VHL and Hypoxia Signaling: Beyond HIF in Cancer


Abstract
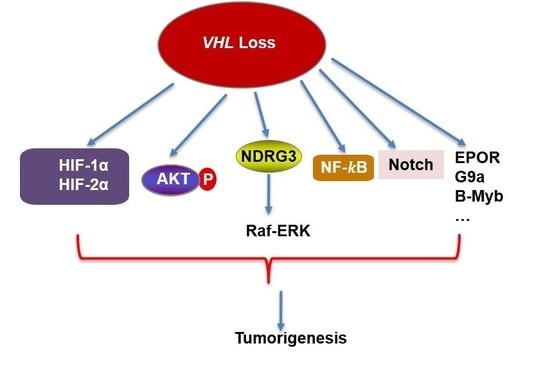
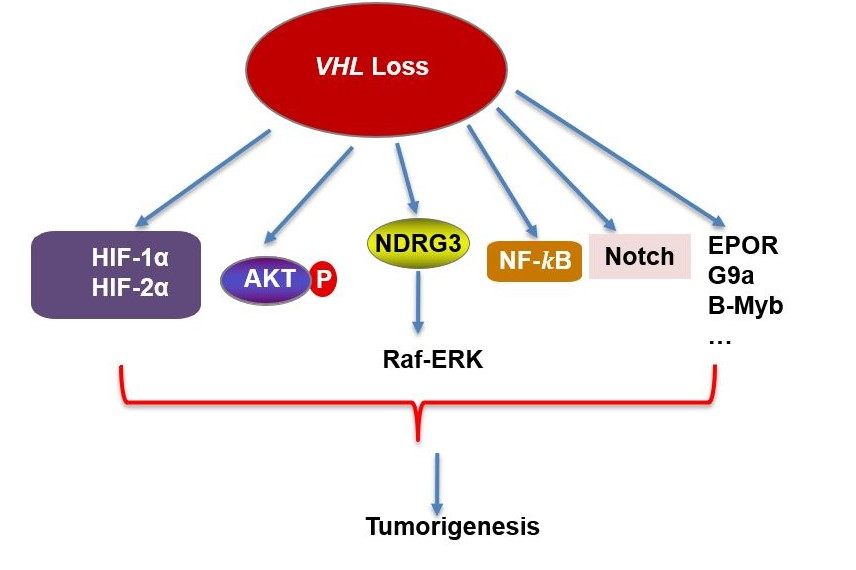{kind=link}
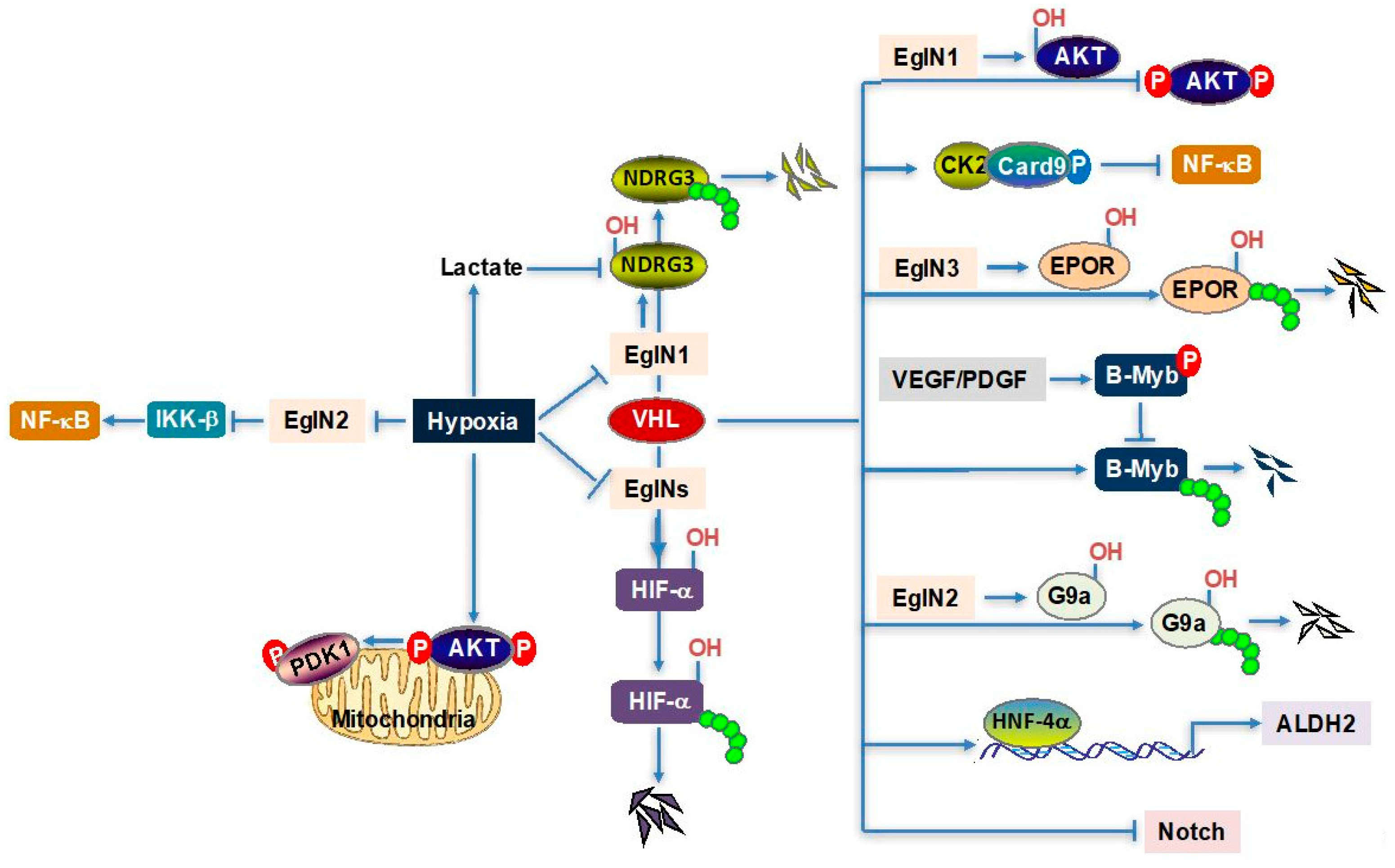{kind=link}
1. Introduction
2. Hypoxia/VHL and Lactate–NDRG3–Raf–ERK Axis
3. Hypoxia/VHL–AKT–mTOR Axis
4. VHL–NF-κB Axis
5. EglN3–VHL–EPOR Axis
6. VHL–B-Myb Axis
7. Other VHL Downstream Regulators
8. VHL and Epigenetic Regulation
9. Targeting VHL Signaling in ccRCC
10. Perspectives
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J. Systemic therapy for metastatic renal-cell carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.E.; Nickerson, M.L.; Brennan, P.; Toro, J.R.; Jaeger, E.; Rinsky, J.; Han, S.S.; Zaridze, D.; Matveev, V.; Janout, V.; et al. Von hippel-lindau (vhl) inactivation in sporadic clear cell renal cancer: Associations with germline vhl polymorphisms and etiologic risk factors. PLoS Genet. 2011, 7, e1002312. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von hippel-lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Kishida, T.; Stackhouse, T.M.; Chen, F.; Lerman, M.I.; Zbar, B. Cellular proteins that bind the von hippel-lindau disease gene product: Mapping of binding domains and the effect of missense mutations. Cancer Res. 1995, 55, 4544–4548. [Google Scholar] [PubMed]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von hippel-lindau tumor suppressor protein to elongin b and c. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.R.; Pause, A.; Burgess, W.H.; Aso, T.; Chen, D.Y.; Garrett, K.P.; Conaway, R.C.; Conaway, J.W.; Linehan, W.M.; Klausner, R.D. Inhibition of transcription elongation by the vhl tumor suppressor protein. Science 1995, 269, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Molecular basis of the vhl hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of hif-alpha to the von hippel-lindau ubiquitylation complex by o2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. Hifalpha targeted for vhl-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. How oxygen makes its presence felt. Genes Dev. 2002, 16, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kaelin, W.G., Jr. Molecular pathogenesis of the von hippel-lindau hereditary cancer syndrome: Implications for oxygen sensing. Cell Growth Differ. 2001, 12, 447–455. [Google Scholar] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. Vhl, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Tan, C.C.; Jones, R.W.; Ratcliffe, P.J. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 10553–10557. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in hypoxia-inducible factor biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.A.; Ohh, M.; Yang, H.; Klco, J.M.; Ivan, M.; Kaelin, W.G., Jr. Von hippel-lindau protein mutants linked to type 2c vhl disease preserve the ability to downregulate hif. Hum. Mol. Genet. 2001, 10, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous chuvash polycythemia vhl mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.C.; Cockman, M.E.; Smallwood, A.C.; Mole, D.R.; Woodward, E.R.; Maxwell, P.H.; Ratcliffe, P.J.; Maher, E.R. Contrasting effects on hif-1alpha regulation by disease-causing pvhl mutations correlate with patterns of tumourigenesis in von hippel-lindau disease. Hum. Mol. Genet. 2001, 10, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Calzada, M.J.; Esteban, M.A.; Feijoo-Cuaresma, M.; Castellanos, M.C.; Naranjo-Suarez, S.; Temes, E.; Mendez, F.; Yanez-Mo, M.; Ohh, M.; Landazuri, M.O. Von hippel-lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006, 66, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G., Jr. PVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappab agonist Card9 by CK2. Mol. Cell 2007, 28, 15–27. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. PVHL suppresses kinase activity of akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Arreola, A.; Payne, L.B.; Julian, M.H.; de Cubas, A.A.; Daniels, A.B.; Taylor, S.; Zhao, H.; Darden, J.; Bautch, V.L.; Rathmell, W.K.; et al. Von hippel-lindau mutations disrupt vascular patterning and maturation via notch. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, G.; Guo, J.; Takada, M.; Wei, W.; Zhang, Q. New insights into protein hydroxylation and its important role in human diseases. Biochim. Biophys. Acta 2016, 1866, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Lee, D.C.; Yeom, Y.I. Ndrg3-mediated lactate signaling in hypoxia. BMB Rep. 2015, 48, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex iii is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Iliopoulos, D.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.; Struhl, K.; Tsichlis, P.N. Akt2 regulates all akt isoforms and promotes resistance to hypoxia through induction of mir-21 upon oxygen deprivation. Cancer Res. 2011, 71, 4720–4731. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial akt regulation of hypoxic tumor reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A pan-cancer proteogenomic atlas of pi3k/akt/mtor pathway alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ohh, M. The von hippel-lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappab-dependent antiapoptotic pathway. Cancer Res. 2003, 63, 7076–7080. [Google Scholar] [PubMed]
- Ghosh, S.; Hayden, M.S. Celebrating 25 years of nf-kappab research. Immunol. Rev. 2012, 246, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-kappab pathway and cancer stem cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-kappab: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Oya, M.; Takayanagi, A.; Horiguchi, A.; Mizuno, R.; Ohtsubo, M.; Marumo, K.; Shimizu, N.; Murai, M. Increased nuclear factor-kappa b activation is related to the tumor development of renal cell carcinoma. Carcinogenesis 2003, 24, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Devarajan, K.; Yang, D.H.; Knudson, A.G.; Balachandran, S. Meta-analysis identifies nf-kappab as a therapeutic target in renal cancer. PLoS ONE 2013, 8, e76746. [Google Scholar] [CrossRef] [PubMed]
- Sourbier, C.; Danilin, S.; Lindner, V.; Steger, J.; Rothhut, S.; Meyer, N.; Jacqmin, D.; Helwig, J.J.; Lang, H.; Massfelder, T. Targeting the nuclear factor-kappab rescue pathway has promising future in human renal cell carcinoma therapy. Cancer Res. 2007, 67, 11668–11676. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates ikappab kinase-beta, giving insight into hypoxia-induced nfkappab activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, C.; Toi, M. Nuclear factor-kappab inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Anti-inflammatory agents for cancer therapy. Mol. Cell. Pharmacol. 2009, 1, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting inflammation in cancer prevention and therapy. Cancer Prev. Res. (Phila) 2016, 9, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Sica, A.; Vannucci, L.; Allavena, P. Inflammation as target in cancer therapy. Curr. Opin. Pharmacol. 2017, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kamba, T.; Yamasaki, T.; Shibasaki, N.; Saito, R.; Terada, N.; Toda, Y.; Mikami, Y.; Inoue, T.; Kanematsu, A.; et al. Junb promotes cell invasion and angiogenesis in vhl-defective renal cell carcinoma. Oncogene 2012, 31, 3098–3110. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflugers Arch. 2005, 450, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. Jak2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Wu, H.; Liu, X.; Jaenisch, R.; Lodish, H.F. Generation of committed erythroid bfu-e and cfu-e progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995, 83, 59–67. [Google Scholar] [CrossRef]
- Gruber, M.; Hu, C.J.; Johnson, R.S.; Brown, E.J.; Keith, B.; Simon, M.C. Acute postnatal ablation of hif-2alpha results in anemia. Proc. Natl. Acad. Sci. USA 2007, 104, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, P.R.; Pei, F.; Lee, R.; Kerestes, H.; Percy, M.J.; Keith, B.; Simon, M.C.; Lappin, T.R.; Khurana, T.S.; Lee, F.S. A knock-in mouse model of human PHD2 gene-associated erythrocytosis establishes a haploinsufficiency mechanism. J. Biol. Chem. 2013, 288, 33571–33584. [Google Scholar] [CrossRef] [PubMed]
- Heir, P.; Srikumar, T.; Bikopoulos, G.; Bunda, S.; Poon, B.P.; Lee, J.E.; Raught, B.; Ohh, M. Oxygen-dependent regulation of erythropoietin receptor turnover and signaling. J. Biol. Chem. 2016, 291, 7357–7372. [Google Scholar] [CrossRef] [PubMed]
- Sala, A. B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur. J. Cancer 2005, 41, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Manak, J.R.; Wen, H.; Van, T.; Andrejka, L.; Lipsick, J.S. Loss of drosophila myb interrupts the progression of chromosome condensation. Nat. Cell Biol. 2007, 9, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Uematsu, K.; Byrne, S.D.; Hirano, M.; Joo-Okumura, A.; Nishikimi, A.; Shuin, T.; Fukui, Y.; Nakatsukasa, K.; Kamura, T. Parallel regulation of von hippel-lindau disease by pvhl-mediated degradation of b-myb and hypoxia-inducible factor alpha. Mol. Cell. Biol. 2016, 36, 1803–1817. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, D.; Zhang, W.; Ouyang, G.; Wang, J.; Liu, X.; Li, S.; Ji, W.; Liu, W.; Xiao, W. Pvhl negatively regulates antiviral signaling by targeting mavs for proteasomal degradation. J. Immunol. 2015, 195, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Segura, I.; Lange, C.; Knevels, E.; Moskalyuk, A.; Pulizzi, R.; Eelen, G.; Chaze, T.; Tudor, C.; Boulegue, C.; Holt, M.; et al. The oxygen sensor phd2 controls dendritic spines and synapses via modification of filamin A. Cell Rep. 2016, 14, 2653–2667. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zheng, L.; Liu, W.; Zhang, D.; Li, W.; Yuan, L. Rootletin prevents Cep68 from VHL-mediated proteasomal degradation to maintain centrosome cohesion. Biochim. Biophys. Acta 2017, 1864, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Dushukyan, N.; Dunn, D.M.; Sager, R.A.; Woodford, M.R.; Loiselle, D.R.; Daneshvar, M.; Baker-Williams, A.J.; Chisholm, J.D.; Truman, A.W.; Vaughan, C.K.; et al. Phosphorylation and ubiquitination regulate protein phosphatase 5 activity and its prosurvival role in kidney cancer. Cell Rep. 2017, 21, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Hasanov, E.; Chen, G.; Chowdhury, P.; Weldon, J.; Ding, Z.; Jonasch, E.; Sen, S.; Walker, C.L.; Dere, R. Ubiquitination and regulation of aurka identifies a hypoxia-independent e3 ligase activity of vhl. Oncogene 2017, 36, 3450–3463. [Google Scholar] [CrossRef] [PubMed]
- Gamper, A.M.; Qiao, X.; Kim, J.; Zhang, L.; DeSimone, M.C.; Rathmell, W.K.; Wan, Y. Regulation of klf4 turnover reveals an unexpected tissue-specific role of pvhl in tumorigenesis. Mol. Cell 2012, 45, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.V.; Meller, J.; Schnell, P.O.; Nash, J.A.; Ignacak, M.L.; Sanchez, Y.; Conaway, J.W.; Conaway, R.C.; Czyzyk-Krzeska, M.F. Von hippel-lindau protein binds hyperphosphorylated large subunit of rna polymerase ii through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 2003, 100, 2706–2711. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylova, O.; Ignacak, M.L.; Barankiewicz, T.J.; Harbaugh, S.V.; Yi, Y.; Maxwell, P.H.; Schneider, M.; Van Geyte, K.; Carmeliet, P.; Revelo, M.P.; et al. The von hippel-lindau tumor suppressor protein and egl-9-type proline hydroxylases regulate the large subunit of rna polymerase ii in response to oxidative stress. Mol. Cell. Biol. 2008, 28, 2701–2717. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Saitoh, K.; Hirai, S.; Iwai, K.; Takaki, Y.; Baba, M.; Minato, N.; Ohno, S.; Shuin, T. The von hippel-lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase c. J. Biol. Chem. 2001, 276, 43611–43617. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Nordquist, K.A.; Gao, X.; Hicks, K.C.; Zhai, B.; Gygi, S.P.; Patel, T.B. Regulation of cellular levels of sprouty2 protein by prolyl hydroxylase domain and von hippel-lindau proteins. J. Biol. Chem. 2011, 286, 42027–42036. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, H. The von hippel-lindau tumor suppressor protein promotes c-cbl-independent poly-ubiquitylation and degradation of the activated egfr. PLoS ONE 2011, 6, e23936. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xiao, K.; Whalen, E.J.; Forrester, M.T.; Freeman, R.S.; Fong, G.; Gygi, S.P.; Lefkowitz, R.J.; Stamler, J.S. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by egln3 and ubiquitylation by pvhl. Sci. Signal. 2009, 2, ra33. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Qiao, M.; Song, M.; Weintraub, S.T.; Shiio, Y. Quantitative proteomics identifies the myb-binding protein p160 as a novel target of the von hippel-lindau tumor suppressor. PLoS ONE 2011, 6, e16975. [Google Scholar] [CrossRef] [PubMed]
- Na, X.; Duan, H.O.; Messing, E.M.; Schoen, S.R.; Ryan, C.K.; di Sant’Agnese, P.A.; Golemis, E.A.; Wu, G. Identification of the RNA polymerase ii subunit hsRPB7 as a novel target of the von hippel-lindau protein. EMBO J. 2003, 22, 4249–4259. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.J.; Park, J.S.; Lee Jeong, A.; Han, S.; Lee, S.; Ka, H.I.; Sumiyasuren, B.; Joo, H.J.; So, S.J.; Park, J.Y.; et al. Von hippel-lindau regulates interleukin-32beta stability in ovarian cancer cells. Oncotarget 2017, 8, 69833–69846. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, J.; Liu, F.; Li, H.; Archacki, S.; Gao, M.; Liu, Y.; Liao, S.; Huang, M.; Wang, J.; Yu, S.; et al. Pvhl interacts with ceramide kinase like (cerkl) protein and ubiquitinates it for oxygen dependent proteasomal degradation. Cell. Signal. 2015, 27, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Arias-Gonzalez, L.; Moreno-Gimeno, I.; del Campo, A.R.; Serrano-Oviedo, L.; Valero, M.L.; Esparis-Ogando, A.; de la Cruz-Morcillo, M.A.; Melgar-Rojas, P.; Garcia-Cano, J.; Cimas, F.J.; et al. Erk5/bmk1 is a novel target of the tumor suppressor vhl: Implication in clear cell renal carcinoma. Neoplasia 2013, 15, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.H.; Wu, Z.X.; Xie, L.Q.; Li, C.X.; Mao, Y.Q.; Duan, Y.T.; Han, B.; Han, S.F.; Yu, Y.; Lu, H.J.; et al. Vhl deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating aldh2. Nat. Commun. 2017, 8, 15337. [Google Scholar] [CrossRef] [PubMed]
- Sjolund, J.; Johansson, M.; Manna, S.; Norin, C.; Pietras, A.; Beckman, S.; Nilsson, E.; Ljungberg, B.; Axelson, H. Suppression of renal cell carcinoma growth by inhibition of notch signaling in vitro and in vivo. J. Clin. Investig. 2008, 118, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.L.; Salama, R.; Smythies, J.; Choudhry, H.; Davies, J.O.; Hughes, J.R.; Ratcliffe, P.J.; Mole, D.R. Capture-c reveals preformed chromatin interactions between hif-binding sites and distant promoters. EMBO Rep. 2016, 17, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Grampp, S.; Platt, J.L.; Lauer, V.; Salama, R.; Kranz, F.; Neumann, V.K.; Wach, S.; Stohr, C.; Hartmann, A.; Eckardt, K.U.; et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects hif binding to a myc enhancer. Nat. Commun. 2016, 7, 13183. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Bardella, C.; Sciesielski, L.K.; Brown, J.M.; Pugh, C.W.; Buckle, V.; Tomlinson, I.P.; Ratcliffe, P.J.; Mole, D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of hif to an enhancer of cyClin. d1 expression. Nat. Genet. 2012, 44, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. Vhl deficiency drives enhancer activation of oncogenes in clear cell renal cell carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of hif binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of jumonji-domain-containing histone demethylases by hypoxia-inducible factor (hif)-1alpha. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases jmjd1a and jmjd2b are transcriptional targets of hypoxia-inducible factor hif. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase jmjd1a by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.A.; Nakamura, E.; Qi, J.; Creech, A.; Jaffe, J.D.; Paulk, J.; Novak, J.S.; Nagulapalli, K.; McBrayer, S.K.; Cowley, G.S.; et al. Hif activation causes synthetic lethality between the vhl tumor suppressor and the ezh1 histone methyltransferase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Choi, K.; Oh, H.; Park, Y.K.; Park, H. Hif-1-dependent induction of jumonji domain-containing protein (jmjd) 3 under hypoxic conditions. Mol. Cells 2014, 37, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone h3 lysine 9 through histone methyltransferase g9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Barsyte-Lovejoy, D.; Li, F.; Xiong, Y.; Korboukh, V.; Huang, X.P.; Allali-Hassani, A.; Janzen, W.P.; Roth, B.L.; Frye, S.V.; et al. Discovery of an in vivo chemical probe of the lysine methyltransferases g9a and glp. J. Med. Chem. 2013, 56, 8931–8942. [Google Scholar] [CrossRef] [PubMed]
- Espana-Agusti, J.; Warren, A.; Chew, S.K.; Adams, D.J.; Matakidou, A. Loss of pbrm1 rescues vhl dependent replication stress to promote renal carcinogenesis. Nat. Commun. 2017, 8, 2026. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate hif1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. Hif-alpha effects on c-myc distinguish two subtypes of sporadic vhl-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of hif2alpha is sufficient to suppress pvhl-defective tumor growth. PLoS Biol. 2003, 1, E83. [Google Scholar] [CrossRef] [PubMed]
- Koehler, A.N. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010, 14, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Li, Q.; Ma, H.W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a hif-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a hif-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase i dose-escalation trial of pt2385, a first-in-class hypoxia-inducible factor-2alpha antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Chan, D.A.; Sutphin, P.D.; Hay, M.P.; Denny, W.A.; Giaccia, A.J. A molecule targeting vhl-deficient renal cell carcinoma that induces autophagy. Cancer Cell 2008, 14, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting glut1 and the warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Sutphin, P.D.; Chan, D.A.; Li, J.M.; Turcotte, S.; Krieg, A.J.; Giaccia, A.J. Targeting the loss of the von hippel-lindau tumor suppressor gene in renal cell carcinoma cells. Cancer Res. 2007, 67, 5896–5905. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Nguyen, Q.H.; Singh, M.; Pavesic, M.W.; Nesterenko, I.; Nelson, L.J.; Liao, A.C.; Razorenova, O.V. Rho-associated kinase 1 inhibition is synthetically lethal with von hippel-lindau deficiency in clear cell renal cell carcinoma. Oncogene 2017, 36, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Bommi-Reddy, A.; Almeciga, I.; Sawyer, J.; Geisen, C.; Li, W.; Harlow, E.; Kaelin, W.G., Jr.; Grueneberg, D.A. Kinase requirements in human cells: Iii. Altered kinase requirements in vhl−/− cancer cells detected in a pilot synthetic lethal screen. Proc. Natl. Acad. Sci. USA 2008, 105, 16484–16489. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.C.; Pavia-Jimenez, A.; Tcheuyap, V.T.; Alexander, S.; Vishwanath, M.; Christie, A.; Xie, X.J.; Williams, N.S.; Kapur, P.; Posner, B.; et al. High-throughput simultaneous screen and counterscreen identifies homoharringtonine as synthetic lethal with von hippel-lindau loss in renal cell carcinoma. Oncotarget 2015, 6, 16951–16962. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and characterization of von hippel-lindau-recruiting proteolysis targeting chimeras (protacs) of tank-binding kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35. https://doi.org/10.3390/biomedicines6010035
Zhang J, Zhang Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines. 2018; 6(1):35. https://doi.org/10.3390/biomedicines6010035
Chicago/Turabian StyleZhang, Jing, and Qing Zhang. 2018. "VHL and Hypoxia Signaling: Beyond HIF in Cancer" Biomedicines 6, no. 1: 35. https://doi.org/10.3390/biomedicines6010035
APA StyleZhang, J., & Zhang, Q. (2018). VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines, 6(1), 35. https://doi.org/10.3390/biomedicines6010035