Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis



Abstract
:1. Introduction
2. Immunologic Background for DAC Efficacy
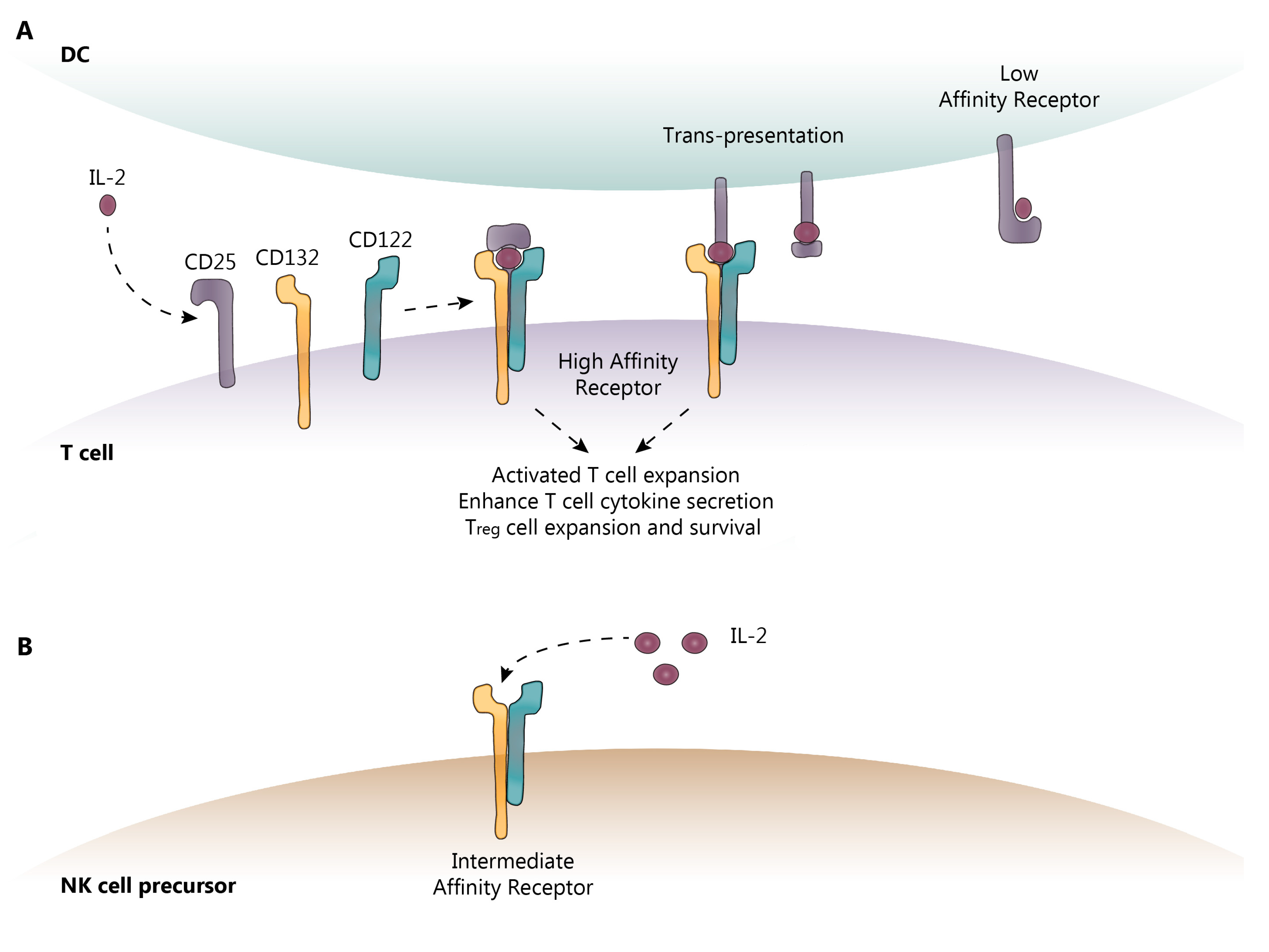
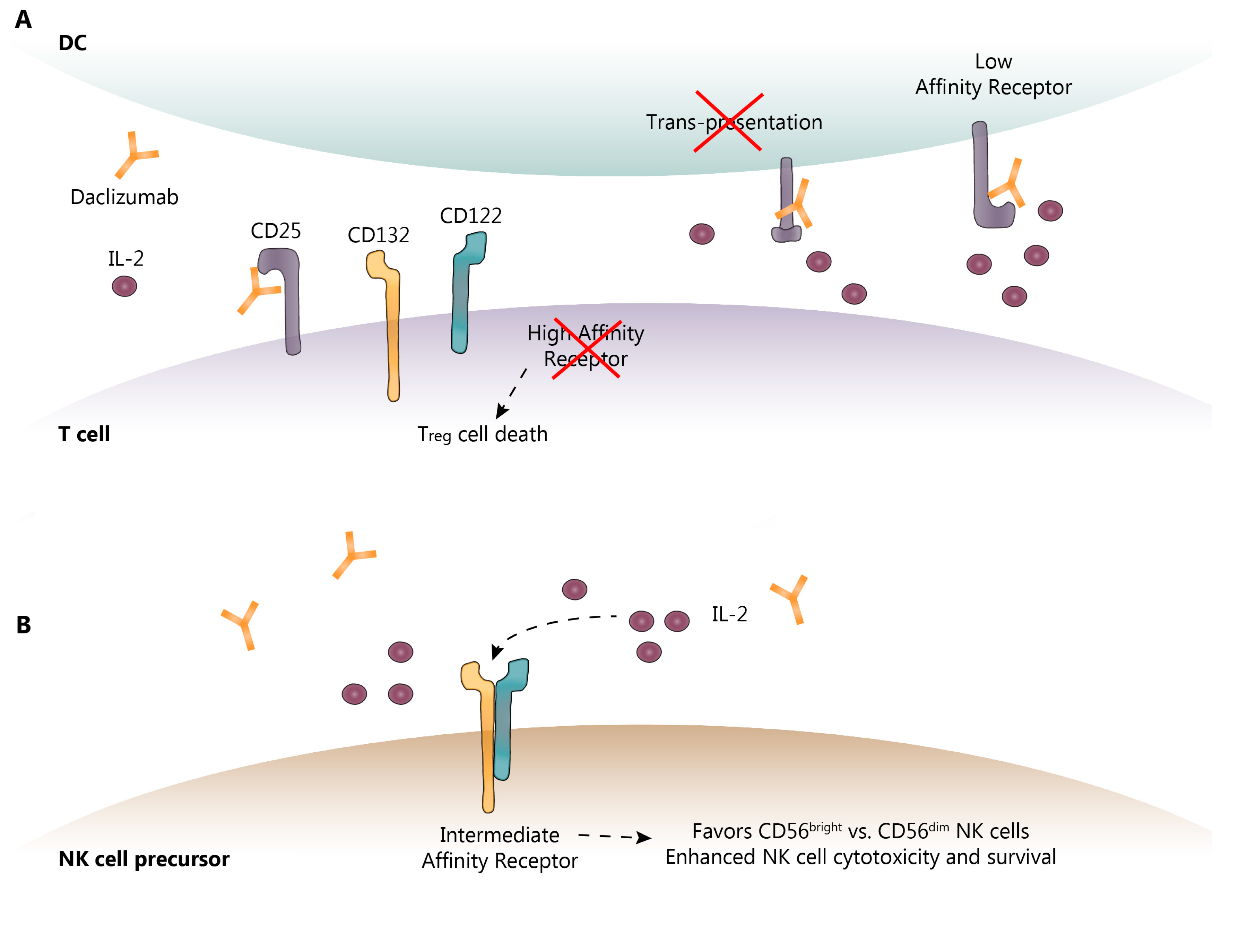
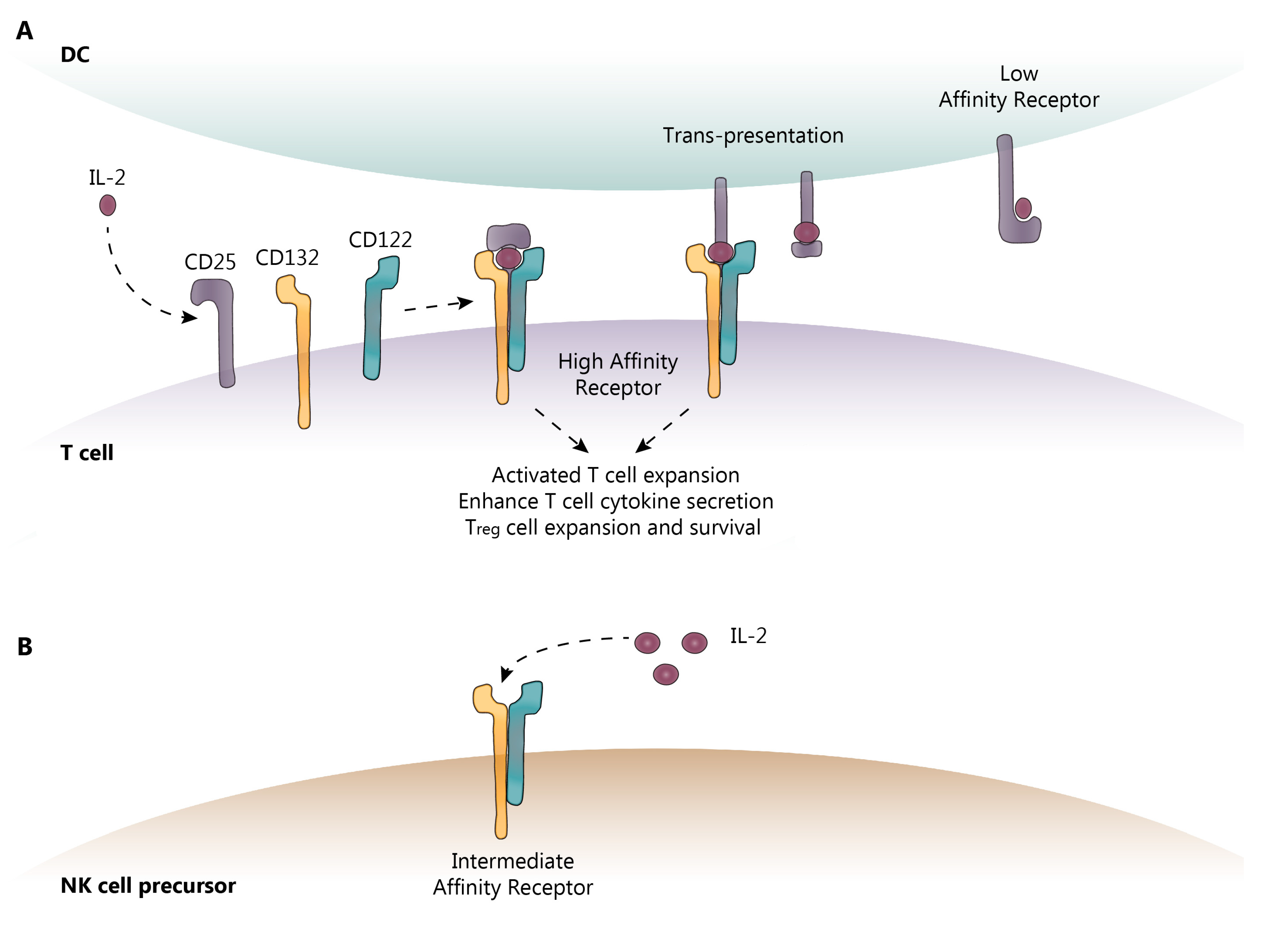
2.1. IL-2 and IL-2R Signaling
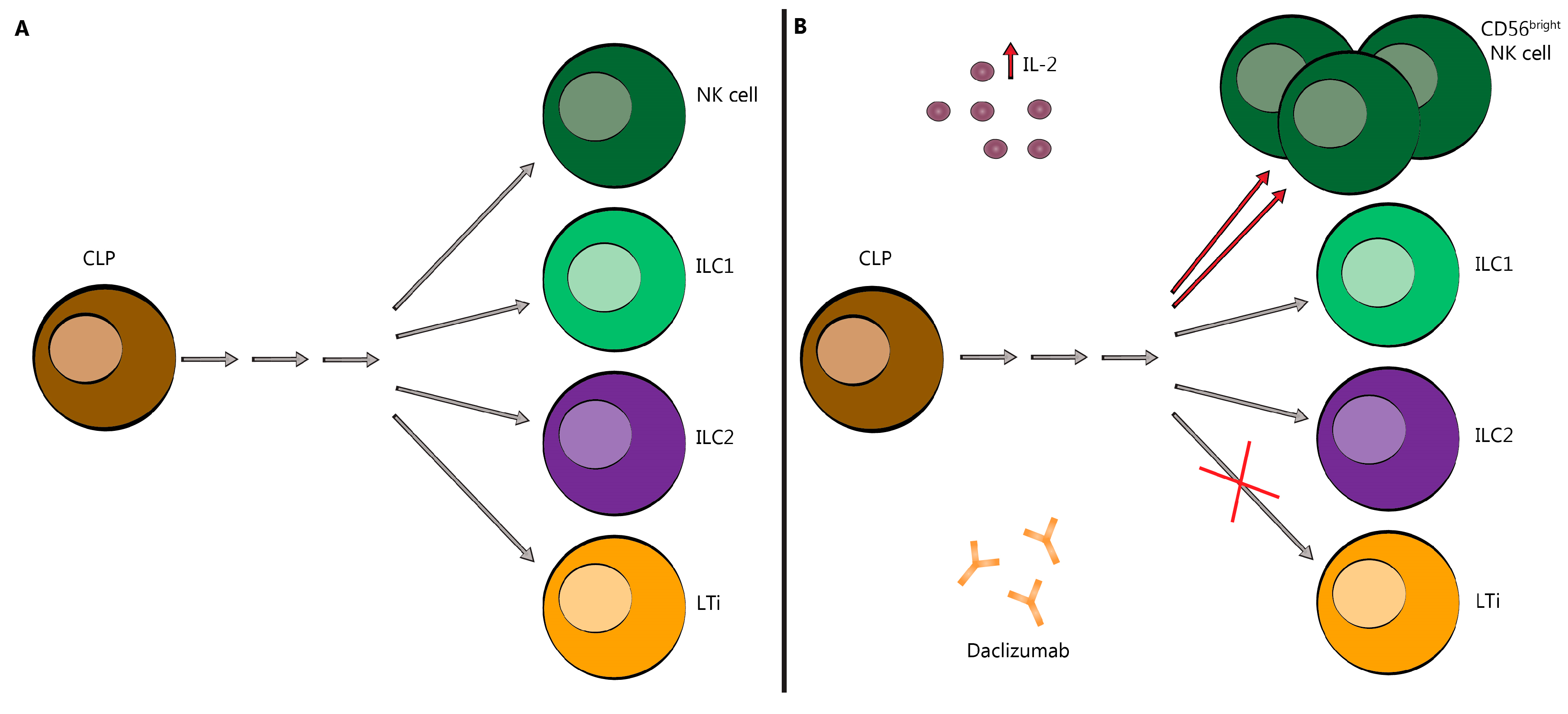
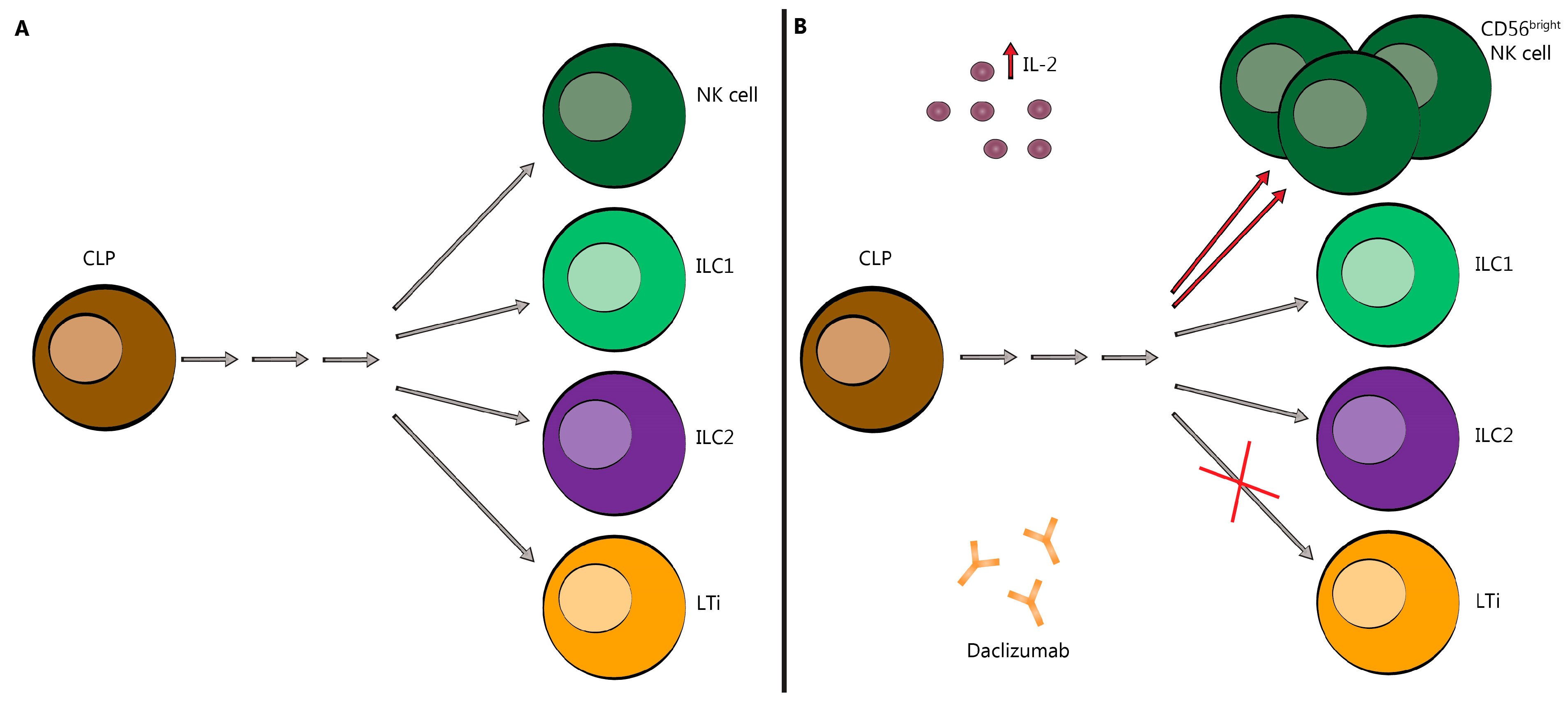
2.2. Innate Lymphoid Cells (ILC)
3. DAC Mechanisms of Action: Known and Proposed
4. Pharmacokinetics and Pharmacodynamics of DAC HYP
5. DAC Efficacy: Controlled Clinical Trial Data
5.1. SELECT
5.2. SELECTION
5.3. DECIDE
5.4. SELECTED
5.5. Patient-Reported Outcomes
6. Adverse Events with DAC: Prospective Clinical Trial Data
6.1. SELECT
6.2. SELECTION
6.3. DECIDE
6.4. SELECTED
7. Adverse Events: DAC HYP Post-Marketing
7.1. Cutaneous Events
7.2. Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS Syndrome)
7.3. Glial Fibrillary Acidic Protein (GFAP)-α Immunoglobulin (IgG)-Associated Encephalitis and Other Severe Encephalopathy Syndromes
7.4. DAC: Is There a Future?
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nussenblatt, R.B.; Fortin, E.; Schiffman, R.; Rizzo, L.; Smith, J.; Van Veldhuisen, P.; Sran, P.; Yaffe, A.; Goldman, C.K.; Waldmann, T.A.; et al. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: A phase I/II clinical trial. Proc. Natl. Acad. Sci. USA 1999, 96, 7462–7466. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Goldman, C.K.; Bongiovanni, K.F.; Sharrow, S.O.; Davey, M.P.; Cease, K.B.; Greenberg, S.J.; Longo, D.L. Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2. Blood 1988, 72, 1805–1816. [Google Scholar] [PubMed]
- Vincenti, F.; Kirkman, R.; Light, S.; Bumgardner, G.; Pescovitz, M.; Halloran, P.; Neylan, J.; Wilkinson, A.; Ekberg, H.; Gaston, R.; et al. Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N. Engl. J. Med. 1998, 338, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Bumgardner, G.L.; Hardie, I.; Johnson, R.W.; Lin, A.; Nashan, B.; Pescovitz, M.D.; Ramos, E.; Vincenti, F. Phase III Daclizumab Study Group. Results of 3-year phase III clinical trials with daclizumab prophylaxis for prevention of acute rejection after renal transplantation. Transplantation 2001, 72, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Wynn, D.; Kaufman, M.; Montalban, X.; Vollmer, T.; Simon, J.; Elkins, J.; O’Neill, G.; Neyer, L.; Sheridan, J.; Wang, C.; et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): A phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010, 9, 381–390. [Google Scholar] [CrossRef]
- Kappos, L.; Wiendl, H.; Selmaj, K.; Arnold, D.L.; Havrdova, E.; Boyko, A.; Kaufman, M.; Rose, J.; Greenberg, S.; Sweetser, M.; et al. Daclizumab HYP versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2015, 373, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Giovannoni, G.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; Robinson, R.; Riester, K.; Rana, J.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): A randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 2167–2175. [Google Scholar] [CrossRef]
- Giovannoni, G.; Gold, R.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; McNeill, M.; Amaravadi, L.; Sweetser, M.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): A multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014, 13, 472–481. [Google Scholar] [CrossRef]
- Lenardo, M.J. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 1991, 353, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Baan, C.C.; Balk, A.H.; van Riemsdijk, I.C.; Vantrimpont, P.J.; Maat, A.P.; Niesters, H.G.; Zondervan, P.E.; van Gelder, T.; Weimar, W. Anti-CD25 monoclonal antibody therapy affects the death signals of graft-infiltrating cells after clinical heart transplantation. Transplantation 2003, 75, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B. Daclizumab therapy for multiple sclerosis. Neurotherapeutics 2013, 10, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.H.; Mollweide, A.; Hanewinkel, K.; Zobywalski, C.; Burdach, S. CD25 blockade protects T cells from activation-induced cell death (AICD) via maintenance of TOSO expression. Scand. J. Immunol. 2009, 70, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.C.; Murakami, M.; Sakamoto, A.; Kappler, J.; Marrack, P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science 2000, 288, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Lenardo, M.; Chan, K.M.; Hornung, F.; McFarland, H.; Siegel, R.; Wang, J.; Zheng, L. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 1999, 17, 221–253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Demirci, G.; Ferrari-Lacraz, S.; Groves, C.; Coyle, A.; Malek, T.R.; Strom, T.B. IL-15 and IL-2: A matter of life and death for T cells in vivo. Nat. Med. 2001, 7, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Feau, S.; Angeli, V.; Trottein, F.; Ricciardi-Castagnoli, P. Early IL-2 production by mouse dendritic cells is the result of microbial-induced priming. J. Immunol. 2003, 170, 5075–5081. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. The multi-subunit interleukin-2 receptor. Annu. Rev. Biochem. 1989, 58, 875–911. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Wuest, S.C.; Edwan, J.H.; Martin, J.F.; Han, S.; Perry, J.S.; Cartagena, C.M.; Matsuura, E.; Maric, D.; Waldmann, T.A.; Bielekova, B. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat. Med. 2011, 17, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Grant, A.; Ju, G.; Tsudo, M.; Sugamura, K.; Waldmann, T.A. Cooperative interactions between the interleukin 2 receptor alpha and beta chains alter the interleukin 2-binding affinity of the receptor subunits. Proc. Natl. Acad. Sci. USA 1994, 91, 3344–3347. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Asao, H.; Ohtani, K.; Ishii, N.; Kumaki, S.; Tanaka, N.; Munakata, H.; Nakamura, M.; Sugamura, K. Cloning of the gamma chain of the human IL-2 receptor. Science 1992, 257, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Signaling domains of the interleukin 2 receptor. Cytokine 2001, 14, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Duprez, V.; Cornet, V.; Dautry-Varsat, A. Down-regulation of high affinity interleukin 2 receptors in a human tumor T cell line. Interleukin 2 increases the rate of surface receptor decay. J. Biol. Chem. 1988, 263, 12860–12865. [Google Scholar] [PubMed]
- Fujii, M.; Sugamura, K.; Nakai, S.; Tanaka, Y.; Tozawa, H.; Hinuma, Y. High- and low-affinity interleukin 2 receptors: Distinctive effects of monoclonal antibodies. J. Immunol. 1986, 137, 1552–1556. [Google Scholar] [PubMed]
- Morelon, E.; Dautry-Varsat, A. Endocytosis of the common cytokine receptor gammac chain. Identification of sequences involved in internalization and degradation. J. Biol. Chem. 1998, 273, 22044–22051. [Google Scholar] [CrossRef] [PubMed]
- Hemar, A.; Subtil, A.; Lieb, M.; Morelon, E.; Hellio, R.; Dautry-Varsat, A. Endocytosis of interleukin 2 receptors in human T lymphocytes: Distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J. Cell Biol. 1995, 129, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Yu, A.; Scibelli, P.; Lichtenheld, M.G.; Codias, E.K. Broad programming by IL-2 receptor signaling for extended growth to multiple cytokines and functional maturation of antigen-activated T cells. J. Immunol. 2001, 166, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Becknell, B.; Roychowdhury, S.; Mao, H.C.; Ferketich, A.K.; Nuovo, G.J.; Hughes, T.L.; Marburger, T.B.; Sung, J.; Baiocchi, R.A.; et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity 2005, 22, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Cupedo, T. Innate lymphoid cells: Emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 2012, 30, 647–675. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Di Santo, J.P. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2011, 12, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Seillet, C.; Belz, G.T. Shaping Innate Lymphoid Cell Diversity. Front. Immunol. 2017, 8, 1569. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Catalfamo, M.; Reichert-Scrivner, S.; Packer, A.; Cerna, M.; Waldmann, T.A.; McFarland, H.; Henkart, P.A.; Martin, R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5941–5946. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Winokur, P.; Blake, A.; Wu, T.; Romm, E.; Bielekova, B. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann. Clin. Transl. Neurol. 2015, 2, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.S.; Han, S.; Xu, Q.; Herman, M.L.; Kennedy, L.B.; Csako, G.; Bielekova, B. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci. Transl. Med. 2012, 4, 145ra106. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Perry, J.S.; Jakhete, N.R.; Wang, X.; Bielekova, B. An IL-2 paradox: Blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells. J. Immunol. 2010, 185, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Fazekas, G.; Hara, H.; Tabira, T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 163, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Benczur, M.; Petranyl, G.G.; Palffy, G.; Varga, M.; Talas, M.; Kotsy, B.; Foldes, I.; Hollan, S.R. Dysfunction of natural killer cells in multiple sclerosis: A possible pathogenetic factor. Clin. Exp. Immunol. 1980, 39, 657–662. [Google Scholar] [PubMed]
- Merrill, J.; Jondal, M.; Seeley, J.; Ullberg, M.; Siden, A. Decreased NK killing in patients with multiple sclerosis: An analysis on the level of the single effector cell in peripheral blood and cerebrospinal fluid in relation to the activity in the disease. Clin. Exp. Immunol. 1982, 47, 419–430. [Google Scholar] [PubMed]
- Neighbour, P.A.; Grayzel, A.I.; Miller, A.E. Endogenous and interferon-augmented natural killer cell activity of human peripheral blood mononuclear cells in vitro. Studies of patients with multiple sclerosis, systemic lupus erythematosus or rheumatoid arthritis. Clin. Exp. Immunol. 1982, 49, 11–21. [Google Scholar] [PubMed]
- Oger, J.; Kastrukoff, L.F.; Li, D.K.; Paty, D.W. Multiple sclerosis: In relapsing patients, immune functions vary with disease activity as assessed by MRI. Neurology 1988, 38, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Howard, T.; Packer, A.N.; Richert, N.; Blevins, G.; Ohayon, J.; Waldmann, T.A.; McFarland, H.F.; Martin, R. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch. Neurol. 2009, 66, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.; Sheridan, J.; Amaravadi, L.; Riester, K.; Selmaj, K.; Bielekova, B.; Parr, E.; Giovannoni, G. CD56(bright) natural killer cells and response to daclizumab HYP in relapsing-remitting MS. Neurol. NeuroImmunol. Neuroinflamm. 2015, 2, e65. [Google Scholar] [CrossRef] [PubMed]
- Oh, U.; Blevins, G.; Griffith, C.; Richert, N.; Maric, D.; Lee, C.R.; McFarland, H.; Jacobson, S. Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis. Arch. Neurol. 2009, 66, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Huss, D.J.; Mehta, D.S.; Sharma, A.; You, X.; Riester, K.A.; Sheridan, J.P.; Amaravadi, L.S.; Elkins, J.S.; Fontenot, J.D. In vivo maintenance of human regulatory T cells during CD25 blockade. J. Immunol. 2015, 194, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Hang, Y.; Othman, A.A.; Mehta, D.; Amaravadi, L.; Nestorov, I.; Tran, J.Q. Population PK-PD analyses of CD25 occupancy, CD56(bright) NK cell expansion, and regulatory T cell reduction by daclizumab HYP in subjects with multiple sclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J., Jr.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 2012, 4, 134ra162. [Google Scholar] [CrossRef] [PubMed]
- Roifman, C.M. Human IL-2 receptor alpha chain deficiency. Pediatr. Res. 2000, 48, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Sharfe, N.; Dadi, H.K.; Shahar, M.; Roifman, C.M. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 3168–3171. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Bluman, E.M.; Porter, M.M.; Mrozek, E.; Cooper, M.A.; VanDeusen, J.B.; Frankel, S.R.; Stock, W.; Caligiuri, M.A. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J. Clin. Investig. 2000, 106, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Zhang, Y.; Riester, K.; Tang, M.T.; Efros, L.; Shi, J.; Harris, J.; Vexler, V.; Elkins, J.S. Intermediate-affinity interleukin-2 receptor expression predicts CD56(bright) natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult. Scler. 2011, 17, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Gillard, G.O.; Saenz, S.A.; Huss, D.J.; Fontenot, J.D. Circulating innate lymphoid cells are unchanged in response to DAC HYP therapy. J. NeuroImmunol. 2016, 294, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Sawa, S.; Cherrier, M.; Lochner, M.; Satoh-Takayama, N.; Fehling, H.J.; Langa, F.; Di Santo, J.P.; Eberl, G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science 2010, 330, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Mnasria, K.; Lagaraine, C.; Velge-Roussel, F.; Oueslati, R.; Lebranchu, Y.; Baron, C. Anti-CD25 antibodies affect cytokine synthesis pattern of human dendritic cells and decrease their ability to prime allogeneic CD4+ T cells. J. Leukoc. Biol. 2008, 84, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.A.; Tran, J.Q.; Tang, M.T.; Dutta, S. Population pharmacokinetics of daclizumab high-yield process in healthy volunteers: Integrated analysis of intravenous and subcutaneous, single- and multiple-dose administration. Clin. Pharm. 2014, 53, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.Q.; Othman, A.A.; Mikulskis, A.; Wolstencroft, P.; Elkins, J. Pharmacokinetics of daclizumab high-yield process with repeated administration of the clinical subcutaneous regimen in patients with relapsing-remitting multiple sclerosis. Clin. Pharmacol. 2016, 8, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; McClellan, M.; Efros, L.; Shi, D.; Bielekova, B.; Tang, M.T.; Vexler, V.; Sheridan, J.P. Daclizumab reduces CD25 levels on T cells through monocyte-mediated trogocytosis. Mult. Scler. 2014, 20, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Soelberg Sorensen, P. Safety concerns and risk management of multiple sclerosis therapies. Acta Neurol. Scand. 2017, 136, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Richert, N.; Howard, T.; Blevins, G.; Markovic-Plese, S.; McCartin, J.; Frank, J.A.; Wurfel, J.; Ohayon, J.; Waldmann, T.A.; et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc. Natl. Acad. Sci. USA 2004, 101, 8705–8708. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.W.; Burns, J.B.; Bjorklund, J.; Klein, J.; Watt, H.E.; Carlson, N.G. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology 2007, 69, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.; Cohan, S.; Lynch, S.G.; Riester, K.; Wang, P.; Castro-Borrero, W.; Elkins, J.; Sabatella, G. Improved cognitive outcomes in patients with relapsing-remitting multiple sclerosis treated with daclizumab beta: Results from the DECIDE study. Mult. Scler. 2018, 24, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Radue, E.W.; Giovannoni, G.; Selmaj, K.; Havrdova, E.; Stefoski, D.; Sprenger, T.; Montalban, X.; Cohan, S.; Umans, K.; et al. Safety and efficacy of daclizumab in relapsing-remitting multiple sclerosis: 3-year results from the SELECTED open-label extension study. BMC Neurol. 2016, 16, 117. [Google Scholar] [CrossRef] [PubMed]
- Cortese, I.; Ohayon, J.; Fenton, K.; Lee, C.C.; Raffeld, M.; Cowen, E.W.; DiGiovanna, J.J.; Bielekova, B. Cutaneous adverse events in multiple sclerosis patients treated with daclizumab. Neurology 2016, 86, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Zawodniak, A.; Lochmatter, P.; Yerly, D.; Kawabata, T.; Lerch, M.; Yawalkar, N.; Pichler, W.J. In vitro detection of cytotoxic T and NK cells in peripheral blood of patients with various drug-induced skin diseases. Allergy 2010, 65, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Porebski, G. In Vitro Assays in Severe Cutaneous Adverse Drug Reactions: Are They Still Research Tools or Diagnostic Tests Already? Int. J. Mol. Sci. 2017, 18, 1737. [Google Scholar] [CrossRef] [PubMed]
- Peter, J.G.; Lehloenya, R.; Dlamini, S.; Risma, K.; White, K.D.; Konvinse, K.C.; Phillips, E.J. Severe Delayed Cutaneous and Systemic Reactions to Drugs: A Global Perspective on the Science and Art of Current Practice. J. Allergy Clin. Immunol. Pract. 2017, 5, 547–563. [Google Scholar] [CrossRef]
- Scheibe, F.; Metz, I.; Radbruch, H.; Siebert, E.; Wolf, S.; Kohnlein, M.; Harms, L.; Meisel, A. Drug reaction with eosinophilia and systemic symptoms after daclizumab therapy in MS. Neurol. NeuroImmunol. Neuroinflamm. 2018, 5, e479. [Google Scholar] [CrossRef] [PubMed]
- Avasarala, J. DRESS Syndrome and Daclizumab Failure-Were Potentially Dangerous Signs Missed in Clinical Trials? Drug Target. Insights 2018, 12, 1177392818785136. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, F.; Molofsky, A.B.; Morar, M.M.; Rosenzwajg, M.; Liang, H.E.; Klatzmann, D.; Locksley, R.M.; Bluestone, J.A. Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood 2014, 124, 3572–3576. [Google Scholar] [CrossRef] [PubMed]
- Luessi, F.; Engel, S.; Spreer, A.; Bittner, S.; Zipp, F. GFAPalpha IgG-associated encephalitis upon daclizumab treatment of MS. Neurol. NeuroImmunol. Neuroinflamm. 2018, 5, e481. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Alemtuzumab vs. Interferon Beta-1a in Early Multiple Sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bates, D.; Hartung, H.P.; Havrdova, E.; Miller, D.; Polman, C.H.; Ravnborg, M.; Hauser, S.L.; Rudick, R.A.; Weiner, H.L.; et al. Natalizumab treatment for multiple sclerosis: Recommendations for patient selection and monitoring. Lancet Neurol. 2007, 6, 431–441. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Hoepner, R.; Kleiter, I. Fingolimod for multiple sclerosis and emerging indications: Appropriate patient selection, safety precautions, and special considerations. Ther. Clin. Risk Manag. 2016, 12, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Cossburn, M.; Pace, A.A.; Jones, J.; Ali, R.; Ingram, G.; Baker, K.; Hirst, C.; Zajicek, J.; Scolding, N.; Boggild, M.; et al. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology 2011, 77, 573–579. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Clinical Trials | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CHOICE | SELECT | SELECTION | DECIDE | |||||||
| DAC 1 | pl | 150 mg | 300 mg | pl | Continuous 2 | Switch 3 | Washout 4 | DAC | IFNβ | |
| Parameters | ||||||||||
| ARR | 0.27 | 0.41 | 0.21 | 0.23 | 0.46 | 0.165 | 0.179 **** | 0.302 | 0.22 | 0.39 |
| Risk Reduction, % | 34 | 54 **** | 50 *** | 45 *** | ||||||
| PPRF, % | 52 | 55 | 81 **** | 80 *** | 64 | 86.4 | 82.4 **** | 75.9 | 67 | 51 |
| CDP, % | ||||||||||
| 12 weeks | NR | NR | 6* | 8 | 13 | 7 | 8 **** | 10 | 16 | 20 |
| 24 weeks | NR | NR | NR | NR | NR | NR | NR | NR | 13 * | 18 |
| New Gd+ | 1.32 ** | 4.75 | 0.3 **** | 0.2 **** | 1.4 | 0.2 | 0.2 **** | 0.2 | 0.4 *** | 1.0 |
| Reduction, % | 72 | 79 | 86 | NR | NR | NR | 60 | |||
| T2 Lesions | ||||||||||
| New/enlarging | 1.1 ** | 3.4 | 2.4 **** | 1.7 **** | 8.1 | 1.2 | 2.1 **** | 3.3 | 4.3 | 9.4 |
| Reduction, % | 68 | 70 | 79 | 54 *** | ||||||
| Volume change, % | ND | −11.1 **** | −12.5 **** | −27.3 | −6.9 | −8.1 **** | −3.1 | 0.2 *** | 8.6 | |
| Brain Volume, % change | NR | NR | −0.79 | −0.70 | −0.74 | −0.536 | −0.830 | −0.551 | −0.559 *** | −0.585 *** |
| NEDA% | NR | NR | NR | NR | NR | NR | NR | NR | 13 *** | 22 |
| Clinical Trials | |||||||
|---|---|---|---|---|---|---|---|
| SELECT | SELECTION | DECIDE | SELECTED | ||||
| Continuous 1 | Switch 2 | Washout 3 | |||||
| DAC Dose | 150 mg | 300 mg | 150 mg, 300 mg | 150 mg, 300 mg | 150 mg, 300 mg | 150 mg | 150 mg |
| AE | |||||||
| Infection (%) | 104 (50) | 112 (54) | 36 (42),36 (41) | 34 (40), 31 (37) | 34 (40), 38 (43) | 595 (65) | 205 (50) |
| Serious Infection (%) | 6 (3) | 3 (1) | 2 (2), 2 (2) | 3 (3), 1 (1) | 3 (3), 2 (2) | 40 (4) | 13 (3) |
| Hepatic TA (%) | NR | NR | NR | NR | NR | 144 (16) | 61 (15) |
| AST/ALT: | |||||||
| 1–3× ULN (%) | 54 (26) | 62 (30) | 30 (35), 30 (34) | 23 (27), 22 (26) | 21 (24), 26 (30) | NR | NR |
| 3–5× ULN (%) | 7 (3) | 6 (3) | 1 (1), 5 (6) | 0, 2 (2) | 2 (2), 0 | 96 (10) | 37 (9) |
| >5× ULN (%) | 9 (4) | 8 (4) | 0, 3 (3) | 1 (1), 1 (1) | 2 (2), 4 (5) | 59 (6) | 18 (4) |
| Hepatic SAE (%) | NR | NR | 0, 0 | 0, 0 | 0, 1 (<1) | 6 (1) | 5 (1) |
| Malignancy (%) | 1 (<1) | 2 (<1) | 0, 0 | 0, 1 (1) | 0, 0 | 7 (1) | 4 (1) |
| Death (%) | 1 (<1) | 0 | 0, 0 | 0, 0 | 0, 1 (<1) | 1 (<1) | 0 |
| Clinical Trials | ||||||||
|---|---|---|---|---|---|---|---|---|
| SELECT | SELECTION | DECIDE | SELECTED | Post | ||||
| Continuous 1 | Switch 2 | Washout 3 | Approval 4 | |||||
| DAC Dose | 150 mg | 300 mg | 150 mg, 300 mg | 150 mg, 300 mg | 150 mg, 300 mg | 150 mg | 150 mg | |
| Cutaneous Events | ||||||||
| AE (%) | 38 (18) | 45 (22) | 15 (17), 21 (24) | 17 (20), 11 (13) | 19 (22), 16 (18) | 344 (37) | 114 (28) | 23 (77) |
| SAE (%) | 2 (<1) | 3 (<1) | 0, 3 (3) | 2 (2), 0 | 1 (1), 0 | 14 (2) | 8 (2) | 6 (19) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohan, S.L.; Lucassen, E.B.; Romba, M.C.; Linch, S.N. Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines 2019, 7, 18. https://doi.org/10.3390/biomedicines7010018
Cohan SL, Lucassen EB, Romba MC, Linch SN. Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines. 2019; 7(1):18. https://doi.org/10.3390/biomedicines7010018
Chicago/Turabian StyleCohan, Stanley L., Elisabeth B. Lucassen, Meghan C. Romba, and Stefanie N. Linch. 2019. "Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis" Biomedicines 7, no. 1: 18. https://doi.org/10.3390/biomedicines7010018
APA StyleCohan, S. L., Lucassen, E. B., Romba, M. C., & Linch, S. N. (2019). Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines, 7(1), 18. https://doi.org/10.3390/biomedicines7010018