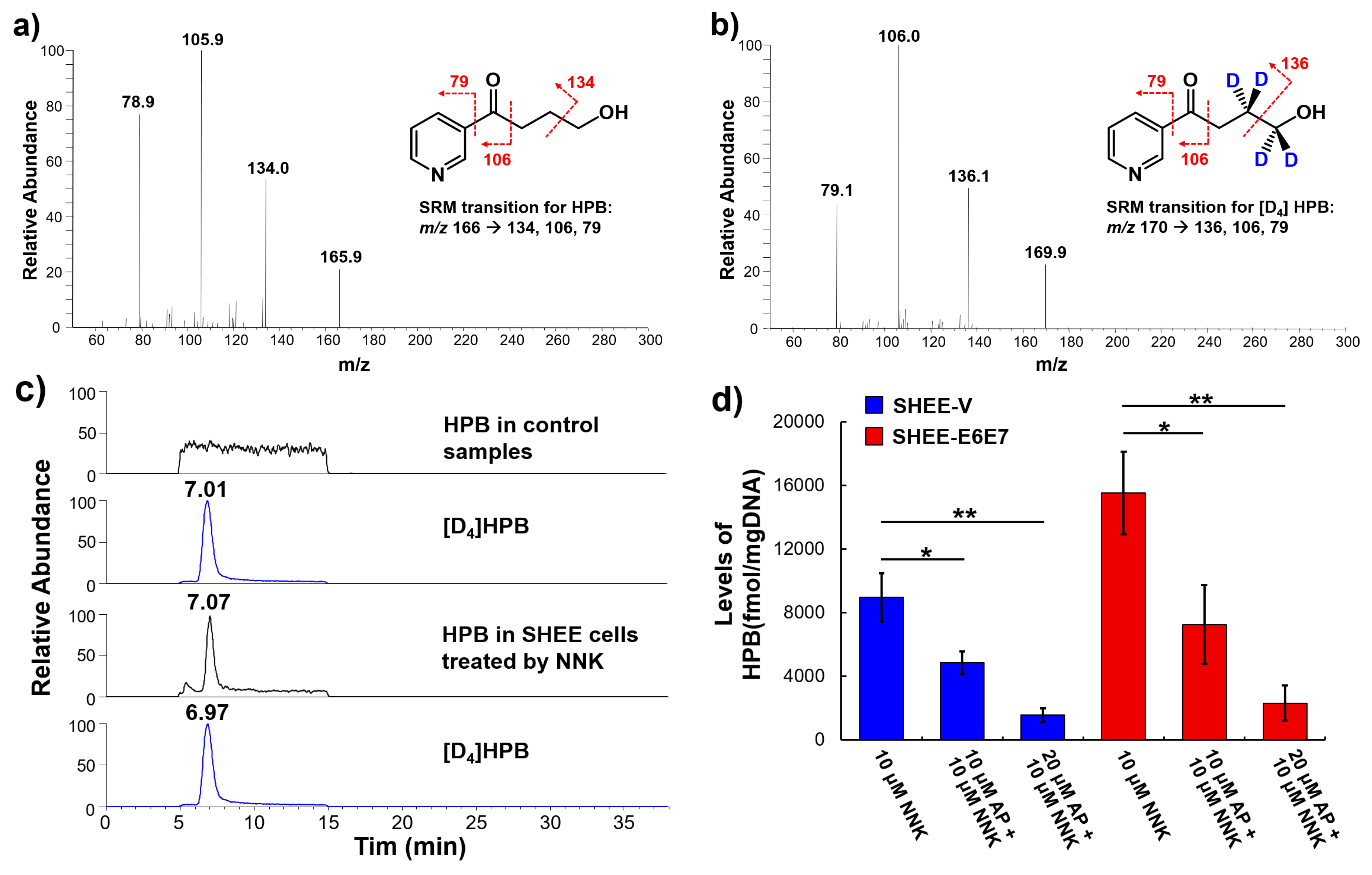
3.6. AP Protected SHEE Cells from DNA Alkylation Induced by NNK
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heppel, C.; Heling, A.K.; Richter, E. Ultrasensitive method for the determination of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts by gas chromatography-high resolution mass spectrometry in mucosal biopsies of the lower esophagus. Anal. Bioanal. Chem. 2009, 393, 1525–1530. [Google Scholar] [CrossRef]
- Smith, C.J.; Perfetti, T.A.; Garg, R.; Hansch, C. IARC carcinogens reported in cigarette mainstream smoke and their calculated log P values. Food Chem. Toxicol. 2003, 41, 807–817. [Google Scholar] [CrossRef]
- Hecht, S.S.; Villalta, P.W.; Sturla, S.J.; Cheng, G.; Yu, N.X.; Upadhyaya, P.; Wang, M.Y. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem. Res. Toxicol. 2004, 17, 588–597. [Google Scholar] [CrossRef]
- Wang, M.Y.; Cheng, G.; Sturla, S.J.; Shi, Y.L.; McIntee, E.J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol. 2003, 16, 616–626. [Google Scholar] [CrossRef]
- Stepanov, I.; Muzic, J.; Le, C.T.; Sebero, E.; Villalta, P.; Ma, B.; Jensen, J.; Hatsukami, D.; Hecht, S.S. Analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing DNA adducts in human exfoliated oral mucosa cells by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2013, 26, 37–45. [Google Scholar] [CrossRef]
- Zhang, N.N.; Sun, X.J.; Sun, M.J.; Zhu, S.T.; Wang, L.; Ma, D.; Wang, Y.J.; Zhang, S.T.; Li, P. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes esophageal squamous cell carcinoma growth via beta-adrenoceptors in vitro and in vivo. PLoS ONE 2015, 10, e0118845. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.L.; Huang, X.B.; Chen, K.L.; Shen, J.; Wang, Z.Y. Involvement of p53 mutation and mismatch repair proteins dysregulation in NNK-induced malignant transformation of human bronchial epithelial cells. BioMed Res. Int. 2014, 2014, 920275. [Google Scholar] [CrossRef]
- Branica, B.V.; Smojver-Jezek, S.; Juros, Z.; Grgic, S.; Srpak, N.; Mitrecic, D.; Gajovic, S. Detection of human papillomaviruses type 16, 18 and 33 in bronchial aspirates of lung carcinoma patients by polymerase chain reaction a study of 84 cases in Croatia. Coll. Antropol. 2010, 34, 159–162. [Google Scholar]
- Carpagnano, G.E.; Koutelou, A.; Natalicchio, M.I.; Martinelli, D.; Ruggieri, C.; Di Taranto, A.; Antonetti, R.; Carpagnano, F.; Foschino-Barbaro, M.P. HPV in exhaled breath condensate of lung cancer patients. Br. J. Cancer 2011, 105, 1183–1190. [Google Scholar] [CrossRef]
- DeFreitas, A.C.; Gurgel, A.P.; de Lima, E.G.; Sao Marcos, B.D.; do Amaral, C.M.M. Human papillomavirus and lung carcinogenesis: An overview. J. Cancer Res. Clin. Oncol. 2016, 142, 2415–2427. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef]
- Laura, Q.M.; Chow, M.D. Head and neck cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Tanaka, T.I.; Alawi, F. Human papillomavirus and oropharyngeal cancer. Dent. Clin. N. Am. 2018, 62, 111–120. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell Biol. 1993, 13, 775–784. [Google Scholar] [CrossRef]
- Lee, J.O.; Russo, A.A.; Letich, N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998, 391, 859–865. [Google Scholar] [CrossRef]
- McIntyre-Seltman, K.; Castle, P.E.; Guido, R.; Schiffman, M.; Wheeler, C.M. Smoking is a risk factor for cervical intraepithelial neoplasia grade 3 among oncogenic human papillomavirus DNA-positive women with equivocal or mildly abnormal cytology. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1165–1170. [Google Scholar] [CrossRef]
- Gunnell, A.S.; Tran, T.N.; Torrang, A.; Dickman, P.W.; Sparen, P.; Palmgren, J.; Ylitalo, N. Synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer in situ development. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 2141–2147. [Google Scholar] [CrossRef]
- Feng, R.M.; Hu, S.Y.; Zhao, F.H.; Zhang, R.; Zhang, X.; Wallach, A.I.; Qiao, Y.L. Role of active and passive smoking in high-risk human papillomavirus infection and cervical intraepithelial neoplasia grade 2 or worse. J. Gynecol. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Madathil, S.; Rousseau, M.C.; Joseph, L.; Coutlée, F.; Schlecht, N.F.; Franco, E.; Nicolau, B. Latency of tobacco smoking for head and neck cancer among HPV-positive and HPV-negative individuals. Int. J. Cancer 2020, 147, 56–64. [Google Scholar] [CrossRef]
- Farsi, N.J.; Rousseau, M.; Schlecht, N.; Castonguay, G.; Allison, P.; Nguyen-Tan, P.F.; Soulieres, D.; Coutlee, F.; Hier, M.; Madathil, S.; et al. Aetiological heterogeneity of head and neck squamous cell carcinomas: The role of human papillomavirus infections, smoking and alcohol. Carcinogenesis 2017, 38, 1188–1195. [Google Scholar] [CrossRef]
- Zhuang, C.C.; Li, J.T.; Sun, G.H.; Cui, X.; Zhang, N.; Zhao, L.J.; Chan, P.K.S.; Zhong, R.G. Synergistic effect between human papillomavirus 18 and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone on malignant transformation of immortalized SHEE cells. Chem. Res. Toxicol. 2020, 33, 470–481. [Google Scholar] [CrossRef]
- Fang, J.; Zhou, Q.; Liu, L.Z.; Xia, C.; Hu, X.W.; Shi, X.L.; Jiang, B.H. Apigenin inhibits tumor angiogenesis through decreasing HIF-1 alpha and VEGF expression. Carcinogenesis 2007, 28, 858–864. [Google Scholar] [CrossRef]
- Salmani, J.M.M.; Zhang, X.P.; Jacob, J.A.; Chen, B.A. Apigenin’s anticancer properties and molecular mechanisms of action: Recent advances and future prospectives. Chin. J. Nat. Med. 2017, 15, 321–329. [Google Scholar] [CrossRef]
- He, J.; Ning, C.W.; Wang, Y.; Ma, T.F.; Huang, H.; Ge, Y.B.; Liu, J.B.; Jiang, Y.Q. Natural plant flavonoid apigenin directly disrupts Hsp90/Cdc37 complex and inhibits pancreatic cancer cell growth and migration. J. Funct. Foods 2015, 18, 10–21. [Google Scholar] [CrossRef]
- Yang, J.L.; Pi, C.C.; Wang, G.H. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Zhu, H.Y.; Zhang, X.H.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol. Cancer 2011, 10, 104. [Google Scholar] [CrossRef]
- Zhu, Y.; Mao, Y.Q.; Chen, H.; Lin, Y.W.; Hu, Z.H.; Wu, J.; Xu, X.; Xu, X.L.; Qin, J.; Xie, L.P. Apigenin promotes apoptosis, inhibits invasion and induces cell cycle arrest of T24 human bladder cancer cells. Cancer Cell Int. 2013, 13, 54. [Google Scholar] [CrossRef]
- Gupta, S.; Afaq, F.; Mukhtar, H. Selective growth-inhibitory, cell-cycle deregulatory and apoptotic response of apigenin in normal versus human prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 287, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Chen, M.; Takahashi, H.; King, J.; Reber, H.A.; Hines, O.J.; Pandol, S.; Eibl, G. Apigenin inhibits NNK-induced focal adhesion kinase activation in pancreatic cancer cells. Pancreas 2012, 41, 1306–1315. [Google Scholar] [CrossRef]
- Souza, R.P.; Bonfim-Mendonca, P.D.S.; Gimenes, F.; Ratti, B.A.; Kaplum, V.; Bruschi, M.L.; Nakamura, C.V.; Silva, S.O.; Maria-Engler, S.S.; Consolaro, M.E.L. Oxidative stress triggered by apigenin induces apoptosis in a comprehensive panel of human cervical cancer-derived cell lines. Oxid. Med. Cell Longev. 2017, 2017, 1512745. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Wang, M.Y.; Villalta, P.W.; Lindgren, B.R.; Upadhyaya, P.; Lao, Y.B.; Hecht, S.S. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 926–936. [Google Scholar] [CrossRef]
- Zhao, L.J.; Balbo, S.; Wang, M.Y.; Upadhyaya, P.; Khariwala, S.S.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)-or (S)-N′-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol. 2013, 26, 1526–1535. [Google Scholar] [CrossRef]
- Smith, E.M.; Rubenstein, L.M.; Haugen, T.H.; Hamsikova, E.; Turek, L.P. Tobacco and alcohol use increases the risk of both HPV-associated and HPV-independent head and neck cancers. Cancer Causes Control 2010, 21, 1369–1378. [Google Scholar] [CrossRef]
- Kero, K.; Rautava, J.; Syrjänen, K.; Willberg, J.; Grenman, S.; Syrjänen, S. Smoking increases oral HPV persistence among men: 7-year follow-up study. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Griego, A.M.; Chu, M.; Ozbun, M.A. Tobacco exposure results in increased E6 and E7 oncogene expression, DNA damage and mutation rates in cells maintaining episomal human papillomavirus 16 genomes. Carcinogenesis 2014, 35, 2373–2381. [Google Scholar] [CrossRef]
- Alam, S.; Bowser, B.S.; Conway, M.J.; Israr, M.; Ryndock, E.J.; Xi, L.F.; Meyers, C. Downregulation of Cdc2/CDK1 kinase activity induces the synthesis of noninfectious human papillomavirus type 31b virions in organotypic tissues exposed to benzo[a]pyrene. J. Virol. 2010, 84, 4630–4645. [Google Scholar] [CrossRef]
- Alam, S.; Conway, M.J.; Chen, H.S.; Meyers, C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J. Virol. 2008, 82, 1053–1058. [Google Scholar] [CrossRef]
- Pena, N.; Carrillo, D.; Munoz, J.P.; Chnaiderman, J.; Urzua, U.; Leon, O.; Tornesello, M.L.; Corvalan, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029. [Google Scholar] [CrossRef]
- Prokopczyk, B.; Sinha, I.; Trushin, N.; Freeman, W.M.; El-Bayoumy, K. Gene expression profiles in HPV-immortalized human cervical cells treated with the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem-Biol. Interact. 2009, 177, 173–180. [Google Scholar] [CrossRef]
- Merne, M.; Rautava, J.; Ruutu, M.; Syrjanen, S. Smokeless tobacco increases aneuploidy in oral HPV16 E6 E7-transformed keratinocytes in vitro. J. Oral Pathol. Med. 2014, 43, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Villepelet, A.; Hugonin, S.; Atallah, S.; Job, B.; Baujat, B.; St Guily, J.L.; Lacave, R. Effects of tobacco abuse on major chromosomal instability in human papilloma virus 16-positive oropharyngeal squamous cell carcinoma. Int. J. Oncol. 2019, 55, 527–535. [Google Scholar] [CrossRef]
- Shi, M.D.; Shiao, C.K.; Lee, Y.C.; Shih, Y.W. Apigenin, a dietary flavonoid, inhibits proliferation of human bladder cancer T-24 cells via blocking cell cycle progression and inducing apoptosis. Cancer Cell Int. 2015, 15, 33. [Google Scholar] [CrossRef]
- Granato, M.; Montani, M.S.G.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef]
- Yan, X.H.; Qi, M.; Li, P.P.; Zhan, Y.H.; Shao, H.J. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Moktar, A.; Ravoori, S.; Vadhanam, M.V.; Gairola, C.G.; Gupta, R.C. Cigarette smoke-induced DNA damage and repair detected by the comet assay in HPV-transformed cervical cells. Int. J. Oncol. 2009, 35, 1297–1304. [Google Scholar] [CrossRef][Green Version]
- Khan, T.H.; Jahangir, T.; Prasad, L.; Sultana, S. Inhibitory effect of apigenin on benzo(a)pyrene-mediated genotoxicity in Swiss albino mice. J. Pharm. Pharmacol. 2006, 58, 1655–1660. [Google Scholar] [CrossRef]
- Das, S.; Das, J.; Paul, A.; Samadder, A.; Khuda-Bukhsh, A.R. Apigenin, a bioactive flavonoid from Lycopodium clavatum, stimulates nucleotide excision repair genes to protect skin keratinocytes from ultraviolet B-induced reactive oxygen species and DNA damage. J. Acupunct. Meridian Stud. 2013, 6, 252–262. [Google Scholar] [CrossRef]
- Lautraite, S.; Musonda, A.C.; Doehmer, J.; Edwards, G.O.; Chipman, J.K. Flavonoids inhibit genetic toxicity produced by carcinogens in cells expressing CYP1A2 and CYP1A1. Mutagenesis 2002, 17, 45–53. [Google Scholar] [CrossRef]
- Boonruang, S.; Prakobsri, K.; Pouyfung, P.; Srisook, E.; Prasopthum, A.; Rongnoparut, P.; Sarapusit, S. Inhibition of human cytochromes P450 2A6 and 2A13 by flavonoids, acetylenic thiophenes and sesquiterpene lactones from Pluchea indica and Vernonia cinerea. J. Enzym. Inhib. Med. Chem. 2017, 32, 1136–1142. [Google Scholar] [CrossRef]
- Wang, F.; Liu, J.C.; Zhou, R.J.; Zhao, X.; Liu, M.; Ye, H.; Xie, M.L. Apigenin protects against alcohol-induced liver injury in mice by regulating hepatic CYP2E1-mediated oxidative stress and PPARα-mediated lipogenic gene expression. Chem. Biol. Interact. 2017, 275, 171–177. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, F.; Zhao, L.; Zhang, L.; Chen, Y.; Sun, G.; Li, J.; Zhang, N.; Xu, Y.; Chan, P.K.-S.; Zhong, R. Chemopreventive Role of Apigenin against the Synergistic Carcinogenesis of Human Papillomavirus and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. Biomedicines 2020, 8, 472. https://doi.org/10.3390/biomedicines8110472
Yin F, Zhao L, Zhang L, Chen Y, Sun G, Li J, Zhang N, Xu Y, Chan PK-S, Zhong R. Chemopreventive Role of Apigenin against the Synergistic Carcinogenesis of Human Papillomavirus and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. Biomedicines. 2020; 8(11):472. https://doi.org/10.3390/biomedicines8110472
Chicago/Turabian StyleYin, Fangzheng, Lijiao Zhao, Lili Zhang, Yuhe Chen, Guohui Sun, Jintao Li, Na Zhang, Yuancong Xu, Paul Kay-Sheung Chan, and Rugang Zhong. 2020. "Chemopreventive Role of Apigenin against the Synergistic Carcinogenesis of Human Papillomavirus and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone" Biomedicines 8, no. 11: 472. https://doi.org/10.3390/biomedicines8110472
APA StyleYin, F., Zhao, L., Zhang, L., Chen, Y., Sun, G., Li, J., Zhang, N., Xu, Y., Chan, P. K.-S., & Zhong, R. (2020). Chemopreventive Role of Apigenin against the Synergistic Carcinogenesis of Human Papillomavirus and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. Biomedicines, 8(11), 472. https://doi.org/10.3390/biomedicines8110472