Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Important Elements of Peptide-Based PPI and PDI Inhibitors
2.1. Incorporation of Non-Protein Amino Acids
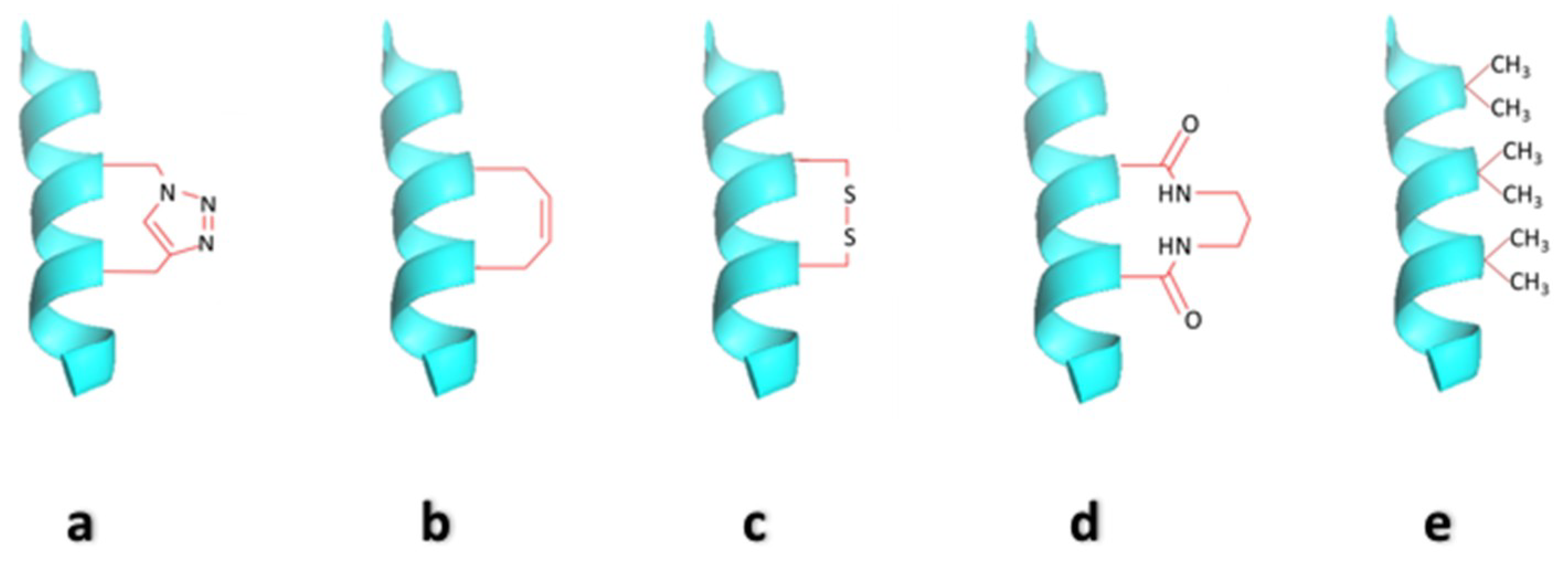
2.2. Side-Chain Cross-Linked α-Helices
2.3. Hydrocarbon Stapled Helices
2.4. Cysteine Bis-Alkylation for Helix Stabilization
2.5. Hydrogen Bond Surrogate Derived α-Helices
2.6. β Peptides
3. Constrained-Helix-Based PPI Inhibitors
3.1. BCL-2 Family Proteins
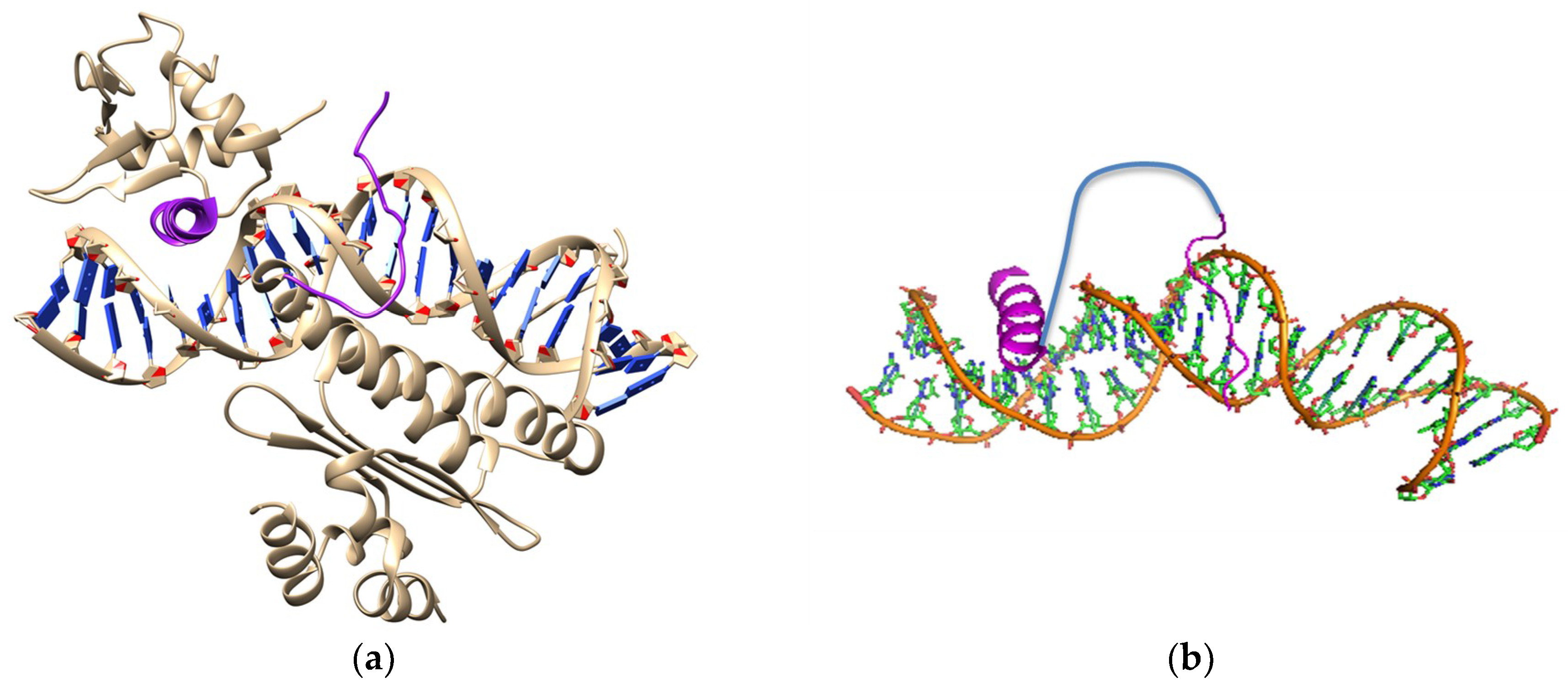
3.2. Inhibition of p53 Binding with MDM2/MDMX
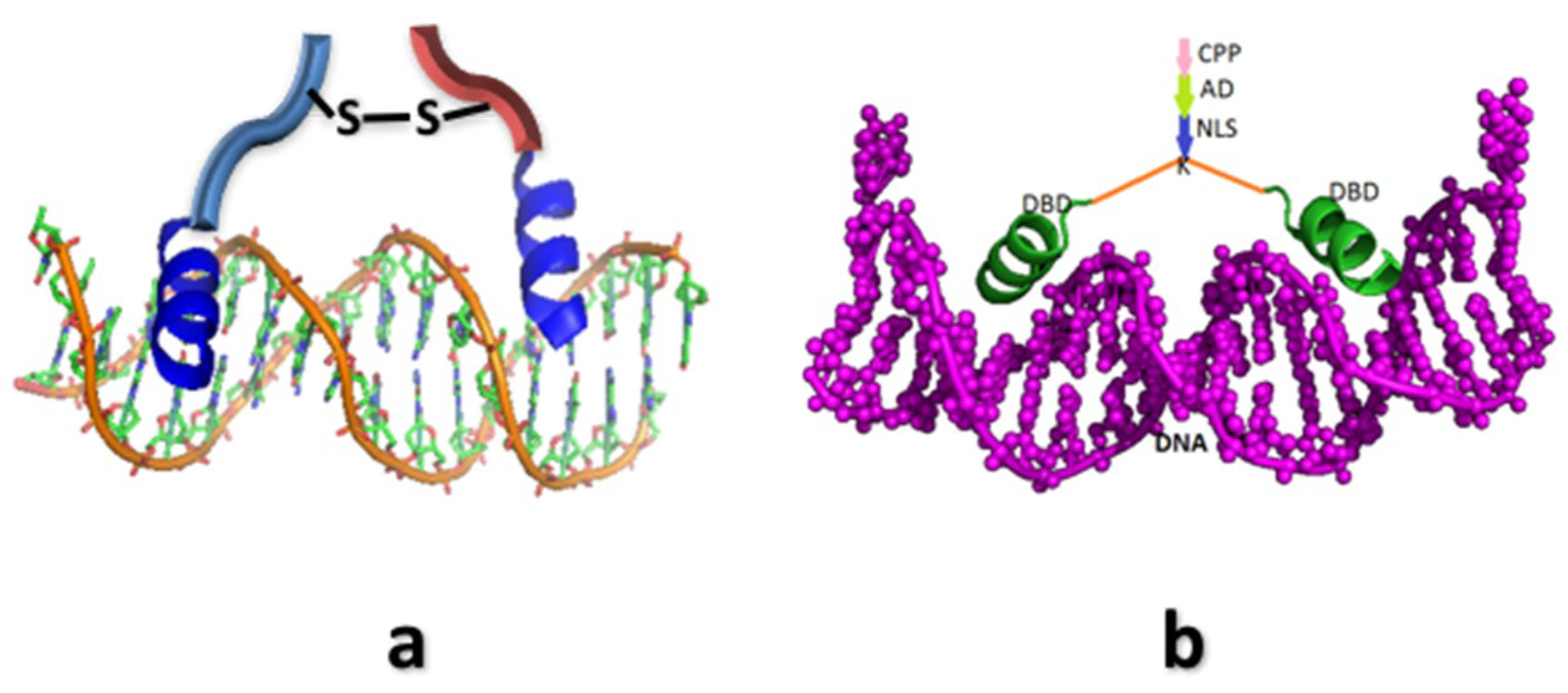
3.3. c-FOS and c-JUN
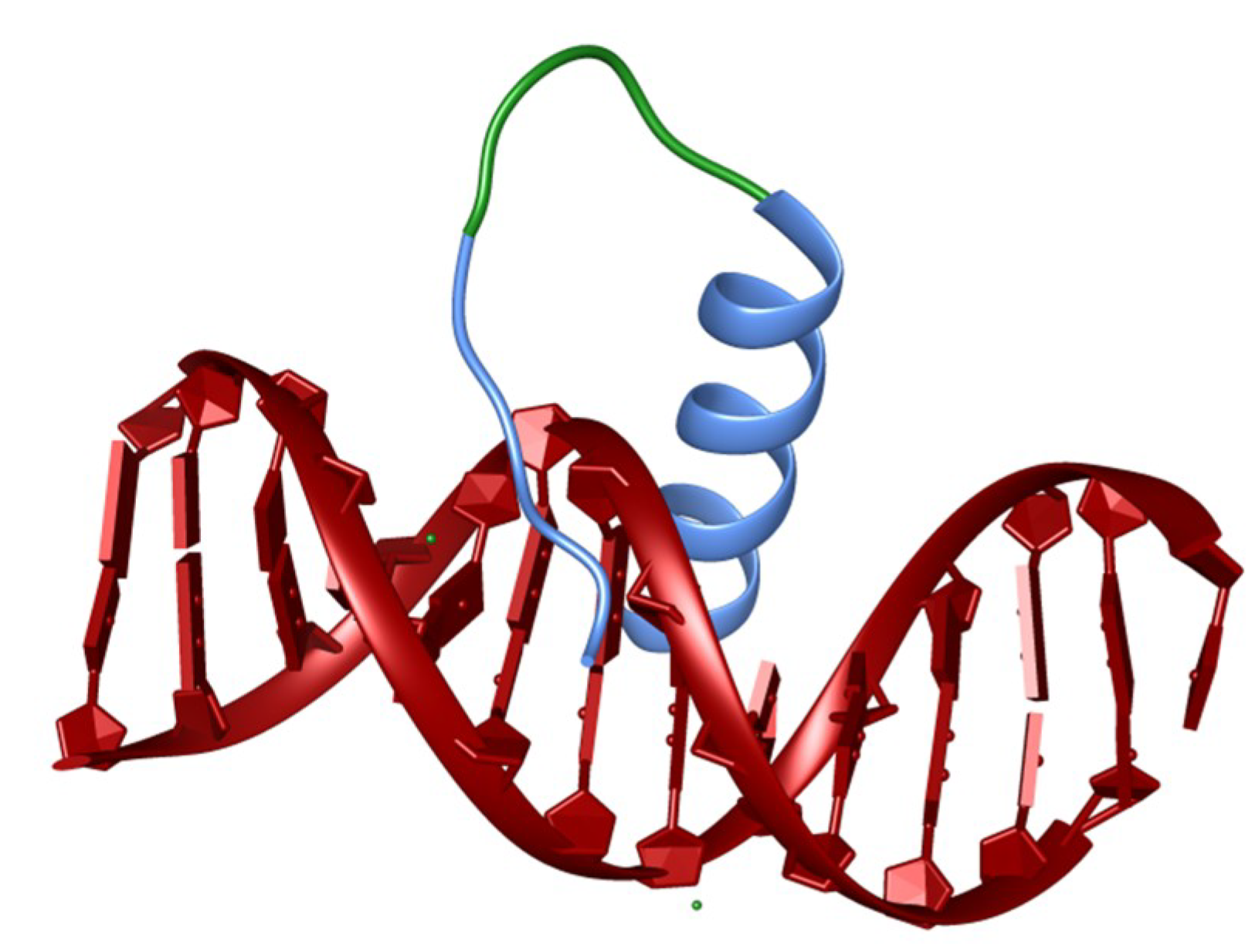
4. Synthetic Transcription Factors from Helical Peptides
5. Strategies for Targeting of Constrained Helical Peptides
5.1. Cell Penetration
5.2. Plasma Stability
5.3. Pharmacokinetics
5.4. Immunogenicity
6. Conclusions
Funding
Conflicts of Interest
References
- Araghi, R.R.; Keating, A.E. Designing helical peptide inhibitors of protein–protein interactions. Curr. Opin. Struct. Biol. 2016, 39, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Badyal, S.K.; Basran, J.; Bhanji, N.; Kim, J.H.; Chavda, A.P.; Jung, H.S.; Craig, R.; Elliott, P.R.; Irvine, A.F.; Barsukov, I.L. Mechanism of the Ca2+-dependent interaction between s100a4 and tail fragments of nonmuscle myosin heavy chain IIA. J. Mol. Biol. 2011, 405, 1004–1026. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the chromosomal breakpoint of t (14; 18) human lymphomas: Clustering around jh on chromosome 14 and near a transcriptional unit on 18. Cell 1985, 41, 899–906. [Google Scholar] [CrossRef]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein-DNA complexes. Genome Biol. 2000, 1. [Google Scholar] [CrossRef] [PubMed]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.-H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMx for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Marqusee, S.; Baldwin, R.L. Helix stabilization by glu-… Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. USA 1987, 84, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Scholtz, J.M.; Qian, H.; Robbins, V.H.; Baldwin, R.L. The energetics of ion-pair and hydrogen-bonding interactions in a helical peptide. Biochemistry 1993, 32, 9668–9676. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Roubini, E.; McKee, R.L.; Gibbons, S.W.; Goldman, M.E.; Caulfield, M.P.; Rosenblatt, M. Cyclic parathyroid hormone-related protein antagonists: Lysine 13 to aspartic acid 17 [i to (i + 4)] side chain to side chain lactamization. Biochemistry 1991, 30, 5968–5974. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.C.; Skelton, N.J.; Braisted, A.C.; McDowell, R.S. A general method for constraining short peptides to an α-helical conformation. J. Am. Chem. Soc. 1997, 119, 455–460. [Google Scholar] [CrossRef]
- Jackson, D.Y.; King, D.S.; Chmielewski, J.; Singh, S.; Schultz, P.G. General approach to the synthesis of short. α-helical peptides. J. Am. Chem. Soc. 1991, 113, 9391–9392. [Google Scholar] [CrossRef]
- Chapman, R.N.; Dimartino, G.; Arora, P.S. A highly stable short α-helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc. 2004, 126, 12252–12253. [Google Scholar] [CrossRef] [PubMed]
- Patgiri, A.; Jochim, A.L.; Arora, P.S. A hydrogen bond surrogate approach for stabilization of short peptide sequences in α-helical conformation. Acc. Chem. Res. 2008, 41, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.S.; Hamilton, A.D. Stabilization of helical domains in short peptides using hydrophobic interactions. Biochemistry 1995, 34, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.R.; Choi, C. Secondary structure nucleation in peptides. Transition metal ion stabilized. α-helices. J. Am. Chem. Soc. 1990, 112, 1630–1632. [Google Scholar] [CrossRef]
- Ruan, F.; Chen, Y.; Hopkins, P.B. Metal ion-enhanced helicity in synthetic peptides containing unnatural, metal-ligating residues. J. Am. Chem. Soc. 1990, 112, 9403–9404. [Google Scholar] [CrossRef]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.-Y.; Wang, S. Design of triazole-stapled bcl9 α-helical peptides to target the β-catenin/b-cell cll/lymphoma 9 (bcl9) protein–protein interaction. J. Med. Chem. 2012, 55, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Kneissl, S.; Loveridge, E.J.; Williams, C.; Crump, M.P.; Allemann, R.K. Photocontrollable peptide-based switches target the anti-apoptotic protein bcl-xl. ChemBioChem 2008, 9, 3046–3054. [Google Scholar] [CrossRef] [PubMed]
- Balaram, P. Non-standard amino acids in peptide design and protein engineering. Curr. Opin. Struct. Biol. 1992, 2, 845–851. [Google Scholar] [CrossRef]
- Karle, I.L.; Balaram, P. Structural characteristics of. α-helical peptide molecules containing aib residues. Biochemistry 1990, 29, 6747–6756. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Cochran, A.G. Antagonists of protein–protein interactions. Chem. Biol. 2000, 7, R85–R94. [Google Scholar] [CrossRef]
- Zutshi, R.; Brickner, M.; Chmielewski, J. Inhibiting the assembly of protein—Protein interfaces. Curr. Opin. Chem. Biol. 1998, 2, 62–66. [Google Scholar] [CrossRef]
- Banerjee, R.; Basu, G.; Chene, P.; Roy, S. Aib-based peptide backbone as scaffolds for helical peptide mimics. J. Pept. Res. 2002, 60, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Judice, J.K.; Tom, J.Y.; Huang, W.; Wrin, T.; Vennari, J.; Petropoulos, C.J.; McDowell, R.S. Inhibition of hiv type 1 infectivity by constrained α-helical peptides: Implications for the viral fusion mechanism. Proc. Natl. Acad. Sci. USA 1997, 94, 13426–13430. [Google Scholar] [CrossRef] [PubMed]
- Sia, S.K.; Carr, P.A.; Cochran, A.G.; Malashkevich, V.N.; Kim, P.S. Short constrained peptides that inhibit hiv-1 entry. Proc. Natl. Acad. Sci. USA 2002, 99, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Werder, M.; Hauser, H.; Abele, S.; Seebach, D. B-peptides as inhibitors of small-intestinal cholesterol and fat absorption. Helv. Chim. Acta 1999, 82, 1774–1783. [Google Scholar] [CrossRef]
- Cummings, C.G.; Hamilton, A.D. Disrupting protein–protein interactions with non-peptidic, small molecule α-helix mimetics. Curr. Opin. Chem. Biol. 2010, 14, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Orner, B.P.; Ernst, J.T.; Hamilton, A.D. Toward proteomimetics: Terphenyl derivatives as structural and functional mimics of extended regions of an α-helix. J. Am. Chem. Soc. 2001, 123, 5382–5383. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.; Robertson, N.S. Regulation of protein–protein interactions using stapled peptides. Rep. Org. Chem. 2018, 5, 65–74. [Google Scholar] [CrossRef]
- Mahalakshmi, R.; Balaram, P. Non-protein amino acids in the design of secondary structure scaffolds. In Protein Design; Springer: Berlin, Germany, 2006; pp. 71–94. [Google Scholar]
- Toniolo, C.; Crisma, M.; Bonora, G.M.; Benedetti, E.; Dl Blasio, B.; Pavone, V.; Pedone, C.; Santini, A. Preferred conformation of the terminally blocked (aib) 10 homo-oligopeptide: A long, regular 310-helix. Biopolymers 1991, 31, 129–138. [Google Scholar] [CrossRef]
- Toniolo, C.; Crisma, M.; Formaggio, F.; Valle, C.; Cavicchioni, G.; Précigoux, G.; Aubry, A.; Kamphuis, J. Structures of peptides from α-amino acids methylated at the α-carbon. Biopolymers 1993, 33, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Ghosh, P.; Sekhar Roy, N.; Mazumder, A.; Roy, K.; Kumar Manna, A.; Mallick, S.; Ahmed, I. Peptide based molecules as protein-protein interaction inhibitors: Tools for chemical genetics and therapy. Curr. Chem. Biol. 2012, 6, 145–163. [Google Scholar] [CrossRef]
- Henchey, L.K.; Jochim, A.L.; Arora, P.S. Contemporary strategies for the stabilization of peptides in the α-helical conformation. Curr. Opin. Chem. Biol. 2008, 12, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.R.; Fernholz, A.K. Peptide architecture. Design of stable. α-helical metallopeptides via a novel exchange-inert ruthenium (III) complex. J. Am. Chem. Soc. 1990, 112, 9633–9635. [Google Scholar] [CrossRef]
- Taylor, J.W. The synthesis and study of side-chain lactam-bridged peptides. Pept. Sci. Orig. Res. Biomol. 2002, 66, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, N.E.; Hoang, H.N.; Desai, V.S.; Letouze, E.; Young, P.R.; Fairlie, D.P. Modular α-helical mimetics with antiviral activity against respiratory syncitial virus. J. Am. Chem. Soc. 2006, 128, 13284–13289. [Google Scholar] [CrossRef] [PubMed]
- Mills, N.L.; Daugherty, M.D.; Frankel, A.D.; Guy, R.K. An α-helical peptidomimetic inhibitor of the hiv-1 rev–rre interaction. J. Am. Chem. Soc. 2006, 128, 3496–3497. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled bh3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Q.; Bhattacharya, S.; Waheed, A.A.; Tong, X.; Hong, A.; Heck, S.; Curreli, F.; Goger, M.; Cowburn, D. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol. 2008, 378, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Kajino, M.; Inouye, M. Development of a series of cross-linking agents that effectively stabilize α-helical structures in various short peptides. Chem. Eur. J. 2008, 14, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Brunel, F.M.; Dawson, P.E. Synthesis of constrained helical peptides by thioether ligation: Application to analogs of GP41. Chem. Commun. 2005, 28, 2552–2554. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chou, D.H.C. A thiolene coupling approach to native peptide stapling and macrocyclization. Angew. Chem. 2015, 54, 10931–10934. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.; Zou, Y.; Ling, J.; Yu, H.; Lin, Y.S.; Pentelute, B. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Meinhardt, N.; Wu, Y.; Kulkarni, S.; Hu, X.; Low, K.E.; Davies, P.L.; DeGrado, W.F.; Greenbaum, D.C. Development of α-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 2005, 6, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Irimia, A.; Ofek, G.; Kwong, P.D.; Wilson, I.A.; Walensky, L.D. Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat. Struct. Mol. Biol. 2014, 21, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.E.; Gavenonis, J.; Kritzer, J.A. Getting in shape: Controlling peptide bio-activity and bioavailability using conformational constraints. ACS Chem. Biol. 2013, 8, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, N.E.; Hoang, H.N.; Abbenante, G.; Fairlie, D.P. Single turn peptide α helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127, 2974–2983. [Google Scholar] [CrossRef] [PubMed]
- Muppidi, A.; Wang, Z.; Li, X.; Chen, J.; Lin, Q. Achieving cell penetration with distance- matching cysteine cross-linkers: A facile route to cell-permeable peptide dual inhibitors of MDM2/MDMx. Chem. Commun. 2011, 47, 9396–9398. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Liu, T.; Liu, Y.Y.; Briesewitz, R.; Barrios, A.M.; Jhiang, S.M.; Pei, D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol. 2013, 8, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Peraro, L.; Siegert, T.R.; Kritzer, J.A. Conformational Restriction of Peptides Using Dithiol Bis-Alkylation. Methods Enzymol. 2016, 580, 303–332. [Google Scholar] [PubMed]
- Cabezas, E.; Satterthwait, A.C. The hydrogen bond mimic approach: Solid-phase synthesis of a peptide stabilized as an α-helix with a hydrazone link. J. Am. Chem. Soc. 1999, 121, 3862–3875. [Google Scholar] [CrossRef]
- Liu, J.; Wang, D.; Zheng, Q.; Lu, M.; Arora, P.S. Atomic structure of a short α-helix stabilized by a main chain hydrogen-bond surrogate. J. Am. Chem. Soc. 2008, 130, 4334–4337. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, M.; Arora, P.S. Inhibition of HIV-1 fusion by hydrogen-bond-surrogate-based α helices. Angew. Chem. Int. Ed. 2008, 47, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liao, W.; Arora, P.S. Enhanced metabolic stability and protein-binding properties of artificial α helices derived from a hydrogen-bond surrogate: Application to bcl-xl. Angew. Chem. Int. Ed. 2005, 117, 6683–6687. [Google Scholar] [CrossRef]
- Boersma, M.D.; Haase, H.S.; Peterson-Kaufman, K.J.; Lee, E.F.; Clarke, O.B.; Colman, P.M.; Smith, B.J.; Horne, W.S.; Fairlie, W.D.; Gellman, S.H. Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: Analogues of the bim bh3 domain. J. Am. Chem. Soc. 2011, 134, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Boersma, M.D.; Windsor, M.A.; Gellman, S.H. Sequence-based design of α/β-peptide foldamers that mimic bh3 domains. Angew. Chem. Int. Ed. 2008, 47, 2853–2856. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A. Stabilized helical peptides: A strategy to target protein–protein interactions. ACS Med. Chem. Lett. 2014, 5, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Appella, D.H.; Christianson, L.A.; Karle, I.L.; Powell, D.R.; Gellman, S.H. B-peptide foldamers: Robust helix formation in a new family of β-amino acid oligomers. J. Am. Chem. Soc. 1996, 118, 13071–13072. [Google Scholar] [CrossRef]
- Hart, S.A.; Bahadoor, A.B.; Matthews, E.E.; Qiu, X.J.; Schepartz, A. Helix macrodipole control of β3-peptide 14-helix stability in water. J. Am. Chem. Soc. 2003, 125, 4022–4023. [Google Scholar] [CrossRef] [PubMed]
- Raguse, T.L.; Lai, J.R.; Gellman, S.H. Environment-independent 14-helix formation in short β-peptides: Striking a balance between shape control and functional diversity. J. Am. Chem. Soc. 2003, 125, 5592–5593. [Google Scholar] [CrossRef] [PubMed]
- Gademann, K.; Kimmerlin, T.; Hoyer, D.; Seebach, D. Peptide folding induces high and selective affinity of a linear and small β-peptide to the human somatostatin receptor 4. J. Med. Chem. 2001, 44, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Lear, J.D.; Hodsdon, M.E.; Schepartz, A. Helical β-peptide inhibitors of the p53-hDM2 interaction. J. Am. Chem. Soc. 2004, 126, 9468–9469. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Luedtke, N.W.; Harker, E.A.; Schepartz, A. A rapid library screen for tailoring β-peptide structure and function. J. Am. Chem. Soc. 2005, 127, 14584–14585. [Google Scholar] [CrossRef] [PubMed]
- Harker, E.A.; Daniels, D.S.; Guarracino, D.A.; Schepartz, A. B-peptides with improved affinity for hDM2 and hDMx. Bioorgan. Med. Chem. 2009, 17, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by MDM2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Popowicz, G.M.; Czarna, A.; Rothweiler, U.; Szwagierczak, A.; Krajewski, M.; Weber, L.; Holak, T.A. Molecular basis for the inhibition of p53 by MDMx. Cell Cycle 2007, 6, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMx and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Müller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The n-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sakaguchi, K.; Shimohigashi, Y.; Samaddar, S.; Banerjee, R.; Basu, G.; Swaminathan, V.; Kundu, T.K.; Roy, S. Effect of phosphorylation on the structure and fold of transactivation domain of p53. J. Biol. Chem. 2002, 277, 15579–15585. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMx. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [PubMed]
- Thean, D.; Ebo, J.; Luxton, T.; Yuen, T.; Ferrer, F.; Johannes, C.; Lane, D.; Brown, C. Enhancing specific disruption of intracellular protein complexes by hydrocarbon stapled peptides using lipid based delivery. Sci. Rep. 2017, 7, 1763. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Ruiz-Gómez, G.; Hill, T.A.; Hoang, H.N.; Fairlie, D.P.; Mason, J.M. Truncated and helix-constrained peptides with high affinity and specificity for the cFos coiled-coil of AP-1. PLoS ONE 2013, 8, e59418. [Google Scholar] [CrossRef] [PubMed]
- Baxter, D.; Perry, S.R.; Hill, T.A.; Kok, W.M.; Zaccai, N.R.; Brady, R.L.; Fairlie, D.P.; Mason, J.M. Downsizing Proto-oncogene cFos to Short Helix-Constrained Peptides That Bind Jun. ACS Chem. Biol. 2017, 12, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.E.; Mapp, A.K. Modulating the masters: Chemical tools to dissect cbp and p300 function. Curr. Opin. Chem. Biol. 2018, 45, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Dervan, P.B.; Edelson, B.S. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol. 2003, 13, 284–299. [Google Scholar] [CrossRef]
- Urbach, A.R.; Dervan, P.B. Toward rules for 1:1 polyamide: DNA recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 4343–4348. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.B.; Kurmis, A.A.; Finn, P.B. Molecular recognition of DNA by py–im polyamides: From discovery to oncology. DNA-Target Mol. Ther. Agents 2018, 7, 298. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Ann. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Talanian, R.V.; McKnight, C.J.; Kim, P.S. Sequence-specific DNA binding by a short peptide dimer. Science 1990, 249, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Zondlo, N.J.; Schepartz, A. Highly specific DNA recognition by a designed miniature protein. J. Am. Chem. Soc. 1999, 121, 6938–6939. [Google Scholar] [CrossRef]
- Rodríguez, J.; Mosquera, J.; Couceiro, J.R.; Vázquez, M.E.; Mascarenas, J.L. The at-hook motif as a versatile minor groove anchor for promoting DNA binding of transcription factor fragments. Chem. Sci. 2015, 6, 4767–4771. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.; Mosquera, J.; García-Fandiño, R.; Vázquez, M.E.; Mascareñas, J.L. A designed DNA binding motif that recognizes extended sites and spans two adjacent major grooves. Chem. Sci. 2016, 7, 3298–3303. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, M.E.; Caamaño, A.M.; Mascarenas, J. From transcription factors to designed sequence-specific DNA-binding peptides. Chem. Soc. Rev. 2003, 32, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Smart, O.S.; Weston, C.J.; Burns, D.C.; Woolley, G.A.; Allemann, R.K. Photochemical regulation of DNA-binding specificity of myod. Angew. Chem. Int. Ed. 2005, 117, 7956–7960. [Google Scholar] [CrossRef]
- Guerrero, L.; Smart, O.S.; Woolley, G.A.; Allemann, R.K. Photocontrol of DNA binding specificity of a miniature engrailed homeodomain. J. Am. Chem. Soc. 2005, 127, 15624–15629. [Google Scholar] [CrossRef] [PubMed]
- Woolley, G.A.; Jaikaran, A.S.; Berezovski, M.; Calarco, J.P.; Krylov, S.N.; Smart, O.S.; Kumita, J.R. Reversible photocontrol of DNA binding by a designed gcn4-bzip protein. Biochemistry 2006, 45, 6075–6084. [Google Scholar] [CrossRef] [PubMed]
- Kajino, M.; Fujimoto, K.; Inouye, M. Side-chain cross-linked short α-helices that behave like original proteins in biomacromolecular interactions. J. Am. Chem. Soc. 2010, 133, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Van Lysebetten, D.; García, Y.R.; Louage, B.; De Geest, B.G.; Madder, A. Stapling monomeric GCN4 peptides allows for DNA binding and enhanced cellular uptake. Org. Biomol. Chem. 2015, 13, 3856–3862. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Mallick, S.; Ghosh, P.; Maiti, A.; Ahmed, I.; Bhattacharya, S.; Mandal, T.; Manna, A.; Roy, K.; Singh, S. Simultaneous inhibition of key growth pathways in melanoma cells and tumor regression by a designed bidentate constrained helical peptide. Pept. Sci. 2014, 102, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Naiya, G.; Kaypee, S.; Kundu, T.K.; Roy, S. A constrained helical peptide against s100a4 inhibits cell motility in tumor cells. Chem. Biol. Drug Des. 2015, 86, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Maiti, A.; Roy, K.; Roy, S. A synthetic peptide mimic of λ-cro shows sequence-specific binding in vitro and in vivo. ACS Chem. Biol. 2012, 7, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Mazumder, A.; Ghosh, P.; Naiya, G.; Ghosh, B.; Roy, S. A peptide-based synthetic transcription factor selectively activates transcription in a mammalian cell. Chem. Commun. 2018, 54, 1611–1614. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Roy, S. A peptide-based synthetic transcription factor selectively down-regulates the proto-oncogene cfos in tumour cells and inhibits proliferation. Chem. Commun. 2017, 53, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Dalton, S.; Treisman, R. Characterization of sap-1, a protein recruited by serum response factor to the c-fos serum response element. Cell 1992, 68, 597–612. [Google Scholar] [CrossRef]
- Deschamps, J.; Meijlink, F.; Verma, I.M. Identification of a transcriptional enhancer element upstream from the proto-oncogene fos. Science 1985, 230, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Boila, L.D.; Choudhury, S.; Mondal, P.; Bhattacharjee, S.; Pal, S.K.; Sengupta, A.; Roy, S. A potent conformation-constrained synthetic peptide mimic of a homeodomain selectively regulates target genes in cells. ACS Chem. Biol. 2018, 13, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.R.; Pau, D.I.; Whiting, A.L.; Kim, Y.J.; Pharoah, B.M.; Moi, C.; Boddy, C.N.; Bernal, F. Inhibition of bacterial gene transcription with an rpon-based stapled peptide. Cell Chem. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Soto, C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem 2002, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, M.; Nizzardo, M.; Zanetta, C.; Ramirez, A.; Corti, S. Therapeutic applications of the cell-penetrating hiv-1 tat peptide. Drug Discov. Today 2015, 20, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Kurrikoff, K.; Gestin, M.; Langel, Ü. Recent in vivo advances in cell-penetrating peptide-assisted drug delivery. Expert Opin. Drug Deliv. 2016, 13, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Futaki, S. Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull. 2016, 64, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gfeller, D.; Buth, S.A.; Michielin, O.; Leiman, P.G.; Heinis, C. Improving binding affinity and stability of peptide ligands by substituting glycines with d-amino acids. ChemBioChem 2013, 14, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic approaches to optimizing peptide adme properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Pollaro, L.; Heinis, C. Strategies to prolong the plasma residence time of peptide drugs. MedChemComm 2010, 1, 319–324. [Google Scholar] [CrossRef]
- Angelini, A.; Morales-Sanfrutos, J.; Diderich, P.; Chen, S.; Heinis, C. Bicyclization and tethering to albumin yields long-acting peptide antagonists. J. Med. Chem. 2012, 55, 10187–10197. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandi, E.S.; Brazeau, P.; High, K.; Procter, B.; Fennell, S.; Dubreuil, P. Non-clinical pharmacology and safety evaluation of th9507, a human growth hormone-releasing factor analogue. Basic Clin. Pharmacol. Toxicol. 2007, 100, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Balas, B.; Charbonnel, B.; Bolli, G.B.; Boldrin, M.; Ratner, R.; Balena, R.; T-emerge 2 Study Group. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes the T-emerge 2 trial. Diabetes Care 2013, 36, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Worobec, A.S. Risk-based approach to immunogenicity concerns of therapeutic protein products, part 2—Considering host specific and product specific factors impacting immunogenicity. Biopharm. Int. 2004, 17, 34–42. [Google Scholar]
- Ho, C.L.; Lin, Y.L.; Chen, W.C.; Rocchi, R.; Piek, T. Comparison of the immunogenicity of wasp venom peptides with or without carbohydrate moieties. Toxicon 1998, 36, 217–221. [Google Scholar] [CrossRef]
- Egrie, J.C.; Browne, J.K. Development and characterization of novel erythropoiesis stimulating protein (NESP). Br. J. Cancer 2001, 84, 3–10. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Ghosh, P.; Ahmed, I.; Chakraborty, M.; Naiya, G.; Ghosh, B. Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions. Biomedicines 2018, 6, 118. https://doi.org/10.3390/biomedicines6040118
Roy S, Ghosh P, Ahmed I, Chakraborty M, Naiya G, Ghosh B. Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions. Biomedicines. 2018; 6(4):118. https://doi.org/10.3390/biomedicines6040118
Chicago/Turabian StyleRoy, Siddhartha, Piya Ghosh, Israr Ahmed, Madhumita Chakraborty, Gitashri Naiya, and Basusree Ghosh. 2018. "Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions" Biomedicines 6, no. 4: 118. https://doi.org/10.3390/biomedicines6040118
APA StyleRoy, S., Ghosh, P., Ahmed, I., Chakraborty, M., Naiya, G., & Ghosh, B. (2018). Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions. Biomedicines, 6(4), 118. https://doi.org/10.3390/biomedicines6040118