Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation


Abstract
:1. Introduction
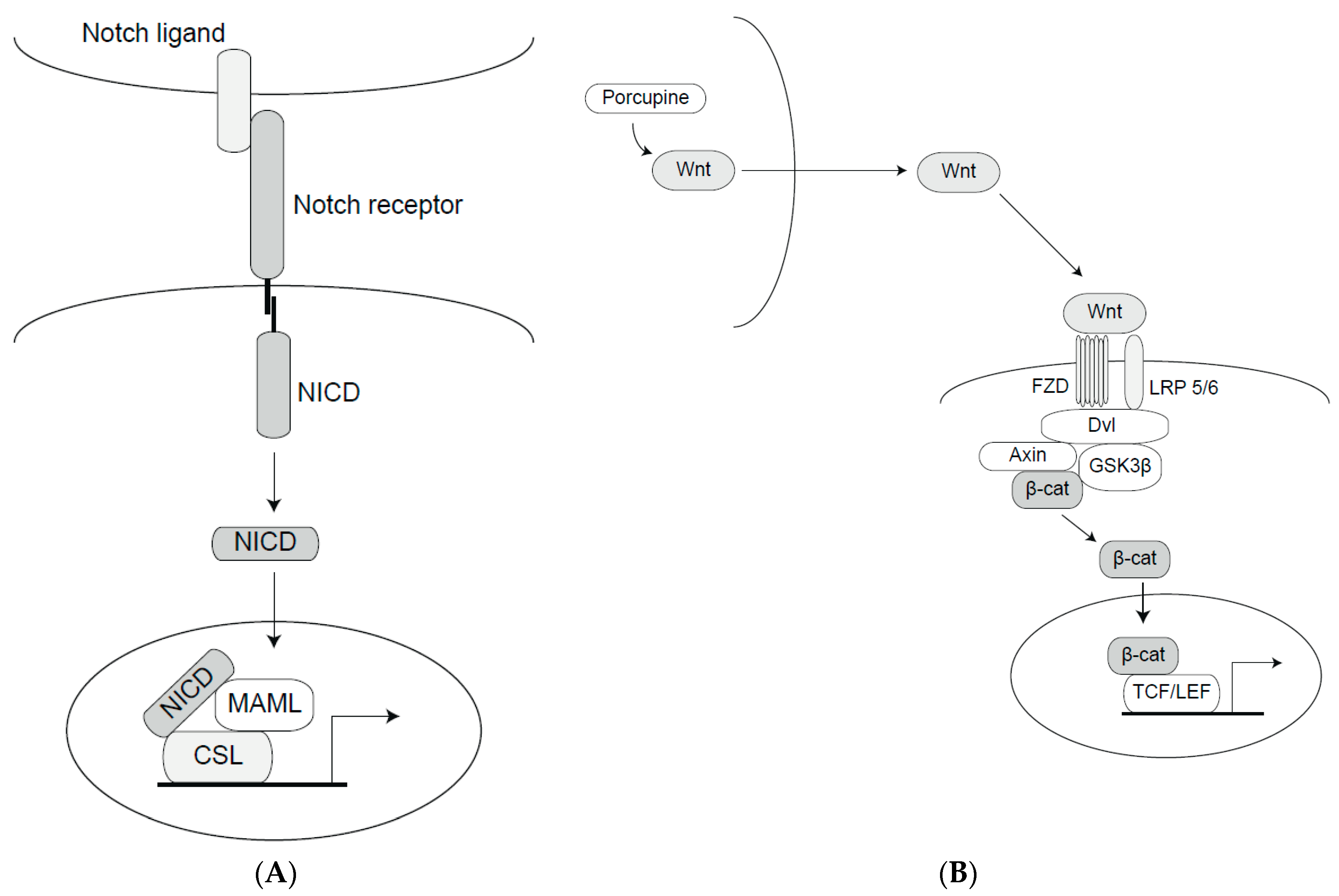
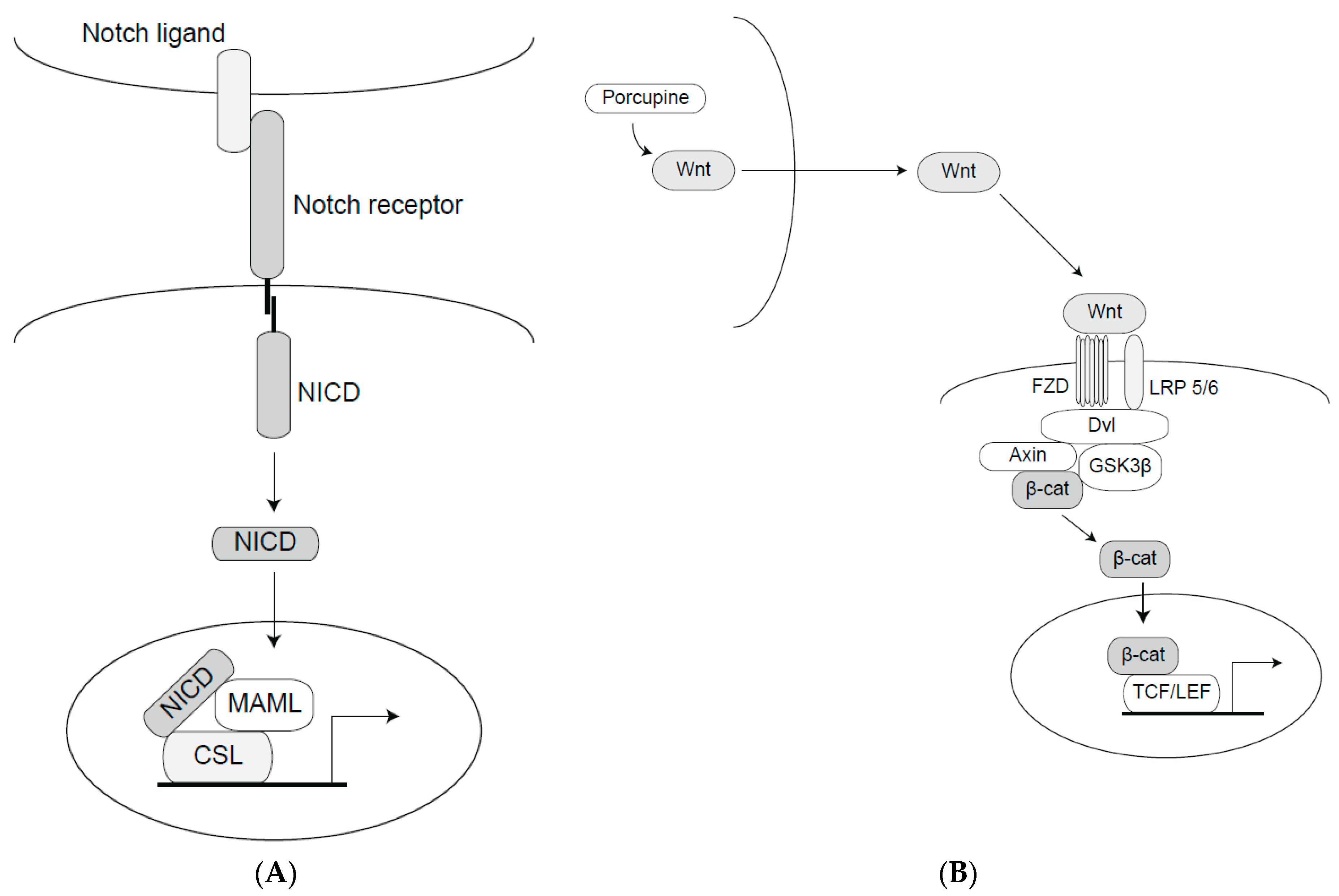
2. The Notch and Wnt Signaling Pathways
2.1. The Notch Signaling Pathway
2.2. The Wnt Signaling Pathway
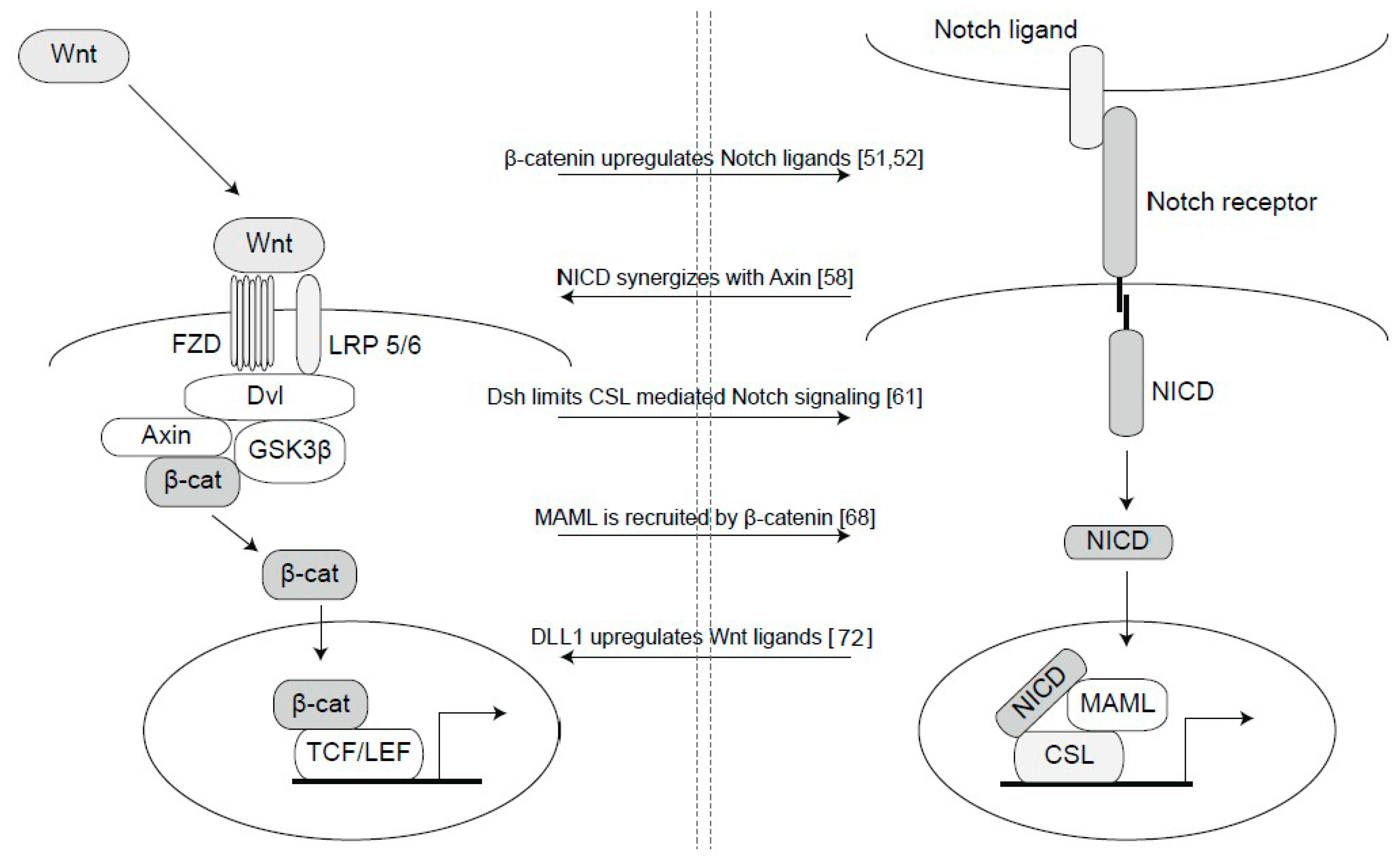
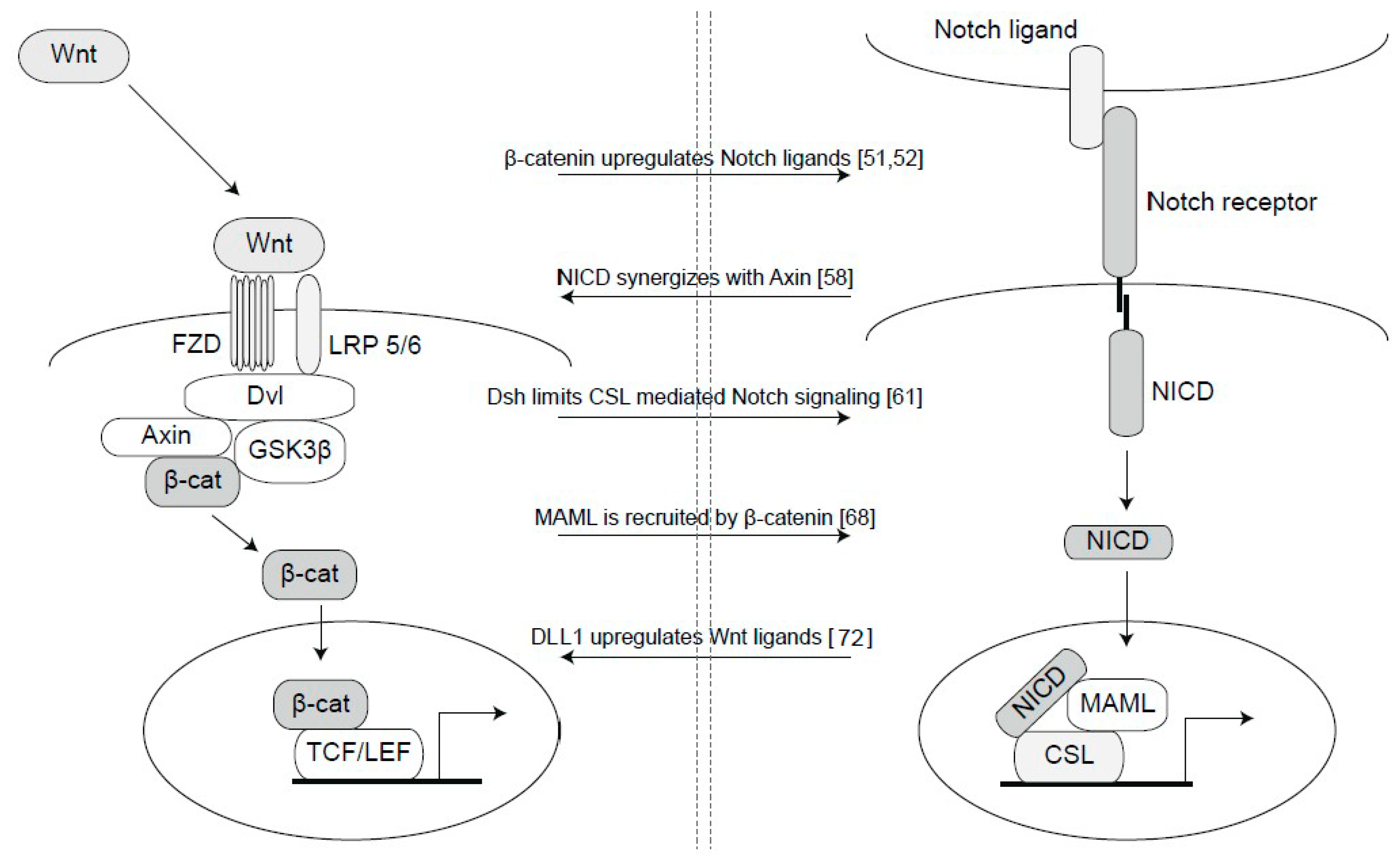
3. Cross-Talk between Notch and Wnt Signaling
4. Notch and Wnt Signaling in Breast Cancer
4.1. Notch Signaling in Breast Cancer
4.2. Wnt Signaling in Breast Cancer
4.3. Synergies between Notch and Wnt Signaling Relevant for Breast Cancer
5. Notch and Wnt Therapy Development
5.1. Notch Therapy Development
5.2. Wnt Therapy Development
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Pantel, K.; Kang, Y. Tumor metastasis: Moving new biological insights into the clinic. Nat. Med. 2013, 19, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiatinig human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Fridriksdottir, A.J.; Villadsen, R.; Morsing, M.; Christine, M.; Kim, J. Proof of region-specific multipotent progenitors in human breast epithelia. Proc. Natl. Acad. Sci. USA 2017, 6, E10102–E10110. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, D.; Liu, D.; Wang, S.; Yu, X.; Dai, E.; Wang, J.; Wang, L.; Jiang, W. Dissecting the origin of breast cancer subtype stem cell and the potential mechanism of malignant transformation. PLoS ONE 2016, 11, e0165001. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.-F.; Wu, L.; Li, Z.-Q.; Wu, M.-L.; Wang, H.-F.; Chan, K.-Y.; Lu, L.-L.; Cai, S.-H.; Wang, H.-S.; Du, J. Nodal signaling modulates the expression of Oxt-4 via nuclear translocation of b-catenin in lung and prostate cancer cells. Arch. Biochem. Biophys. 2016, 608, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Leis, O.; Eguiara, A.; Lopoez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2011, 31, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, S.; Wang, P.; Zhao, S.; Wang, F.; Bing, L.; Zhang, Y.; Ling, E.A.; Gao, J.; Hao, A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 2011, 59, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Hermann, P.C.; Mueller, M.; Huber, S.; Balic, A.; Miranda-lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Cufí, S.; Oliveras-Ferraros, C.; Cuyàs, E.; López-Bonet, E.; Lupu, R.; Alarcón, T.; Vellon, L.; Manuel Iglesias, J.; Leis, O.; et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013, 12, 3109–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchuluun, K.; Azuma, M.; Fujiwara, K.; Yashiro, T.; Kikuchi, M. Notch signaling and maintenance of Sox2 expression in rat anterior pituitary cells. Acta Histochem. Cytochem. 2017, 50, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Azzam, D.J.; Zhao, D.; Sun, J.; Minn, A.J.; Ranganathan, P.; Drews-elger, K.; Han, X.; Picon-Ruiz, M.; Gilbert, C.A.; Wander, S.A.; et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in g-secretase inhibitor drug responses. EMBO Mol. Med. 2013, 5, 1502–1522. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, I.K.; Gacquer, D.; Heurck, R.; Van Polleux, F.; Detours, V.; Vanderhaeghen, P. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 2018, 173, 1370–1384. [Google Scholar] [CrossRef] [PubMed]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-specific NOTCH2NL genes affect Notch signaling and cortical neurogenesis. Cell 2018, 173, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Santio, N.M.; Landor, S.K.-J.; Vahtera, L.; Ylä-Pelto, J.; Paloniemi, E.; Imanishi, S.Y.; Corthals, G.; Varjosalo, M.; Manoharan, G.B.; Uri, A.; et al. Phosphorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget 2016, 7, 43220–43228. [Google Scholar] [CrossRef] [PubMed]
- Sjöqvist, M.; Antfolk, D.; Ferraris, S.; Rraklli, V.; Haga, C.; Antila, C.; Mutvei, A.; Imanishi, S.Y.; Holmberg, J.; Jin, S.; et al. PKCζ regulates Notch receptor routing and activity in a Notch signaling-dependent manner. Cell Res. 2014, 24, 433–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, L.; Wong, E.Y.M.; Tsang, S.L.; Xu, P.; Lendahl, U.; Sham, M.H. An Eya1-Notch axis specifies bipotential epibranchial differentiation in mammalian craniofacial morphogenesis. eLife 2017, 6, e30126. [Google Scholar] [CrossRef] [PubMed]
- Tveriakhina, L.; Schuster-Gossler, K.; Jarrett, S.M.; Andrawes, M.B.; Rorhbach, M.; Blacklow, S.C.; Gossler, A. The ectodomains determine ligand function in vivo and selectivity of DLL1 and DLL4 toward NOTCH1 and NOTCH2 in vitro. eLife 2018, 7, e40045. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Boyle, S.; Zhu, Y.; Zhang, A.; Piwnica-Worms, D.R.; Ilagan, M.X.; Kopan, R. The extracellular domain of Notch2 increases its cell-surface abundance and ligand responsiveness during kidney development. Dev. Cell 2013, 25, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/b-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Cselenyi, C.S.; Jernigan, K.K.; Tahinci, E.; Thorne, C.A.; Lee, L.A.; Lee, E. LRP6 transduces a canonical Wnt signal independently of Axin degradation by inhibiting GSK3’s phosphorylation of b-catenin. Proc. Natl. Acad. Sci. USA 2008, 105, 8032–8037. [Google Scholar] [CrossRef] [PubMed]
- Bilic, J.; Huang, Y.; Davidson, G.; Zimmermann, T.; Cruciat, C.; Bienz, M.; Niehrs, C. Wnt Induces LRP6 Signalosomes and Promotes Dishevelled-Dependent LRP6 Phosphorylation. Science 2007, 316, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Cong, F.; Varmus, H. Nuclear-cytoplasmic shuttling of Axin regulates subcellular localization of b-catenin. Proc. Natl. Acad. Sci. USA 2004, 101, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibáñez, C.F. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Dahlqvist, C.; Blokzijl, A.; Chapman, G.; Falk, A.; Dannaeus, K.; Ibâñez, C.F.; Lendahl, U. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development 2003, 130, 6089–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastronardi, F.G.; Min, W.; Wang, H.; Winer, S.; Dosch, M.; Boggs, J.M.; Moscarello, M.A. Attenuation of experimental autoimmune encephalomyelitis and nonimmune demyelination by IFN-b plus Vitamin B12, Treatment to modify Notch-1/Sonic hedgehog balance. J. Immunol. 2004, 172, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.H.; Yang, L.; Dessaud, E.; Chuang, K.; Moore, D.M.; Rohatgi, R.; Briscoe, J.; Novitch, B.G. Notch activity modulates the responsiveness of neural progenitors to Sonic hedgehog signaling. Dev. Cell 2015, 33, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.; Gao, B.; et al. Hippo signaling interactions with Wnt/b -catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Manderfield, L.J.; Aghajanian, H.; Engleka, K.A.; Lim, L.Y.; Lui, F.; Jain, R.; Li, L.; Olson, E.N.; Epstein, J.A. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development 2015, 142, 2962–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Nguyen, Q.; Wan, Y.; Zhou, T.; Venter, J.; Frampton, G.A.; DeMorrow, S.; Pan, D.; Meng, F.; Glaser, S.; et al. The Hippo signaling functions through the Notch signaling to regulate intrahepatic bile duct development in mammals. Lab. Investig. 2017, 97, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires Notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, E.-B.; Tsoi, Y.L.; Phoon, Y.P.; Landor, S.; Silva Cascales, H.; Ramsköld, D.; Deng, Q.; Lindqvist, A.; Lian, X.; Sahlgren, C.; et al. Loss of CSL unlocks a hypoxic response and enhanced tumor growth potential in breast cancer cells. Stem Cell Rep. 2016, 6, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Mutvei, A.P.; Landor, S.K.-J.; Fox, R.; Braune, E.-B.; Tsoi, Y.L.; Phoon, Y.P.; Sahlgren, C.; Hartman, J.; Bergh, J.; Jin, S.; et al. Notch signaling promotes a HIF2a-driven hypoxic response in multiple tumor cell types. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Landor, S.K.-J.; Lendahl, U. The interplay between the cellular hypoxic response and Notch signaling. Exp. Cell Res. 2017, 356, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Estrach, S.; Ambler, C.A.; Lo Celso, C.; Hozumi, K.; Watt, F.M. Jagged 1 is a beta-catenin target gene required for ectopic hair follicle formation in adult epidermis. Development 2006, 133, 4427–4438. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/β-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Ungerbäck, J.; Elander, N.; Grüneberg, J.; Sigvardsson, M.; Söderkvist, P. The Notch-2 Gene Is Regulated by Wnt Signaling in Cultured Colorectal Cancer Cells. PLoS ONE 2011, 6, e17957. [Google Scholar] [CrossRef] [PubMed]
- Ammeux, N.; Housden, B.E.; Georgiadis, A.; Hu, Y.; Perrimon, N. Mapping signaling pathway cross-talk in Drosophila cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9940–9945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rallis, C.; Pinchin, S.M.; Ish-horowicz, D. Cell-autonomous integrin control of Wnt and Notch signalling during somitogenesis. Development 2010, 3601, 3591–3601. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, J.A.; Sansom, O.J. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res. 2008, 68, 4963–4966. [Google Scholar] [CrossRef] [PubMed]
- Hayward, P.; Balayo, T.; Arias, A.M. Notch synergizes with axin to regulate the activity of Armadillo in Drosophila. Dev. Dyn. 2006, 235, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.D.; Matsuno, K.; Artavanis-Tsakonas, S.; Perrimon, N. Interaction Between Wingless and Notch Signaling Pathways Mediated by Dishevelled. Science 1996, 271, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-descalzo, S.; Sanders, P.G.; Montagne, C.; Ruth, I.; Balayo, T.; Arias, A.M. Wingless modulates the ligand independent traffic of Notch through Dishevelled. Fly 2010, 6934, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Collu, G.M.; Hidalgo-Sastre, A.; Acar, A.; Bayston, L.; Gildea, C.; Leverentz, M.K.; Mills, C.G.; Owens, T.W.; Meurette, O.; Dorey, K.; et al. Dishevelled limits Notch signalling through inhibition of CSL. Development 2012, 139, 4405–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Descalzo, S.; Tkocz, K.; Balayo, T.; Arias, A.M. Modulation of the ligand-independent traffic of Notch by Axin and Apc contributes to the activation of Armadillo in Drosophila. Development 2011, 138, 1501–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, P.; Brennan, K.; Sanders, P.; Balayo, T.; DasGupta, R.; Perrimon, N.; Arias, A.M. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 2005, 132, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.G.; Munoz-Descalzo, S.; Balayo, T.; Wirtz-Peitz, F.; Hayward, P.; Arias, A.M. Ligand-Independent Traffic of Notch Buffers Activated Armadillo in Drosophila. PLoS Biol. 2009, 7, e1000169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Kagawa, T.; Inoue, T.; Nonaka, A.; Takada, S.; Aburatani, H.; Taga, T. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell Biol. 2008, 28, 7427–7441. [Google Scholar] [CrossRef] [PubMed]
- Yamamizu, K.; Matsunaga, T.; Uosaki, H.; Fukushima, H.; Katayama, S.; Hiraoka-Kanie, M.; Mitani, K.; Yamashita, J.K. Convergence of Notch and β-catenin signaling induces arterial fate in vascular progenitors. J. Cell Biol. 2010, 189, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Saint Just Ribeiro, M.; Hansson, M.L.; Lindberg, M.J.; Popko-Scibor, A.E.; Wallberg, A.E. GSK3 b is a negative regulator of the transcriptional coactivator MAML1. Nucleic Acids Res. 2009, 37, 6691–6700. [Google Scholar] [CrossRef] [PubMed]
- Alves-Guerra, M.C.; Ronchini, C.; Capobianco, A.J. Mastermind-like 1 is a specific coactivator of b-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res. 2007, 67, 8690–8698. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Ingles-Esteve, J.; Aguilera, C.; Bigas, A. Phosphorylation by Glycogen Synthase Kinase-3b Down-regulates Notch Activity, a Link for Notch and Wnt Pathways. J. Biol. Chem. 2003, 278, 32227–32235. [Google Scholar] [CrossRef] [PubMed]
- Foltz, D.R.; Santiago, M.C.; Berechid, B.E.; Nye, J.S. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr. Biol. 2002, 12, 1006–1011. [Google Scholar] [CrossRef]
- Zheng, L.; Conner, S.D. Glycogen synthase kinase 3 β inhibition enhances Notch1 recycling. Mol. Biol. Cell 2018, 29, 289–395. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Celia-Terrassa, T.; Kumar, S.; Hang, X.; Wei, Y.; Choudhury, A.; Hwang, J.; Peng, J.; Nixon, B.; Grady, J.J.; et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 2018, 4153, eaan4153. [Google Scholar] [CrossRef] [PubMed]
- Braune, E.-B.; Lendahl, U. Notch—A goldilocks signaling pathway in disease and cancer therapy. Discov. Med. 2016, 21, 189–196. [Google Scholar] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Jhappan, C.; Gallahan, D.; Stahle, C.; Chu, E.; Smith, G.H.; Merlino, G.; Callahan, R. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. 1992, 6, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of Notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Simin, K.; Liu, Q.; Plescia, J.; Guha, M.; Khan, A.; Hsieh, C.C.; Altieri, D.C. A functional Notch—Survivin gene signature in basal breast cancer. Breast Cancer Res. 2008, 10, R97. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Oyama, T.; Ito, E.; Satoh, H.; Azuma, S.; Hayashi, M.; Shimizu, K.; Honma, R.; Yanagisawa, Y.; Nishikawa, A.; et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res. 2008, 68, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Choy, L.; Hagenbeek, T.J.; Solon, M.; French, D.; Finkle, D.; Shelton, A.; Venook, R.; Brauer, M.J.; Siebel, C.W. Constitutive NOTCH3 signaling promotes the growth of basal breast cancers. Cancer Res. 2017, 77, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clay, M.R.; Varma, S.; West, R.B. MAST2 and NOTCH1 translocations in breast carcinoma and associated pre-invasive lesions. Hum. Pathol. 2013, 44, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.T.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Ikeda, S.; Fahey, F.; Bielenberg, D.; Smits, P.; Hauschka, P.V. Notch3 in human breast cancer cell lines regulates osteoblast-cancer cell interactions and osteolytic bone metastasis. Am. J. Pathol. 2010, 177, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Leontovich, A.A.; Jalalirad, M.; Salisbury, J.L.; Mills, L.; Haddox, C.; Schroeder, M.; Tuma, A.; Guicciardi, M.E.; Zammataro, L.; Gambino, M.W.; et al. NOTCH3 expression is linked to breast cancer seeding and distant metastasis. Breast Cancer Res. 2018, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Szabolcs, M.; Politi, K.; Kiaris, H.; Artavanis-tsakonas, S.; Efstratiadis, A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9262–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Mutvei, A.P.; Chivukula, I.V.; Andersson, E.R.; Ramsköld, D.; Sandberg, R.; Lee, K.L.; Kronqvist, P.; Mamaeva, V.; Östling, P.; et al. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKα/IKKβ. Oncogene 2012, 32, 4893. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guturi, K.K.N.; Gautreau, B.; Patel, P.S.; Saad, A.; Morii, M.; Mateo, F.; Palomero, L.; Barbour, H.; Gomez, A.; et al. Ubiquitin ligase RNF8 suppresses Notch signaling to regulate mammary development and tumorigenesis. J. Clin. Investig. 2018, 128, 4525–4542. [Google Scholar] [CrossRef] [PubMed]
- Granit, R.Z.; Masury, H.; Condiotti, R.; Fixler, Y.; Gabai, Y.; Glikman, T.; Dalin, S.; Winter, E.; Nevo, Y.; Carmon, E.; et al. Regulation of cellular heterogeneity and rates of symmetric and asymmetric divisions in triple negative breast cancer. Cell Rep. 2018, 24, 3237–3250. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, R.; Al-Khaldi, S.; Sleiman, G.M.; Sarkar, A.; Al-Dhfyan, A.; Al-Mohanna, F.; Ghebeh, H.; Al-Alwan, M. Fascin is critical for the maintenance of breast cancer stem cell pool predominantly via the activation of the Notch self-renewal pathway. Stem Cells 2016, 34, 2799–2813. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.M.; Hess, D.; Lala, P.K. COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Morata-tarifa, C.; Jiménez, G.; García, M.A.; Entrena, J.M.; Grinan-Lison, C.; Aguilera, M.; Picon-Ruiz, M.; Marchal, J.A. Low adherent cancer cell subpopulations are enriched in tumorigenic and metastatic epithelial-to-mesenchymal cancer stem-like cells. Sci. Rep. 2016, 6, 18772. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, M.J.; Serra, R.; Hermance, N.; Kelliher, M.A. NOTCH1 inhibition in vivo results in mammary tumor regression and reduced mammary tumorsphere-forming activity in vitro. Breast Cancer Res. 2012, 14, R126. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Gupta, A.; Chattopadhyay, D.; Chatterji, U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci. Rep. 2017, 7, 9170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simões, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, T.; Nakamura, M.; Yanai, K.; Nagai, S.; Wada, J.; Koga, K.; Nakashima, H.; Sato, N.; Tanaka, M.; Katano, M. γ-Secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology 2008, 134, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; Hao, L.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res 2008, 68, 5226–5235. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, A.S.; Grosschedl, R.; Guzman, R.C.; Parslow, T.; Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988, 55, 619–625. [Google Scholar] [CrossRef]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, J.; Gaspar, C.; Richer, W.; Franken, P.F.; Sacchetti, A.; Joosten, R.; Idali, A.; Brandao, J.; Decraene, C.; Fodde, R. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis 2014, 35, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axis2-GSK3b cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Kang, Y. Complex interplay between tumor microenvironment and cancer therapy. Front. Med. 2018, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Bae, J.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived Jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudoran, O.; Soritau, O.; Balacescu, L. Regulation of stem cells-related signaling pathways in response to doxorubicin treatment in Hs578T triple-negative breast cancer cells. Mol. Cell Biochem. 2015, 409, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling-are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-human study of LY3039478, an oral notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Dumbrava, E.I.; Mills, G.B.; Yap, T.A. Targeting gamma secretase: Has progress moved up a notch? Ann. Oncol. 2018, 29, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Simoes, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alférez, D.G.; Spence, K.; Santiago-Gómez, A.; Chemi, F.; et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, L.; Yan, W.; Zhai, J.; Pizzo, D.; Chu, P.; Chin, A.R.; Shen, M.; Dong, C.; Ruan, X.; et al. Chemotherapy induces breast cancer stemness in association with dysregulated monocytosis. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Begicevic, R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, C.; Yang, Y.; Wang, C.; Yang, T.; Yang, X.; Liu, S.C. Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4378–4387. [Google Scholar] [PubMed]
- Shen, Q.; Cohen, B.; Zheng, W.; Rahbar, R.; Martin, B.; Murakami, K.; Lamorte, S.; Thompson, P.; Berman, H.; Zúñiga-Pflücker, J.C.; et al. Notch Shapes the Innate Immunophenotype in Breast Cancer. Cancer Discov. 2017, 7, 1320–1335. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Tran, I.T.; Sandy, A.R.; Carulli, A.J.; Ebens, C.; Chung, J.; Shan, G.T.; Radojcic, V.; Friedman, A.; Gridley, T.; Shelton, A.; et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J. Clin. Investig. 2013, 123, 1590–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafkas, D.; Shelton, A.; Chiu, C.; de Leon Boenig, G.; Chen, Y.; Stawicki, S.S.; Siltanen, C.; Reichelt, M.; Zhou, M.; Wu, X.; et al. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 2015, 528, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.; Berger, C.; Moll, I.; Adam, M.G.; Schwarz, F.; Mohr, K.; Augustin, H.G.; Fischer, A. Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiovasc. Res. 2015, 107, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamagnone, L.; Zacchigna, S.; Rehman, M. Taming the Notch Transcriptional Regulator for Cancer Therapy. Molecules 2018, 23, 431. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, L.; Da Silva, T.G.; Wang, Z.; Han, X.; Jin, K.; VanWye, J.; Zhu, X.; Weaver, K.L.; Oashi, T.; Lopes, P.E.; et al. The small molecule IMR-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016, 76, 3593–3603. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2010, 462, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kulic, I.; Robertson, G.; Chang, L.; Baker, J.H.; Lockwood, W.W.; Mok, W.; Fuller, M.; Fournier, M.; Wong, N.; Chou, V.; et al. Loss of the Notch effector RBPJ promotes tumorigenesis. J. Exp. Med. 2014, 212, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, J.; Li, Q.S.; Fan, J.Q.; Dong, X.M.; Xu, J.P.; Wang, X.M.; Yang, G.W.; Yan, P.; Wen, G.Z.; et al. Suppression of PPN MG61 attenuates Wnt/b-catenin signaling pathway and induces apoptosis in human lung cancer. Oncogene 2008, 27, 3483–3488. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically de fi ned cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A first-in-human phase I study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation—Driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3145. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Arques, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argiles, G.; Dienstmann, R.; Fernández, N.; Caratù, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase inhibition blocks Wnt/β-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Yang, Y.; Ueberheide, B.; Smith, S. Whole proteome analysis of human tankyrase knockout cells reveals targets of tankyrase-mediated degradation. Nat. Commun. 2017, 8, 2214. [Google Scholar] [CrossRef] [PubMed]
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braune, E.-B.; Seshire, A.; Lendahl, U. Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation. Biomedicines 2018, 6, 101. https://doi.org/10.3390/biomedicines6040101
Braune E-B, Seshire A, Lendahl U. Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation. Biomedicines. 2018; 6(4):101. https://doi.org/10.3390/biomedicines6040101
Chicago/Turabian StyleBraune, Eike-Benjamin, Anita Seshire, and Urban Lendahl. 2018. "Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation" Biomedicines 6, no. 4: 101. https://doi.org/10.3390/biomedicines6040101
APA StyleBraune, E.-B., Seshire, A., & Lendahl, U. (2018). Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation. Biomedicines, 6(4), 101. https://doi.org/10.3390/biomedicines6040101