Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics

Abstract
1. Introduction
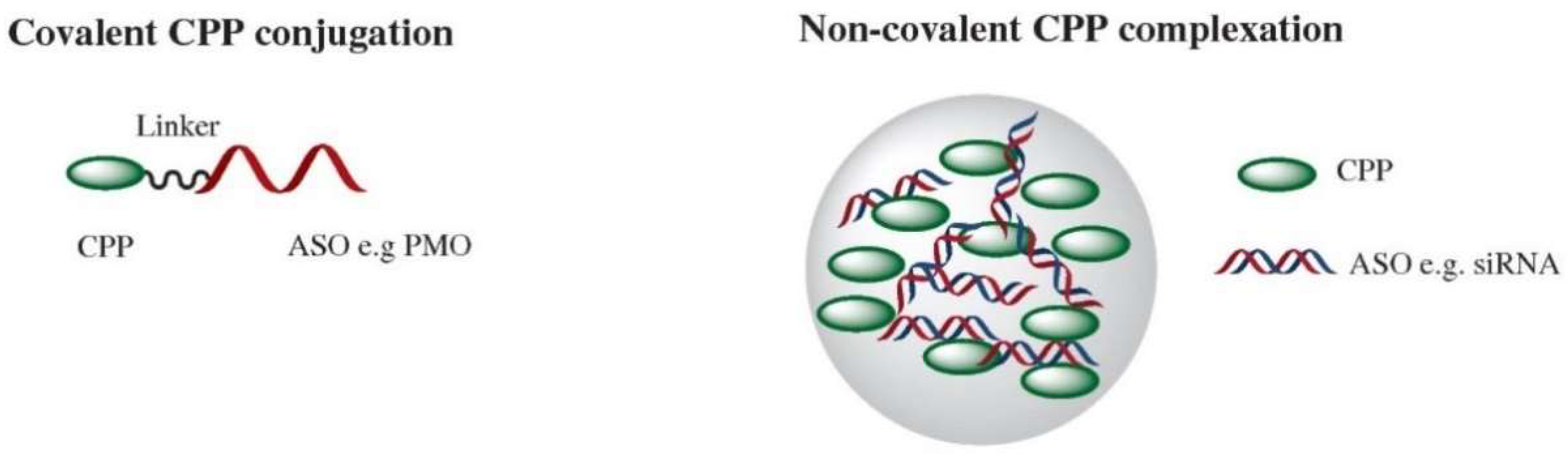
2. Direct CPP Conjugation for Nucleic Acid Delivery
3. Noncovalent CPP Delivery of Nucleic acids
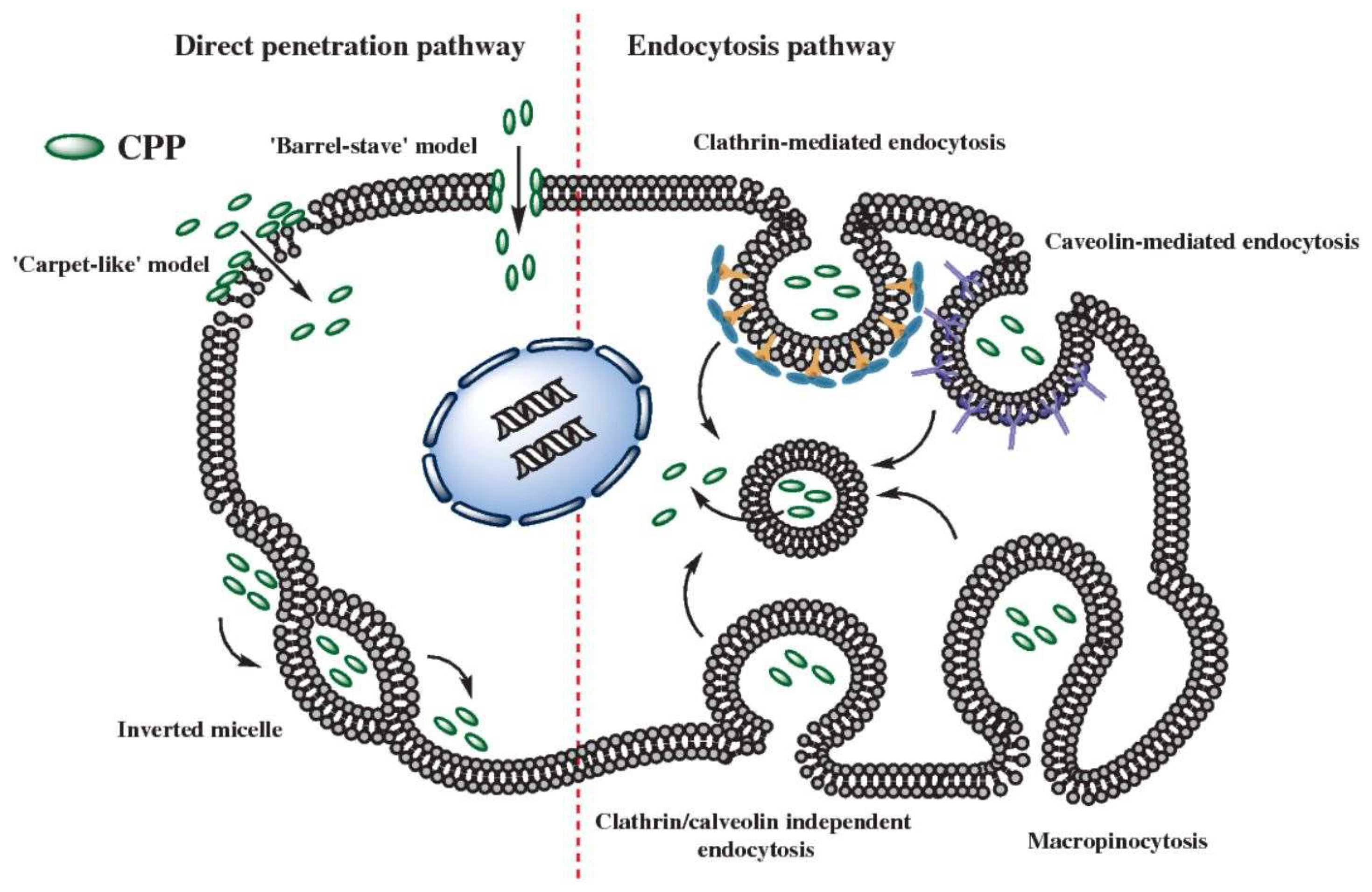
4. Mechanism of CPP Mediated Uptake
5. Future Perspectives of Clinical Application of CPP-ASOs
Acknowledgments
Conflicts of Interest
References
- Nayerossadat, N.; Ali, P.; Maedeh, T. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn. Res. 2015, 9, GE01-6. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; EL Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.-H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.L.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J.A. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, J.; Han, G.; Zhang, Y.; Dong, X.; Cao, L.; Wang, Q.; Moulton, H.M.; Yin, H. Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 2014, 22, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Shabanpoor, F.; Hammond, S.M.; Abendroth, F.; Hazell, G.; Wood, M.J.A.; Gait, M.J. Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy. Nucleic Acid Ther. 2017, 27, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.M.G.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ‘t Hoen, P.A.C.; et al. Peptide Conjugation of 2′-O-methyl Phosphorothioate Antisense Oligonucleotides Enhances Cardiac Uptake and Exon Skipping in mdx Mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Burrer, R.; Neuman, B.W.; Ting, J.P.C.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kuhn, P.; Buchmeier, M.J. Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models. J. Virol. 2007, 81, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Geller, B.L.; Marshall-Batty, K.; Schnell, F.J.; McKnight, M.M.; Iversen, P.L.; Greenberg, D.E. Gene-silencing antisense oligomers inhibit acinetobacter growth in vitro and in vivo. J. Infect. Dis. 2013, 208, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc Master, G.; Coll, J.-L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J. Nanobiotechnol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Mäe, M.; EL Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, Ü. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [PubMed]
- Veiman, K.L.; Mäger, I.; Ezzat, K.; Margus, H.; Lehto, T.; Langel, K.; Kurrikoff, K.; Arukuusk, P.; Suhorutsenko, J.; Padari, K.; et al. PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol. Pharm. 2013, 10, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; El Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Hee Kim, M.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Attaway, C.C.; Butler, B.; Andino, L.M.; Sokolosky, M.L.; Jakymiw, A. Fusogenic-Oligoarginine Peptide-Mediated Delivery of siRNAs Targeting the CIP2A Oncogene into Oral Cancer Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Bryant, A.A.; Zhang, H.; Attaway, C.C.; Pugh, W.; Eggart, L.; Sansevere, R.M.; Andino, L.M.; Dinh, L.; Cantini, L.P.; Jakymiw, A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral Oncol. 2017, 72, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Scaria, P.; Lu, P.; Woodle, M.C.; Mixson, A.J. Systemic delivery of HK Raf-1 siRNA polyplexes inhibits MDA-MB-435 xenografts. Cancer Gene Ther. 2008, 15, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Bendifallah, N.; Rasmussen, F.W.; Zachar, V.; Ebbesen, P.; Nielsen, P.E.; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug. Chem. 2006, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Boffa, L.C.; Cutrona, G.; Cilli, M.; Matis, S.; Damonte, G.; Mariani, M.R.; Millo, E.; Moroni, M.; Roncella, S.; Fedeli, F.; et al. Inhibition of Burkitt’s lymphoma cells growth in SCID mice by a PNA specific for a regulatory sequence of the translocated c-myc. Cancer Gene Ther. 2007, 14, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Fabani, M.M.; Abreu-Goodger, C.; Williams, D.; Lyons, P.A.; Torres, A.G.; Smith, K.G.C.; Enright, A.J.; Gait, M.J.; Vigorito, E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010, 38, 4466–4475. [Google Scholar] [CrossRef] [PubMed]
- Stirchak, E.P.; Summerton, J.E.; Weller, D.D. Uncharged Stereoregular Nucleic Acid Analogues. 1. Synthesis of a Cytosine-Containing Oligomer with Carbamate Internucleoside Linkages. J. Org. Chem. 1987, 52, 4202–4206. [Google Scholar] [CrossRef]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide—Morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.A.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Adams, A.M.; Johnsen, R.D.; Greer, K.; Moulton, H.M.; Wilton, S.D. Dystrophin isoform induction in vivo by antisense-mediated alternative splicing. Mol. Ther. 2010, 18, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.K.; Malerba, A.; Popplewell, L.; Foster, K.; Dickson, G. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa guanidine morpholino oligomer treatment. Mol. Ther. 2011, 19, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Kang, J.K.; McClorey, G.; Saleh, A.F.; Popplewell, L.; Gait, M.J.; Wood, M.J.A.; Dickson, G. Dual myostatin and dystrophin exon skipping by morpholino nucleic acid oligomers conjugated to a cell-penetrating peptide is a promising therapeutic strategy for the treatment of duchenne muscular dystrophy. Mol. Ther. Nucleic Acids 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Malerba, A.; Popplewell, L.; Schnell, F.; Hanson, G.; Dickson, G. Systemic Antisense Therapeutics for Dystrophin and Myostatin Exon Splice Modulation Improve Muscle Pathology of Adult mdx Mice. Mol. Ther. Nucleic Acids 2017, 6, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Shabanpoor, F.; McClorey, G.; Saleh, A.F.; Jarver, P.; Wood, M.J.A.; Gait, M.J. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of Duchenne muscular dystrophy. Nucleic Acids Res. 2015, 43, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Mao, X.G.; Zhou, Y.; Chen, Z.; Hu, Y.; Hou, Z.; Li, M.-K.; Meng, J.-R.; Luo, X.-X. Advances in the delivery of antisense oligonucleotides for combating bacterial infectious diseases. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Goodman, M. Recent developments in retro peptides and proteins—An ongoing topochemical exploration. Trends Biotechnol. 1995, 13, 438–445. [Google Scholar] [CrossRef]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched HK peptides are effective carriers of siRNA. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Järver, P.; Zaghloul, E.M.; Arzumanov, A.A.; Saleh, A.F.; McClorey, G.; Hammond, S.M.; Hällbrink, M.; Langel, Ü.; Smith, C.I.; Wood, M.J.; et al. Peptide Nanoparticle Delivery of Charge-Neutral Splice-Switching Morpholino Oligonucleotides. Nucleic Acid Ther. 2015, 25, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific siRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Teixidó, M.; Giralt, E. Building cell selectivity into CPP-mediated strategies. Pharmaceuticals 2010, 3, 1456–1490. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. (Camb.) 2009, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Jia, X.L.; Qi, J.L.; Yang, L.P.; Sun, W.H.; Yan, X.; Yang, S.-K.; Cao, D.-Y.; Du, Q.; Qi, X.-R. Enhancing siRNA-based cancer therapy using a new pH-responsive activatable cell-penetrating peptide-modified liposomal system. Int. J. Nanomed. 2017, 12, 2385–2405. [Google Scholar] [CrossRef] [PubMed]
- Luneberg, J.; Martin, I.; Nussler, F.; Ruysschaert, J.M.; Herrmann, A. Structure and topology of the influenza virus fusion peptide in lipid bilayers. J. Biol. Chem. 1995, 270, 27606–27614. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Rydström, A.; Deshayes, S.; Konate, K.; Crombez, L.; Padari, K.; Boukhaddaoui, H.; Aldrian, G.; Pooga, M.; Divita, G. Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Takeuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Elmquist, A.; Hansen, M.; Padari, K.; Saar, K.; Viht, K.; Langel, U.; Pooga, M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug. Chem. 2004, 15, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppé, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell Membrane Lipid Rafts Mediate Caveolar Endocytosis of HIV-1 Tat Fusion Proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed]
- Gestin, M.; Dowaidar, M.; Langel, Ü. Uptake mechanism of cell-penetrating peptides. Adv. Exp. Med. Biol. 2017, 1030, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hällbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta Biomembr. 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Lehto, T.; Alvarez, A.C.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.A.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.-B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef] [PubMed]
- Helmfors, H.; Lindberg, S.; Langel, Ü. SCARA involvement in the uptake of nanoparticles formed by cell-penetrating peptides. Methods Mol. Biol. 2015, 1324, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; El-Andaloussi, S.; Pooga, M.; Langel, U. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012, 26, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Juks, C.; Lorents, A.; Arukuusk, P.; Langel, Ü.; Pooga, M. Cell-penetrating peptides recruit type A scavenger receptors to the plasma membrane for cellular delivery of nucleic acids. FASEB J. 2017, 31, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed]
- Dowaidar, M.; Gestin, M.; Cerrato, C.P.; Jafferali, M.H.; Margus, H.; Kivistik, P.A.; Ezzat, K.; Hallberg, E.; Pooga, M.; Hällbrink, M.; et al. Role of autophagy in cell-penetrating peptide transfection model. Sci. Rep. 2017, 7, 12635. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Luzio, J.P. Delivery of endocytosed membrane proteins to the lysosome. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 615–624. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Su, R.; Shi, G.; Ma, Z.S.; Zou, Q. CPPred-RF: A Sequence-based Predictor for Identifying Cell-Penetrating Peptides and Their Uptake Efficiency. J. Proteome Res. 2017, 16, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Arukuusk, P.; Langel, U.; Pooga, M. Characteristics of Cell-Penetrating Peptide/Nucleic Acid Nanoparticles. Mol. Pharm. 2016, 13, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.; Lau, C.Y.; Tosh, D.; Ward, S.G.; Mrsny, R.J. Cell penetrating peptides fail to induce an innate immune response in epithelial cells in vitro: Implications for continued therapeutic use. Eur. J. Pharm. Biopharm. 2013, 85, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Peptide | Sequence | Application |
|---|---|---|
| Covalent conjugated CPPs | ||
| B | (RXRRBR)2XB | SSO for DMD, DM1 [9,10,11] |
| B-MSP | (RXRRBR)2XBASSLNIA | SSO for DMD [12] |
| Pip6 | RXRRBRRXR YQFLI RXRBRXRB | SSO for DMD, SMA [13,14,15,16] |
| M12 | RRQPPRSISSHP | SSO for DMD [17] |
| Br-ApoE(K→A) | Ac-LRALRARLLRGGAc-LRALRARLLRGGKX-Bpg-G | SSO for SMA [18] |
| P4 | LGAQSNF | SSO (2OMe) for DMD [19] |
| (RXR)4 | RXRRXRRXRRXR | anti-viral anti-bacterial [20,21] |
| Nanoparticle forming CPPs | ||
| MPG-8 | βAFLGWLGAWGTMGWSPKKKRK-Cya | siRNA for xenograft tumor model [22] |
| CADY | Ac-GLWRALWRLLRSLWRLLWRA-Cya | siRNA, cell lines (various) [23] |
| RICK | KWLLRWLSRLLRWLARWLG | siRNA, human glioblastoma cells [24] |
| Pepfect 3 | stearyl-AGYLLGKINLKALAALAKKIL-NH2 | Plasmid DNA, cell lines, intramuscular [25] |
| Pepfect 6 | See reference | siRNA, cell lines (various), systemic IV [26] |
| Pepfect 14 | stearyl-AGYLLGKLLOOLAAAALOOLL-NH2 | Plasmid DNA, SSO [27,28] |
| RVG-9R | YTIWMPENPRPGTPCDIFTNSRGKRASNGGGGRRRRRRRRR | siRNA, brain-targeting disease models [29] |
| 599 | GLFEAIEGFIENGWEGMIDGWYGGGGRRRRRRRRRK | siRNA, oral cancer [30,31] |
| H3K(+H)4b | Branched KHHHKHHHKHHHHKHHHK | siRNA, tumor xenograft [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. https://doi.org/10.3390/biomedicines6020051
McClorey G, Banerjee S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines. 2018; 6(2):51. https://doi.org/10.3390/biomedicines6020051
Chicago/Turabian StyleMcClorey, Graham, and Subhashis Banerjee. 2018. "Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics" Biomedicines 6, no. 2: 51. https://doi.org/10.3390/biomedicines6020051
APA StyleMcClorey, G., & Banerjee, S. (2018). Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines, 6(2), 51. https://doi.org/10.3390/biomedicines6020051