Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Study
2.2. Study Participants
2.3. Study Protocol
2.4. Supplements and Monitoring
2.5. Outcome Measures
2.5.1. Glutathione Concentrations and Oxidative Stress
2.5.2. Mitochondrial Fuel Oxidation
2.5.3. Protein Isolation and Immunoblot Analyses
2.5.4. Tracer Studies (at 0 w and 12 w Only)
2.5.5. Physical Function
2.5.6. Cognitive Function
2.5.7. Insulin Resistance
2.5.8. Plasma Biomarkers of Inflammation, Endothelial Function, and DNA Damage
2.5.9. Body Composition and Anthropometry
2.6. Statistics
3. Results
3.1. Adverse Effects or Participant Withdrawals
3.2. HIV Parameters
3.3. Plasma Biochemistry
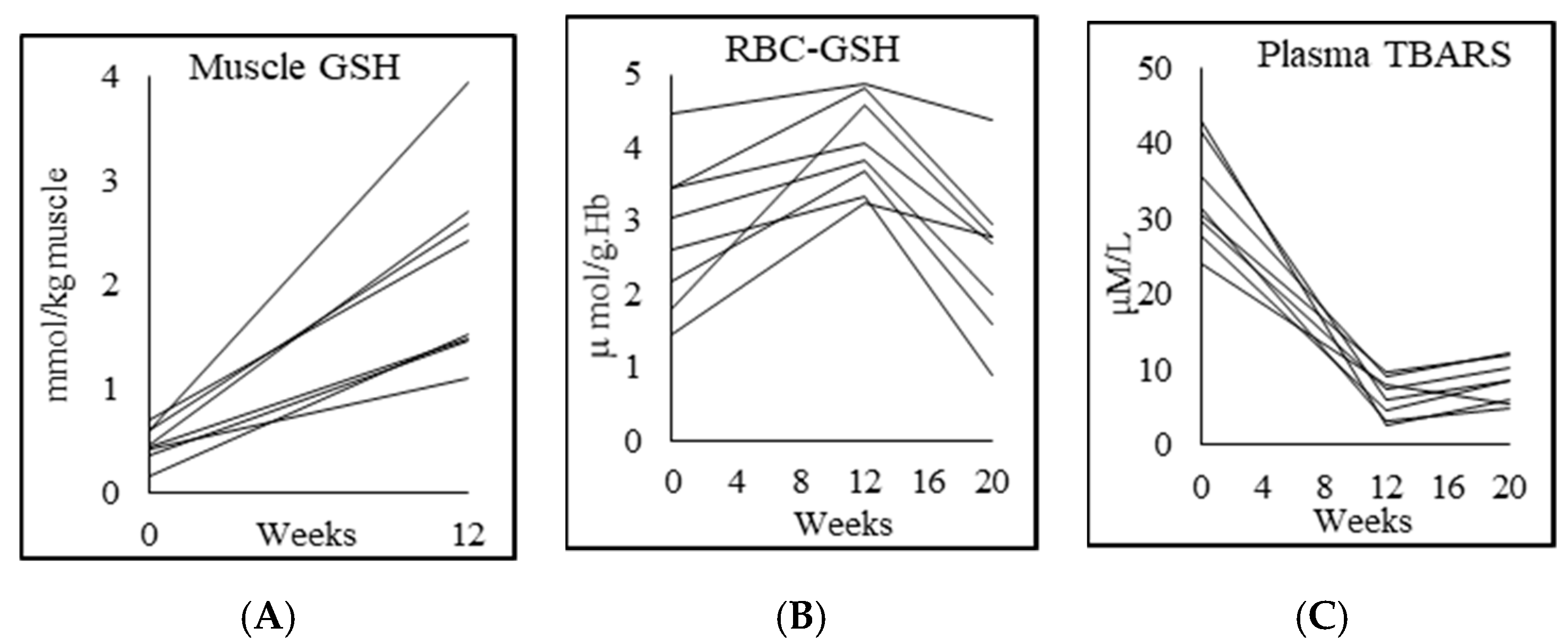
3.4. Glutathione and Oxidative Stress
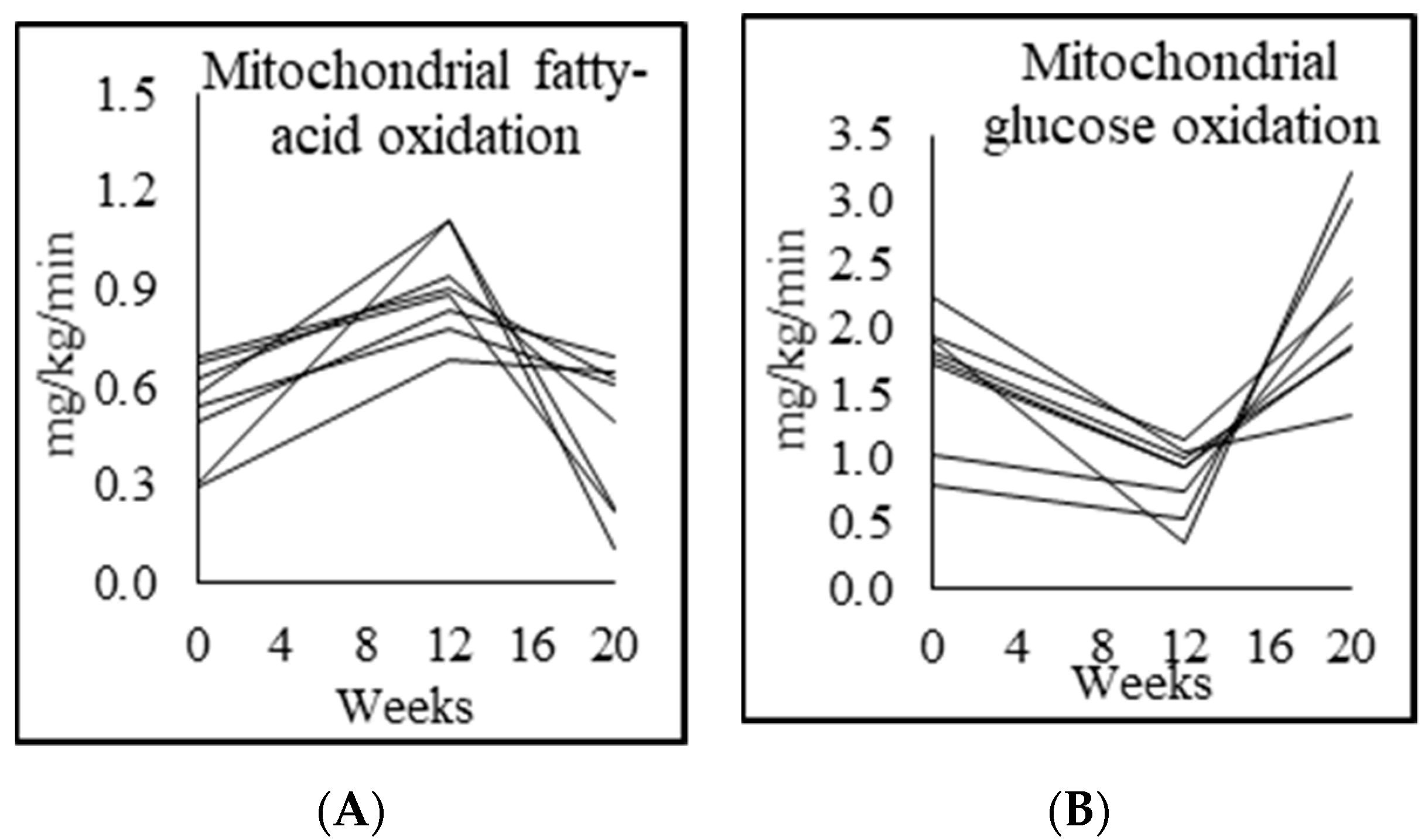
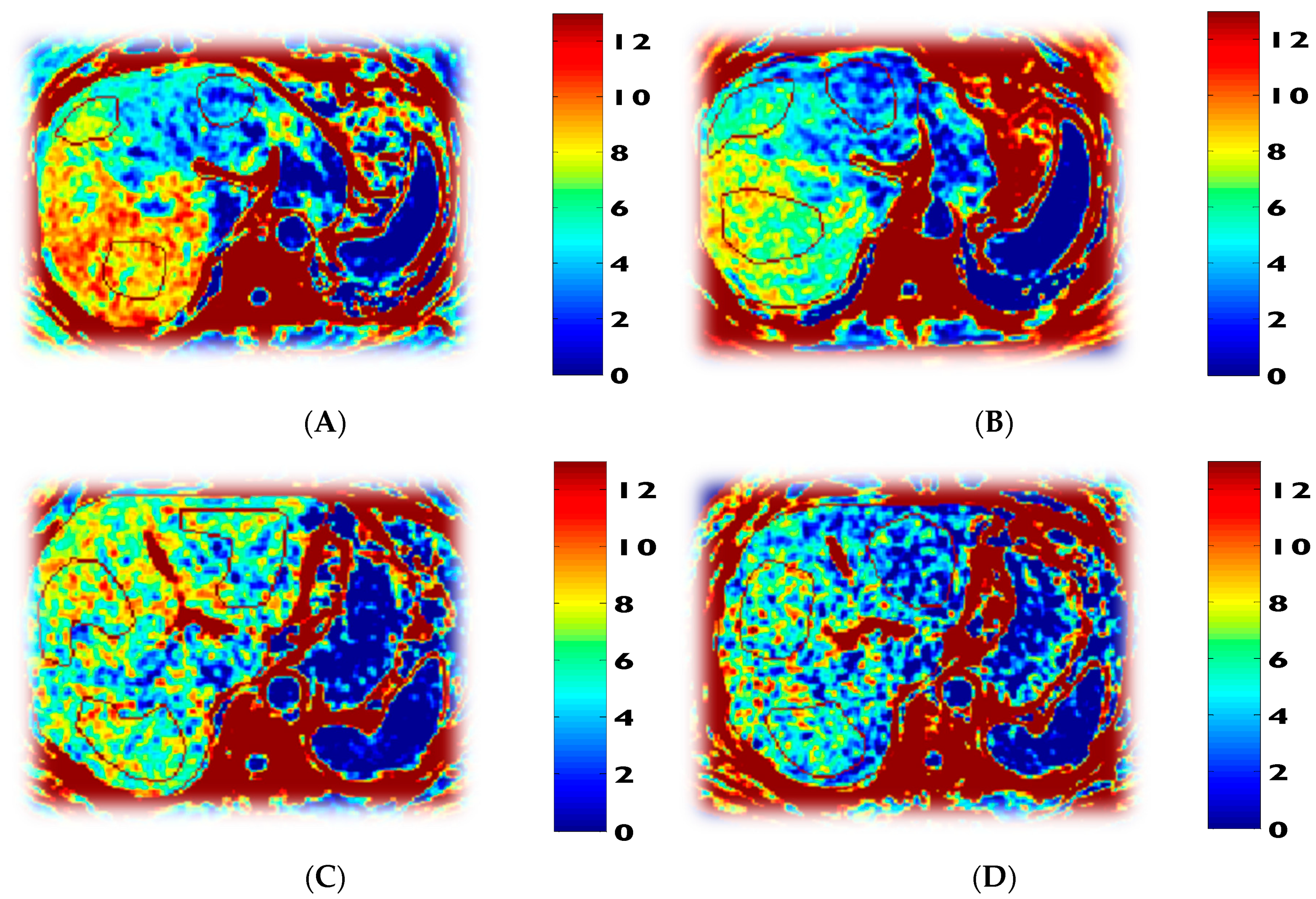
3.5. Mitochondrial Fuel Oxidation
3.6. Tracer Kinetic Data and Glucose Metabolism
3.7. Inflammation, Endothelial Function, and Genotoxicity
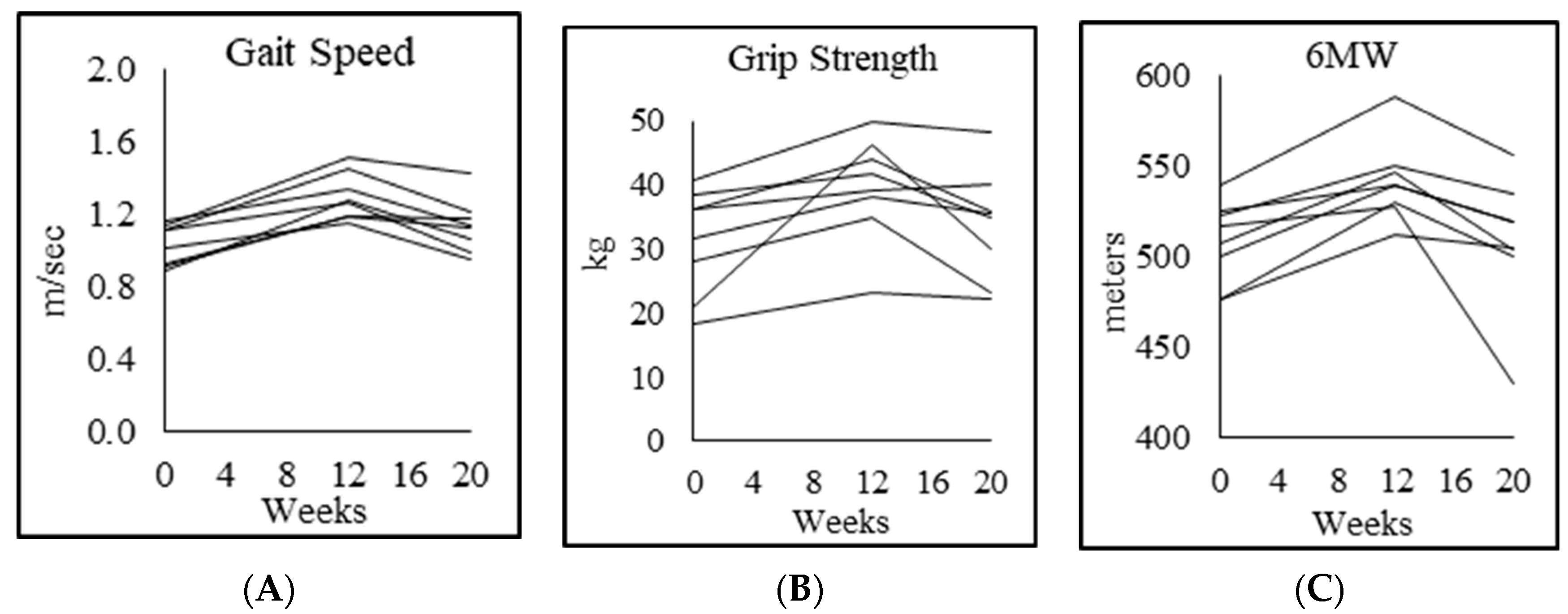
3.8. Physical Function
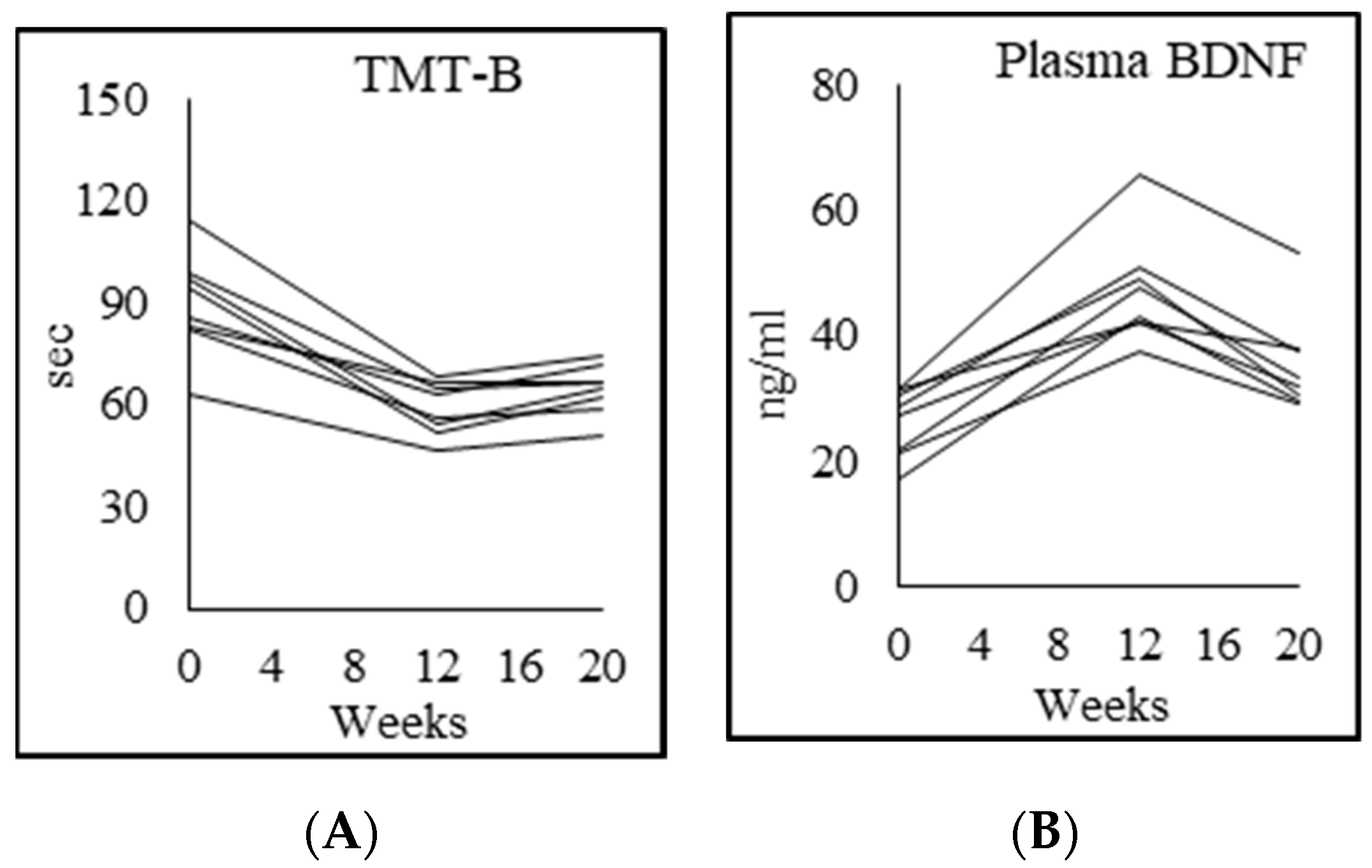
3.9. Cognition
3.10. Body Composition
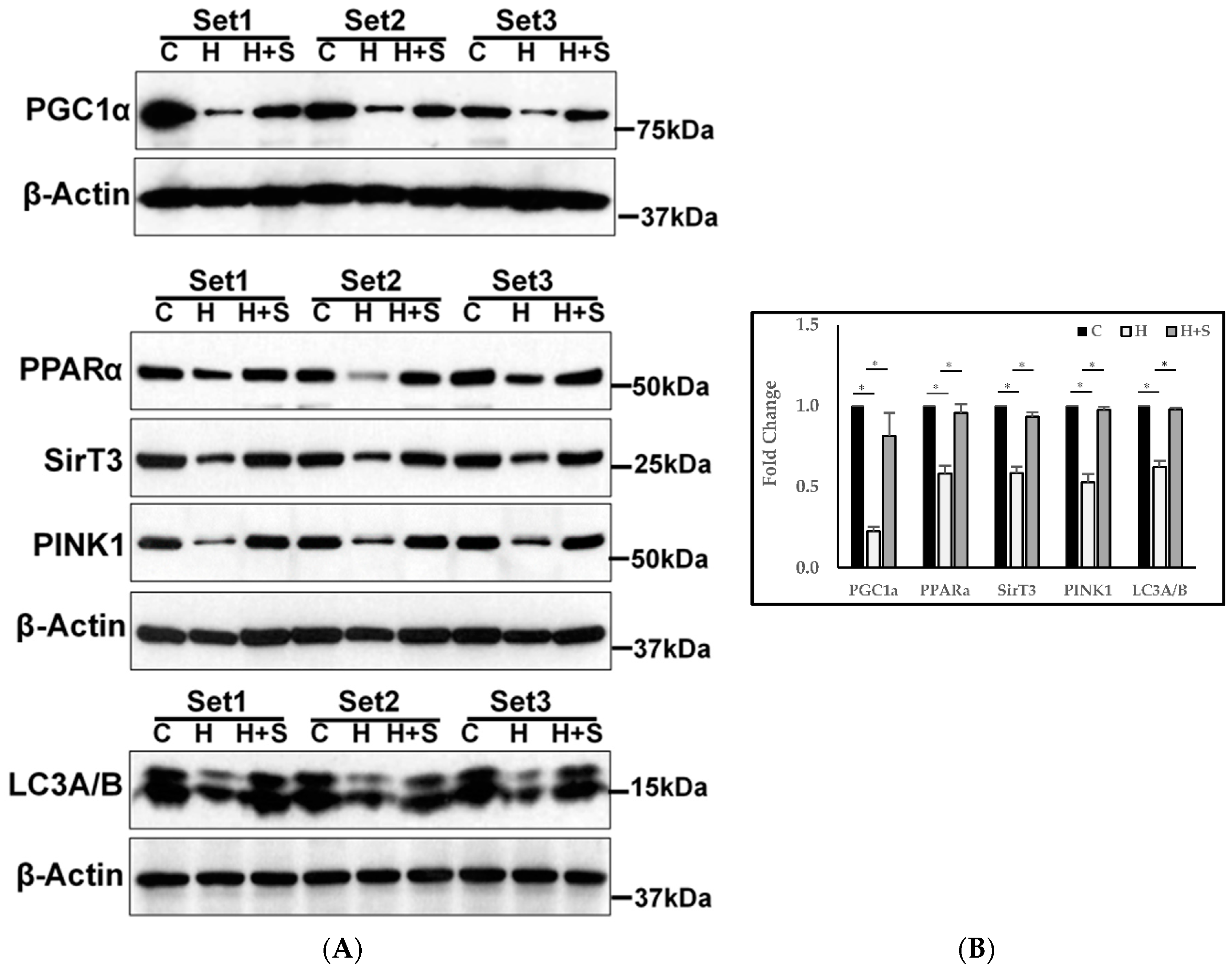
3.11. Molecular Regulation of Energy Metabolism, Autophagy, and Mitophagy
4. Discussion
4.1. Effect of GlyNAC on GSH Deficiency and OxS
4.2. Effect of GlyNAC on Mitochondrial Fuel Oxidation and Molecular Regulation of Energy Metabolism
4.3. Effect of GlyNAC on Rate of Muscle Protein Breakdown
4.4. Effect of GlyNAC on Muscle Strength and Exercise Capacity
4.5. Effect of GlyNAC Supplementation on Improving Insulin Resistance
4.6. Effects of GlyNAC on Cognitive Decline
4.7. Effect of GlyNAC on Total Body Fat, Waist Circumference, and Liver Fat
4.8. Effect of GlyNAC on Inflammation, Endothelial Function, and Cardiovascular Risk
4.9. Effect of GlyNAC on Genomic Damage
4.10. Hallmarks of Aging
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 8OHdG | 8-hydroxy-deoxyguanosine |
| BMI | Body mass index |
| CRP | C-reactive protein |
| DEXA | Dual-energy X-ray absorptiometry |
| DSST | Digital symbol-substitution test |
| FDG-PET | Fluorodeoxyglucose Positron Emission Tomography |
| GlyNAC | Combination of glycine plus N-acetylcysteine |
| GSH | Glutathione |
| GSSG | oxidized glutathione |
| HDL | High density lipoprotein |
| IL6 | Interleukin 6 |
| LDL | low density lipoprotein |
| LM | Lean mass |
| LC3A/B | Microtubule-associated protein 1A/1B-light chain 3 |
| MFO | Mitochondrial fatty-acid oxidation |
| MGO | Mitochondrial glucose oxidation |
| MoCA | Montreal cognitive assessment |
| MPBR | Muscle protein breakdown rate |
| OxS | Oxidative stress |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK | Phosphatase and tensin homolog (PTEN) induced kinase 1 |
| PPARα | Peroxisome proliferator-activated receptor 1-alpha |
| PWH | Patients with HIV |
| RBC | Red-blood cell |
| ROS | Reactive-oxygen species |
| sICAM1 | soluble intracellular adhesion molecule 1 |
| sVCAM1 | soluble vascular-cell adhesion molecule 1 |
| SIRT3 | Sirtuin 3 |
| TBARS | thiobarbituric acid reducing substances |
| TNFα | Tumor-necrosis factor alpha |
References
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. The HIV Outpatient Study Investigators. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef]
- Mack, K.A.; Ory, M.G. AIDS and older Americans at the end of the twentieth century. J. Acquir. Immune Defic. Syndr. 2003, 33, S68–S75. [Google Scholar] [CrossRef]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Ryscavage, P.; Taiwo, B. Accelerated aging and human immunodeficiency virus infection: Emerging challenges of growing older in the era of successful antiretroviral therapy. J. Neurovirol. 2012, 18, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Önen, N.F.; Overton, E.T. A review of premature frailty in HIV-infected persons; another manifestation of HIV-related accelerated aging. Curr. Aging Sci. 2011, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Rajasuriar, R.; Chong, M.L.; Ahmad Bashah, N.S.; Abdul Aziz, S.A.; Mcstea, M.; Lee, E.C.; Wong, P.L.; Azwa, I.; Omar, S.F.S.; Lai, P.S.M.; et al. Major health impact of accelerated aging in young HIV-infected individuals on antiretroviral therapy. AIDS 2017, 31, 1393–1403. [Google Scholar] [CrossRef]
- Jiménez, Z.; Sánchez-Conde, M.; Brañas, F. HIV infection as a cause of accelerated aging and frailty. Rev. Esp. Geriatr. Gerontol. 2018, 53, 105–110. [Google Scholar] [CrossRef]
- Schrack, J.A.; Althoff, K.N.; Jacobson, L.P.; Erlandson, K.M.; Jamieson, B.D.; Koletar, S.L.; Phair, J.; Ferrucci, L.; Brown, T.T.; Margolick, J.B. Accelerated Gait Speed Decline in HIV-Infected Men. J. Acquir. Immune Defic. Syndr. 2015, 70, 370–376. [Google Scholar] [CrossRef]
- Khoury, A.L.; Morey, M.C.; Wong, T.C.; McNeil, D.L.; Humphries, B.; Frankey, K.; Pieper, C.F.; Hicks, C.B.; Huffman, K.; McKellar, M.S. Diminished physical function in older HIV-infected adults in the Southeastern, U.S. despite successful antiretroviral therapy. PLoS ONE 2017, 12, e0179874. [Google Scholar] [CrossRef]
- Kuhn, T.; Jin, Y.; Huang, C.; Kim, Y.; Nir, T.M.; Gullett, J.M.; Jones, J.D.; Sayegh, P.; Chung, C.; Dang, B.H.; et al. The joint effect of aging and HIV infection on microstructure of white matter bundles. Hum. Brain Mapp. 2019, 40, 4370–4380. [Google Scholar] [CrossRef]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef]
- Payne, B.A.; Wilson, I.J.; Hateley, C.A.; Horvath, R.; Santibanez-Koref, M.; Samuels, D.C.; Price, D.A.; Chinnery, P.F. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat. Genet. 2011, 43, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Effect of increasing glutathione with cysteine and glycine supplementation on mitochondrial fuel oxidation, insulin sensitivity, and body composition in older HIV-infected patients. J. Clin. Endocrinol. Metab. 2014, 99, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; Liu, C.W.; Rice, S. Increasing glutathione concentrations with cysteine and glycine supplementation lowers inflammation in HIV patients. AIDS 2015, 29, 1899–1900. [Google Scholar] [CrossRef] [PubMed]
- Monczor, A.N.; Li, X.; Palella, F.J., Jr.; Erlandson, K.M.; Wiley, D.; Kingsley, L.A.; Post, W.S.; Jacobson, L.P.; Brown, T.T.; Lake, J.E. Systemic Inflammation Characterizes Lack of Metabolic Health in Nonobese HIV-Infected Men. Mediat. Inflamm. 2018, 2018, 5327361. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.; Moyo, S.; Gabaitiri, L.; Gaseitsiwe, S.; Bussmann, H.; Koethe, J.R.; Musonda, R.; Makhema, J.; Novitsky, V.; Marlink, R.G.; et al. Persistently elevated serum interleukein-6 predicts mortality among adults receiving combination antiretroviral therapy in Botswana: Results from a clinical trial. AIDS Res. Hum. Retrovir. 2013, 29, 993–999. [Google Scholar] [CrossRef]
- Pathai, S.; Lawn, S.D.; Shiels, P.G.; Weiss, H.A.; Cook, C.; Wood, R.; Gilbert, C.E. Corneal endothelial cells provide evidence of accelerated cellular senescence associated with HIV infection: A case-control study. PLoS ONE 2013, 8, e57422. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Harman, D. The biological clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Ghosh, S.; Pulinilkunnil, T.; Yuen, G.; Kewalramani, G.; An, D.; Qi, D.; Abrahani, A.; Rodrigues, B. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H768–H776. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Restivo, F.; Vercellinatto, I.; Danni, O.; Brignardello, E.; Aragno, M.; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Samson, S.L.; Donzalez, E.V.; Reddy, V.T.; Sekhar, R.V. Impaired mitochondrial fuel oxidation and insulin resistance in aging—Novel protective role of glutathione. Aging Cell 2013, 12, 415–425. [Google Scholar] [CrossRef]
- Frayn, K.N. Calculation of substrate oxidation rates in vivo from gaseous exchange. J. Appl. Physiol. 1983, 55, 628–634. [Google Scholar] [CrossRef]
- Chacko, S.K.; Sunehag, A.L.; Sharma, S.; Sauer, P.J.; Haymond, M.W. Measurement of gluconeogenesis using glucose fragments and mass spectrometry after ingestion of deuterium oxide. J. Appl. Physiol. 2008, 104, 944–951. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Seth, A.; Sherman, K.E. Fatty liver disease in persons with HIV infection. Top. Antivir. Med. 2019, 27, 75–82. [Google Scholar]
- Mazzuca, P.; Caruso, A.; Caccuri, F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New Microbiol. 2016, 39, 167–173. [Google Scholar]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Echeverría, P.; Bonjoch, A.; Puig, J.; Estany, C.; Ornelas, A.; Clotet, B.; Negredo, E. High Prevalence of Sarcopenia in HIV-Infected Individuals. Biomed. Res. Int. 2018, 2018, 5074923. [Google Scholar] [CrossRef]
- Studenski, S.; Perera, S.; Patel, K.; Rosano, C.; Faulkner, K.; Inzitari, M.; Brach, J.; Chandler, J.; Cawthon, P.; Connor, E.B.; et al. Gait speed and survival in older adults. JAMA 2011, 305, 50–58. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Mouzannar, R.; McCafferty, J.; Benedetto, G.; Richardson, C. Transcriptional and phosphor-proteomic screens reveal stem cell activation of insulin resistance and transformation pathways following a single minimally toxic episode of ROS. Int. J. Genom. Proteom. 2011, 2, 34–49. [Google Scholar]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Bekinschtein, P.; Cammarota, M.; Katche, C.; Slipczuk, L.; Rossato, J.I.; Goldin, A.; Izquierdo, I.; Medina, J.H. BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. USA 2008, 105, 2711–2716. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, D.A.; Sinharay, S.; Steinbach, S.; Wakim, P.G.; Geannopoulos, K.; Traino, K.; Dey, A.K.; Tramont, E.; Rapoport, S.I.; Snow, J.; et al. Global and regional brain hypometabolism on FDG-PET in treated HIV-infected individuals. Neurology 2018, 91, e1591–e1601. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, F.; Lillioja, S.; Esposito-Del Puente, A.; Nyomba, B.L.; Raz, I.; Saad, M.F.; Swinburn, B.A.; Knowler, W.C.; Bogardus, C.; Ravussin, E. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: Study of 24-h RQ. Am. J. Physiol. 1990, 259, E650–E657. [Google Scholar] [CrossRef]
- Kolgiri, V.; Nagar, V.; Patil, V. Association of Metabolic Syndrome and Oxidative DNA Damage in HIV/AIDS Patients. Indian J. Clin. Biochem. 2018, 33, 273–281. [Google Scholar] [CrossRef]
- Kolgiri, V.; Patil, V.W. Protein carbonyl content: A novel biomarker for aging in HIV/AIDS patients. Braz. J. Infect. Dis. 2017, 21, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kallianpur, K.J.; Gerschenson, M.; Mitchell, B.I.; LiButti, D.E.; Umaki, T.M.; Ndhlovu, L.C.; Nakamoto, B.K.; Chow, D.C.; Shikuma, C.M. Oxidative mitochondrial DNA damage in peripheral blood mononuclear cells is associated with reduced volumes of hippocampus and subcortical gray matter in chronically HIV-infected patients. Mitochondrion 2016, 28, 8–15. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Controls | PWH: 0 weeks before Supplementation | PWH: 12 weeks after Supplementation | p-Value Con vs. HIV 0 w; HIV 0 w vs. HIV 12 w |
|---|---|---|---|---|
| Hemoglobin (g/L) | 13.8 ± 1.3 | 14.0 ± 0.6 | 13.3 ± 0.8 | p = 0.8; p = 0.07 |
| Total bilirubin (mg/dL) | 0.7 ± 0.3 | 1.2 ± 1.2 | 1.2 ± 1.4 | p = 0.6; p = 0.8 |
| Alanine transaminase (U/L) | 24.8 ± 12.3 | 27.5 ± 11.1 | 19.9 ± 5.5 | p = 0.7; p = 0.08 |
| Aspartate transaminase (U/L) | 24.3 ± 11.2 | 24.3 ± 3.5 | 21.1 ± 2.4 | p = 1.0; p = 0.1 |
| Alkaline phosphatase (U/L) | 70.8 ± 30.1 | 87.1 ± 24.1 | 93.4 ± 23.6 | p = 0.3; p = 0.2 |
| Blood urea nitrogen (mmol/L) | 12.8 ± 2.7 | 11.8 ± 3.0 | 9.8 ± 2.7 | p = 0.4; p = 0.1 |
| Creatinine (mg/dL) | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.2 | p = 0.5; p = 0.8 |
| Estimated glomerular filtration rate (eGFR mL/min) | 94.4 ± 7.0 | 92.6 ± 14.9 | 107.3± 20.3 | p = 0.8; p = 0.004 |
| HbA1c (%) | 5.7 ± 0.4 | 5.8 ± 0.3 | 5.8 ± 0.4 | p = 0.8; p = 0.5 |
| Plasma glucose (mmol/L) | 4.8 ± 0.4 | 5.5 ± 0.7 | 4.8 ± 0.6 | p = 0.048; p = 0.000 |
| Total cholesterol (mg/dL) | 192.8 ± 53.5 | 219.5 ± 42.8 | 210.6 ± 54.2 | p = 0.3; p = 0.6 |
| Triglycerides (mg/dL) | 91.5 ± 38.8 | 194.4 ± 140.7 | 138.9 ± 67.4 | p = 0.1; p = 0.3 |
| High density lipoprotein-cholesterol (mg/dL) | 57.9 ± 14.8 | 41.9 ± 10.6 | 43.4 ± 15.5 | p = 0.028; p = 0.2 |
| Low density lipoprotein-cholesterol (mg/dL) | 116.8 ± 42.6 | 136.9 ± 28.3 | 137.5 ± 41.3 | p = 0.3; p = 0.8 |
| Thyroid stimulating hormone (mIU/L) | 1.9 ± 1.0 | 1.6 ± 0.8 | 1.4 ± 0.7 | p = 0.6; p = 0.6 |
| Free T4 (ng/L) | 1.1 ± 0.1 | 1.1 ± 0.2 | 1.0 ± 0.1 | p = 1; p = 0.5 |
| Cortisol (mg/dL) | 10.5 ± 4.8 | 7.5 ± 3.1 | p = 0.2 |
| Parameters | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Matching parameters: | ||||
| Age (y) | 55.0 ± 3.6 | 54.9 ± 4.4 p = 0.9 | ||
| Gender (M:F) | 6:2 | 6:2 | ||
| Body-mass index (BMI) | 28.9 ± 2.7 | 29.5 ± 2.3 p = 0.7 | ||
| Glutathione and oxidative stress: | ||||
| RBC-GSH (μmol/g Hb) | 4.5 ± 0.6 | 2.8 ± 1.0 p = 0.002 | 4.1 ± 0.6 p = 0.003 | 2.5 ± 1.0 p = 0.001 |
| RBC-GSSG (μmol/g Hb) | 1.0 ± 1.0 | 0.4 ± 0.4 p = 0.2 | 0.5 ± 0.4 p = 0.8 | 1.0 ± 0.5 p = 0.004 |
| Skeletal muscle GSH (mmol/kg muscle) | 2.2 ± 0.8 | 0.5 ± 0.2 p = 0.000 | 2.2 ± 0.9 p = 0.001 | |
| Plasma TBARS (μM/L) | 2.7 ± 1.3 | 32.9 ± 6.6 p = 0.000 | 6.4 ± 2.7 p = 0.000 | 8.5 ± 2.9 p = 0.025 |
| Plasma F2-isoprostane (pg/mL) | 50.7 ± 3.7 | 267.7 ± 102.9 p = 0.001 | 53.3 ± 3.7 p = 0.001 | 195.1 ± 73.2 p = 0.001 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Fasted respiratory quotient (RQ) | 0.76 ± 0.01 | 0.86 ± 0.04 p = 0.000 | 0.78 ± 0.03 p = 0.001 | 0.90 ± 0.05 p = 0.004 |
| Fasted fatty acid oxidation (mg/kg lean mass/min) | 1.39 ± 0.30 | 0.50 ± 0.20 p = 0.007 | 1.39 ± 0.09 p = 0.001 | |
| Fasted carbohydrate oxidation (mg/kg lean mass/min) | 0.80 ± 0.12 | 2.15 ± 0.34 p = 0.001 | 1.30 ± 0.45 p = 0.001 | |
| Fasted fatty-acid oxidation (mg/kg/min) | 0.96 ± 0.25 | 0.30 ± 0.11 p = 0.005 | 0.90 ± 0.15 p = 0.001 | 0.45 ± 0.23 p = 0.009 |
| Fasted carbohydrate oxidation (mg/kg/min) | 0.55 ± 0.11 | 1.32 ± 0.12 p = 0.001 | 0.84 ± 0.28 p = 0.001 | 1.24 ± 0.28 p = 0.002 |
| Energy expenditure (kcal/d) | 1637 ± 299 | 1538 ± 269 p = 0.4 | 1509 ± 232 p = 0.6 | 1592 ± 296 p = 0.2 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Tracer kinetic data: | ||||
| Muscle protein breakdown rate (mg/kg LBM/h) | 104.6 ± 23.7 | 145.4 ± 36.8 p = 0.003 | 96.3 ± 41.6 p = 0.027 | |
| Glucose production rate (μmol/kg/min) | 7.8 ± 1.4 | 7.7 ± 0.6 p = 0.83 | 7.5 ± 0.4 p = 0.4 | |
| Rate of gluconeogenesis (μmol/kg/min) | 5.6 ± 0.9 | 5.0 ± 0.8 p = 0.079 | 5.0 ± 0.4 p = 0.96 | |
| Rate of glycogenolysis (μmol/kg/min) | 2.2 ± 0.7 | 2.7 ± 0.5 p = 0.16 | 2.5 ± 0.3 p = 0.54 | |
| Glycemia and insulin resistance: | ||||
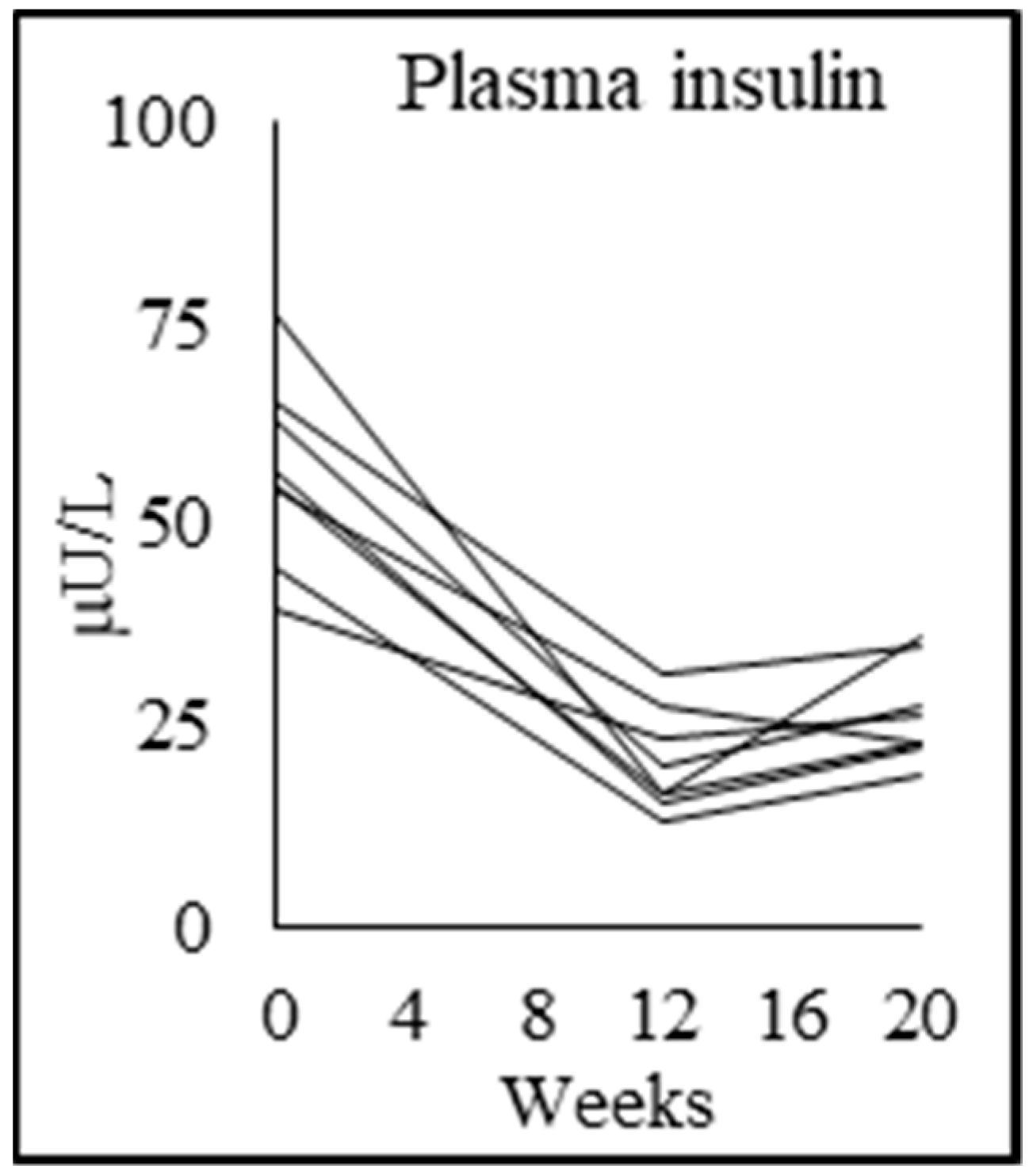
| Fasting plasma glucose (mmol/L) | 4.8 ± 0.4 | 5.5 ± 0.7 p = 0.048 | 4.8 ± 0.6 p = 0.000 | 5.5 ± 0.7 p = 0.002 |
| Fasting plasma insulin (mU/L) | 10.0 ± 2.7 | 56.8 ± 11.6 p = 0.000 | 20.4 ± 6.5 p = 0.000 | 26.3 ± 6.2 p = 0.039 |
| Insulin resistance (HOMA-IR) | 2.2 ± 0.7 | 13.7 ± 2.6 p = 0.000 | 4.3 ± 1.3 p = 0.000 | 6.3 ± 1.6 p = 0.002 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Plasma high-sensitivity IL-6 (pg/mL) | 0.8 ± 0.2 | 4.9 ± 1.0 p = 0.000 | 1.7 ± 0.7 p = 0.000 | 2.5 ± 1.0 p = 0.003 |
| Plasma TNFα (pg/mL) | 33.8 ± 10.5 | 89.9 ± 15.1 p = 0.000 | 59.1 ± 8.0 p = 0.000 | 64.5 ± 7.4 p = 0.006 |
| Plasma high-sensitivity C-Reactive Protein (ng/mL) | 0.4 ± 0.0 | 2.2 ± 0.3 p = 0.000 | 1.1 ± 0.3 p = 0.000 | 1.3 ± 0.3 p = 0.034 |
| Plasma sICAM1 (ng/mL) | 364.9 ± 114.5 | 994.1 ± 222.8 p = 0.000 | 445.4 ± 99.9 p = 0.000 | 658.8 ± 104.8 p = 0.000 |
| Plasma sVCAM1 (ng/mL) | 627.1 ± 132.7 | 1643.9 ± 289.7 p = 0.000 | 976.5 ± 263.4 p = 0.000 | 1247.4 ± 245.1 p = 0.001 |
| Plasma E-Selectin (ng/mL) | 28.2 ± 7.6 | 62.8 ± 12.4 p = 0.000 | 41.1 ± 9.3 p = 0.000 | 52.1 ± 9.6 p = 0.000 |
| Plasma 8-OHdG (pg/mL) | 53.4 ± 4.8 | 182.0 ± 44.4 p = 0.000 | 54.9 ± 4.7 p = 0.000 | 115.3 ± 49.7 p = 0.01 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Gait speed (m/s) | 1.3 ± 0.1 | 1.0 ± 0.1 p = 0.005 | 1.3 ± 0.2 p = 0.003 | 1.1 ± 0.2 p = 0.016 |
| Rapid 6-min walk (m) | 644 ± 62 | 508 ± 23 p = 0.001 | 542 ± 23 p = 0.000 | 509 ± 37 p = 0.012 |
| Chair-rise test (s) | 18.9 ± 3.5 | 28.8 ± 3.8 p = 0.003 | 23.0 ± 3.0 p = 0.002 | 25.0 ± 2.6 p = 0.044 |
| Muscle strength, dominant forearm (kg) | 46.0 ± 13.0 | 31.3 ± 8.3 p = 0.004 | 39.0 ± 8.7 p = 0.023 | 33.7 ± 8.6 p = 0.028 |
| Muscle strength, nondominant forearm (kg) | 41.8 ± 12.5 | 27.3 ± 8.6 p = 0.009 | 35.3 ± 7.8 p = 0.022 | 30.4 ± 8.0 p = 0.061 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Montreal cognitive assessment test (MoCA) | 28.0 ± 1.6 | 21.1 ± 3.8 p = 0.002 | 25.0 ± 2.0 p = 0.005 | 26.0 ± 1.8 p = 0.17 |
| Trails test A (sec) | 34.6 ± 10.2 | 62.6 ± 17.4 p = 0.001 | 46.4 ± 12.6 p = 0.008 | 40.4 ± 11.2 p = 0.068 |
| Trails test B (sec) | 53.8 ± 20.4 | 117.5 ± 42.4 p = 0.003 | 69.8 ± 15.3 p = 0.007 | 86.0 ± 30.2 p = 0.13 |
| Verbal-fluency test | 41.0 ± 9.5 | 28.9 ± 9.2 p = 0.053 | 34.6 ± 5.6 p = 0.025 | 36.6 ± 5.5 p = 0.28 |
| DSST: % completed | 44.9 ± 7.7 | 31.3 ± 8.0 p = 0.012 | 37.1 ± 7.5 p = 0.015 | 36.1 ± 7.2 p = 0.47 |
| DSST: % accuracy | 99.0 ± 1.5 | 95.1 ± 4.2 p = 0.058 | 97.7 ± 3.2 p = 0.033 | 97.4 ± 4.3 p = 0.7 |
| Plasma BDNF (ng/mL) | 47.9 ± 9.6 | 26.3 ± 5.4 p = 0.003 | 47.2 ± 8.7 p = 0.000 | 36.7 ± 9.1 p = 0.005 |
| Outcome Measures | Controls | PWH: 0 Weeks p-Value: HIV 0 w vs. Con | PWH: 12 Weeks p-Value: HIV 0 w vs. HIV 12 w | PWH: 20 Weeks p-Value: HIV 12 w vs. HIV 20 w |
|---|---|---|---|---|
| Weight (kg) | 91.5 ± 14.5 | 87.6 ± 8.6 p = 0.5 | 84.2 ± 9.7 p = 0.041 | 84.8 ± 11.6 p = 0.7 |
| BMI | 28.9 ± 2.7 | 29.5 ± 2.3 p = 0.7 | 28.3 ± 3.0 p = 0.04 | 28.3 ± 3.4 p = 0.6 |
| Fat-mass (kg) | 25.8 ± 6.7 | 29.9 ± 3.6 p = 0.2 | 27.6 ± 3.8 p = 0.047 | |
| Truncal fat mass (kg) | 12.7 ± 3.5 | 16.7 ± 2.7 p = 0.017 | 15.1 ± 2.7 p = 0.038 | |
| Lean mass (kg) | 63.1 ± 13.8 | 55.2 ± 9.9 p = 0.079 | 54.1 ± 10.8 p = 0.21 | |
| Waist circumference (cm) | 97.5 ± 6.7 | 105.4 ± 7.7 p = 0.10 | 98.1 ± 7.9 p = 0.000 | 100.3 ± 8.4 p = 0.031 |
| Hip circumference (cm) | 107.5 ± 8.6 | 106.4 ± 5.6 p = 0.81 | 106.4 ± 4.9 p = 0.96 | 106.4 ± 5.2 p = 0.99 |
| Waist:Hip ratio | 0.9 ± 0.0 | 1.0 ± 0.1 p = 0.016 | 0.9 ± 0.1 p = 0.000 | 0.9 ± 0.1 p = 0.053 |
| Systolic BP (mm Hg) | 120.8 ± 11.8 | 131.9 ± 8.8 p = 0.003 | 119.4 ± 14.3 p = 0.009 | 117.3 ± 14.8 p = 0.65 |
| Diastolic BP (mm Hg) | 75.6 ± 10.0 | 84.1 ± 8.5 p = 0.003 | 76.6 ± 9.9 p = 0.039 | 76.1 ± 9.6 p = 0.89 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Liu, C.; Suliburk, J.W.; Minard, C.G.; Muthupillai, R.; Chacko, S.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial. Biomedicines 2020, 8, 390. https://doi.org/10.3390/biomedicines8100390
Kumar P, Liu C, Suliburk JW, Minard CG, Muthupillai R, Chacko S, Hsu JW, Jahoor F, Sekhar RV. Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial. Biomedicines. 2020; 8(10):390. https://doi.org/10.3390/biomedicines8100390
Chicago/Turabian StyleKumar, Premranjan, Chun Liu, James W. Suliburk, Charles G. Minard, Raja Muthupillai, Shaji Chacko, Jean W. Hsu, Farook Jahoor, and Rajagopal V. Sekhar. 2020. "Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial" Biomedicines 8, no. 10: 390. https://doi.org/10.3390/biomedicines8100390
APA StyleKumar, P., Liu, C., Suliburk, J. W., Minard, C. G., Muthupillai, R., Chacko, S., Hsu, J. W., Jahoor, F., & Sekhar, R. V. (2020). Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial. Biomedicines, 8(10), 390. https://doi.org/10.3390/biomedicines8100390