Survival Comparison between Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines and Other Immunotherapies Based on Extent of Disease at the Time of Treatment

Abstract
1. Introduction
2. Experimental Section
2.1. Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines
2.2. Patient-Specific Dendritic Cell Vaccines
2.3. Comparator Populations of Melanoma Patients Treated with Immunotherapy
2.4. Statistical Methods
3. Results
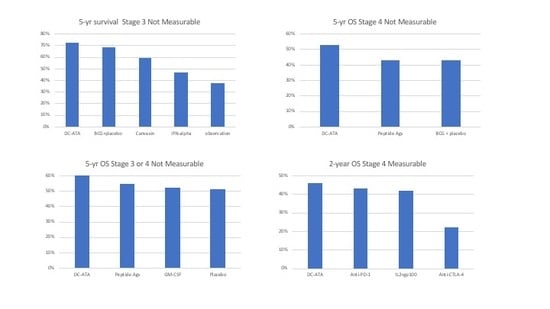
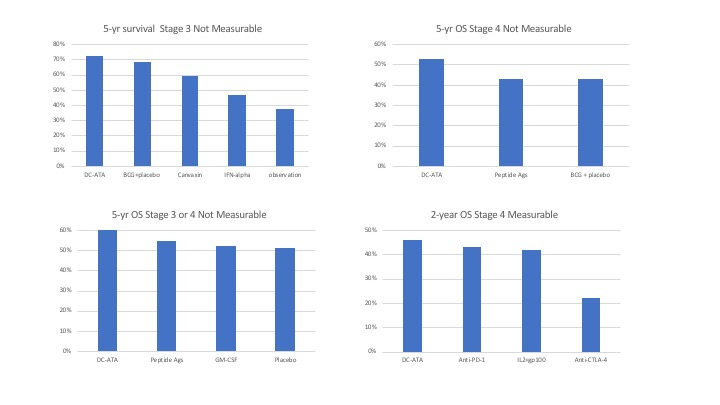
3.1. Stage 3 with No Measurable Disease
3.2. Stage 4 with No Measurable Disease
3.3. Stage 3 or 4 with No Measurable Disease
3.4. Distant Stage 4 Measurable Disease
3.5. Results for Immunotherapies Recently Approved for the Treatment of Melanoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Comprehensive Cancer Network, Inc. Cutaneous Melanoma, Version 2., 2019, NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Available online: nccn.org (accessed on 12 March 2019).
- Dillman, R.O. An update on the relevance of vaccine research for the treatment of metastatic melanoma. Melanoma Manaag. 2017, 4, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O. Is there a role for therapeutic cancer vaccines in the age of checkpoint inhibitors? Hum. Vaccin. Immunother. 2017, 13, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Gulley, J.L. Vaccines as an integral component of cancer immunotherapy. JAMA 2018, 320, 2195–2196. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O. Melanoma vaccines: Trials and tribulations. Vaccine Dev. Ther. 2013, 3, 57–78. [Google Scholar] [CrossRef]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Cornforth, A.N.; Nistor, G.I. Dendritic cell vaccines for melanoma: Past, present, and future. Melanoma Manag 2016, 3, 267–283. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Feuerstein, B.; Berger, T.G.; Maczek, C.; Röder, C.; Schreiner, D.; Hirsch, U.; Haendle, I.; Leisgang, W.; Glaser, A.; Kuss, O.; et al. A method for the production of cryopreserved aliquots of antigen-preloaded, mature dendritic cells ready for clinical use. J. Immunol. Methods 2000, 245, 15–29. [Google Scholar] [CrossRef]
- Dillman, R.O.; Cornforth, A.N.; Nistor, G. Cancer stem cell antigen-based vaccines: The preferred strategy for active specific immunotherapy of metastatic melanoma? Expert Opin. Biol. Ther. 2013, 13, 643–656. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the heterogeneity of cancer with individualized neoepitope vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.; Selvan, S.; Schiltz, P.; Peterson, C.; Allen, K.; Depriest, C.; McClay, E.F.; Barth, N.; Sheehy, P.; de Leon, C.; et al. Phase I/II trial of melanoma patient-specific vaccine of proliferating autologous tumor cells, dendritic cells, and GM-CSF: Planned interim analysis. Cancer Biother. Radiopharm. 2004, 19, 658–665. [Google Scholar] [PubMed]
- Dillman, R.O.; Selvan, S.R.; Schiltz, P.M. Patient-specific dendritic cell vaccines for metastatic melanoma. N. Engl. J. Med. 2006, 355, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Selvan, S.R.; Schiltz, P.M.; McClay, E.F.; Barth, N.M.; Depriest, C.; de Leon, C.; Mayorga, C.; Cornforth, A.N.; Allen, K. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific anti-tumor vaccines in patients with metastatic melanoma: Final Report. Cancer Biother. Radio. 2009, 24, 311–319. [Google Scholar]
- Dillman, R.O.; Cornforth, A.N.; Depriest, C.; McClay, E.F.; Amatruda, T.T.; de Leon, C.; Ellis, R.E.; Mayorga, C.; Carbonell, D.; Cubellis, J.M. Tumor stem cell antigens as consolidative active specific immunotherapy: A randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J. Immunother 2012, 35, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Cornforth, A.N.; Nistor, G.I.; McClay, E.F.; Amatruda, T.T.; Depriest, C. Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in patients with metastatic melanoma: 5-year follow up and additional analyses. J. Immunother Cancer 2018, 6, 19. [Google Scholar] [CrossRef]
- Dillman, R.O.; Cornforth, A.N.; McClay, E.F.; Depriest, C. Patient-specific dendritic cell vaccines with autologous tumor antigens in 72 patients with metastatic melanoma. Melanoma Manag. 2019, 6, MMT20. [Google Scholar] [CrossRef]
- Dillman, R.O.; Hsieh, C.; Nistor, G.I. A A clinical odyssey: Cancer stem cells as the antigen source for autologous cancer vaccines. Adv. Stem Cell Res. 2019, 1, 1–13. [Google Scholar]
- Morton, D.L.; Mozzillo, N.; Thompson, J.F.; Kelley, M.C.; Faries, M.; Wagner, J.; Schneebaum, S.; Schuchter, L.; Gammon, G.; Elashoff, R. An international, randomized, phase III trial of bacillus Calmette-Guerin (BCG) plus allogeneic melanoma vaccine (MCV) or placebo after complete resection of melanoma metastatic to regional or distant sites. Presented at the 43rd Annual Meeting of the American Society for Clinical Oncology, Chicago, IL. USA. J. Clin. Oncol. 2007, 25, 8508. [Google Scholar]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AjCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Ibrahim, J.G.; Sosman, J.A.; Sondak, V.K.; Agarwala, S.S.; Ernstoff, M.S.; Rao, U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared to the GM2-KLH-QS-21 vaccine in patients with stage IIB and III melanoma: Results of intergroup trial E1694/S9512/C509801. J. Clin. Oncol. 2001, 19, 2370–2380. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Lee, S.J.; Yip, W.; Rao, U.N.; Tarhini, A.A.; Choen, G.I.; Reintgen, D.S.; Evans, T.L.; Brell, J.M.; Albertini, M.R.; et al. Phase III randomized study of 4 weeks of high-dose interferon-α-2b in stage T2bN0, T3a-bN0, Tra-bN0, and T1-4N1a-2a (microscopic melanoma: A trial of the Eastern Cooperative Oncology Group-Amerian College of Radiology Imaging Network Cancer Research Group (E1697). J. Clin. Oncol. 2017, 35, 885–892. [Google Scholar] [PubMed]
- Tagawa, S.T.; Cheung, E.; Banta, W.; Gee, C.; Weber, J.S. Survival analysis after resection of metastatic disease followed by peptide vaccines in patients with stage IV melanoma. Cancer 2006, 106, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Faries, M.B.; Mozzilo, N.; Kashani-Sabet, M.; Thompson, J.F.; Kelley, M.C.; DeConti, R.C.; Lee, J.E.; Huth, J.F.; Wagner, J.; Dalgleish, A.; et al. MMAIT-IV Clinical Trial Group. Long-term survival after complete surgical resection and adjuvant immunotherapy for distant melanoma metastases. Ann. Surg. Oncol. 2017, 24, 3991–4000. [Google Scholar] [CrossRef]
- Lawson, D.H.; Lee, S.; Zhao, F.; Tarhini, A.A.; Margolin, K.A.; Ernstoff, M.S.; Atkins, M.B.; Cohen, G.I.; Whiteside, T.L.; Butterfield, L.H.; et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) versus peptide vaccination versus GM-CSF Plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV Melanoma: A trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J. Clin. Oncol. 2015, 33, 4066–4076. [Google Scholar]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 4077–4084. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; Dronca, R.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotern, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicenter, randomised, controlled, phase 3 study. Lancet Oncol. 2019. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. CheckMate 238 Collaborators. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [PubMed]
- Dillman, R.O.; Nanci, A.A.; Williams, S.T.; Kim, R.B.; Hafer, R.L.; Coleman, C.L.; Wang, P.C.; Duma, C.M.; Chen, P.V.; Selvan, S.R.; et al. Durable complete response of refractory, progressing metastatic melanoma after treatment with a patient-specific vaccine. Cancer Biother. Radio. 2010, 25, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Van Roey, M.; Wang, C.; Chen, T.H.; Korman, A.; Jooss, K. Anti-programmed death-1 synergizs with granulocyte macrophage colony-stimulating factor secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin. Cancer Res. 2009, 15, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab verus ipilimumab alone in patients with advanced, unresectable melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; Mcnamara, M.; Marcela Diaz-Montero, C.; Gastman, B. Talimogene laherparepvec combined with anti-PD-1 immmunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 36. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Stage 3 Non-Measurable | DC-ATA Vaccine [19] | Allogeneic Tumor Cell Vaccine + BCG [21] | BCG [21] | Interferon Alpha [23] | Observation [23] |
|---|---|---|---|---|---|
| Median survival in months | > 60 | > 60 | >60 | 45.8 | 33.4 |
| 5-year survival | 72% | 59% | 68% | 46% | 37% |
| Stage 4 Non-Measurable | DC-ATA Vaccine [19] | Various Peptide Antigens [26] | Allogeneic Tumor Cell Vaccine + BCG [21,27] | BCG [21,27] |
|---|---|---|---|---|
| Median survival in months | > 60 | 46 | 35 | 39 |
| 5-year OS | 53% | 43% | 42% | 43% |
| Stage 3 or 4 Non-Measurable | DC-ATA Vaccine [19] | Multiple Peptides [28] | GM-CSF [28] | Placebo [28] |
|---|---|---|---|---|
| Median OS | >60 mos | >60 mos | >60 mos | >60 mos |
| 5- yr OS | 60% | 54% | 52% | 51% |
| Stage 4 Measurable | DC-ATA Vaccine [19] | IL-2 [29] | IL-2 + GP-100 [29] | Anti-CTLA4 [30] | Anti-PD-1 [31] |
|---|---|---|---|---|---|
| Objective Response Rate | 0% | 10% | 20% | ≈12% | 31% |
| Median survival in months | 18.5 | 11.1 | 25.8 | 11.4 | 16.8 |
| 2-year survival | 46% | 18% | 42% | 22% | 43% |
| Variables | Pembro [31,32] | Pembro vs. Ipi [34,35] | Nivo [33] | Nivo [36] | Nivo + Ipi [36] |
|---|---|---|---|---|---|
| Number of patients | 655 Measurable disease = 581 | 556 | 210 | 316 | 314 |
| Prior systemic therapies (n) | 0 (159) | 0–1 | 0 | 0 | 0 |
| 1 (205) | |||||
| 2 (178) | |||||
| ≥ 3 (113) | |||||
| PFS | 8.3 mos | 8.4 mos | 5.1 mos | 6.9 mos (2-yr 37%) | 11.5 mos (2-yr 43%) |
| OS | 23.8 mos | 32.7 mos | 1-yr 70% | 2-yr 59% 3-yr 52% | 2-yr 64% 3-yr 58% |
| 2-yr 49% | |||||
| 5-yr 34% | |||||
| ORR | 41% (267/655) | 33% | 40% | 44% | 58% |
| 33% (194/581) | |||||
| 0 prior therapies 45% (60/133) | |||||
| >1 prior therapy 30% (134/438) |
| Variables | Nivo vs. Ipi [37] | Pembro vs. Placebo [38] |
|---|---|---|
| Eligible | Resected stage 3 or 4 (3B, 3C, or 4 but not 3A) | Completely resected stage 3 including 3A |
| Number of patients | 453 vs. 453 | 514 vs. 505 |
| PFS | 1-year 70.5% vs. 60.8% | 1-year 75.4% vs. 61.0% |
| OS | Too early | Too early |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dillman, R.O.; Hsieh, C. Survival Comparison between Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines and Other Immunotherapies Based on Extent of Disease at the Time of Treatment. Biomedicines 2019, 7, 80. https://doi.org/10.3390/biomedicines7040080
Dillman RO, Hsieh C. Survival Comparison between Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines and Other Immunotherapies Based on Extent of Disease at the Time of Treatment. Biomedicines. 2019; 7(4):80. https://doi.org/10.3390/biomedicines7040080
Chicago/Turabian StyleDillman, Robert Owen, and Candace Hsieh. 2019. "Survival Comparison between Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines and Other Immunotherapies Based on Extent of Disease at the Time of Treatment" Biomedicines 7, no. 4: 80. https://doi.org/10.3390/biomedicines7040080
APA StyleDillman, R. O., & Hsieh, C. (2019). Survival Comparison between Melanoma Patients Treated with Patient-Specific Dendritic Cell Vaccines and Other Immunotherapies Based on Extent of Disease at the Time of Treatment. Biomedicines, 7(4), 80. https://doi.org/10.3390/biomedicines7040080