The Role of Angiogenesis in Cancer Treatment



Abstract
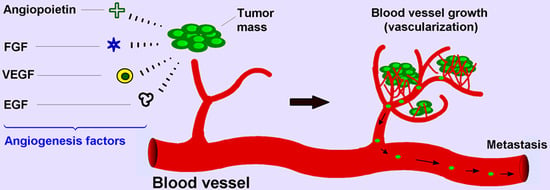
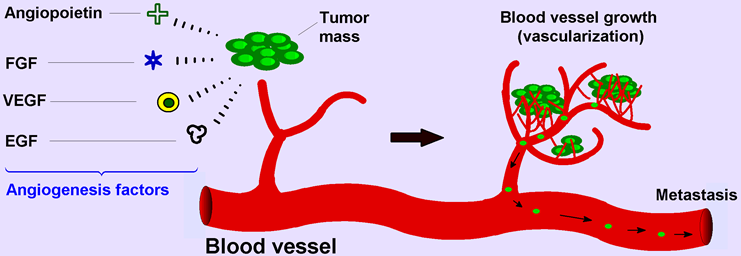
:
1. Introduction
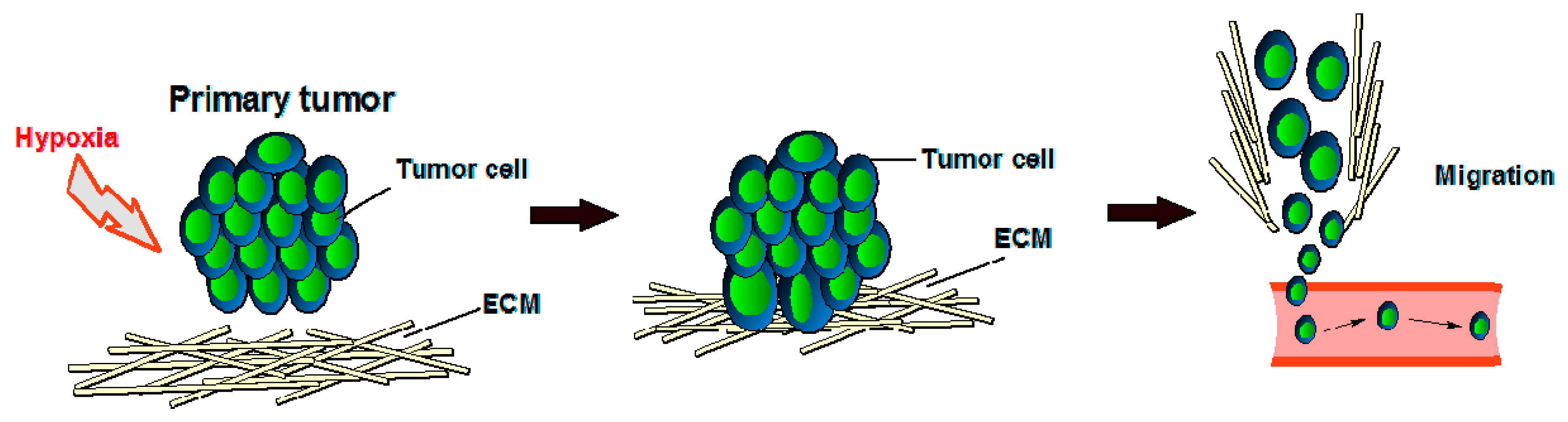
2. Angiogenesis Mechanism in Cancer
3. Side Effects in Anti-Angiogenic Therapy
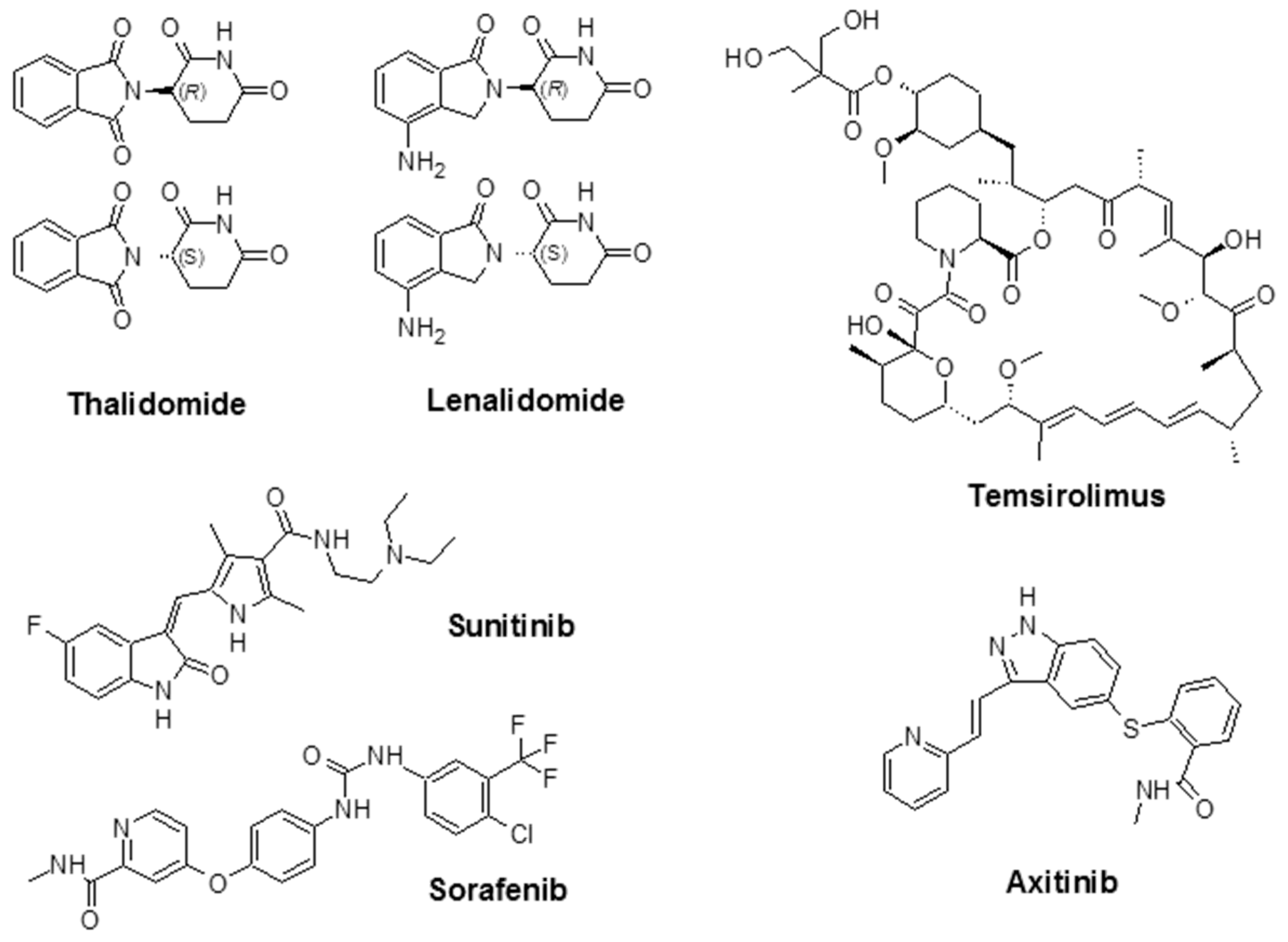
4. Examples of Angiogenesis Inhibitors for Cancer Therapy
5. Conclusions
Conflicts of Interest
References
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Distler, O. Vasculopathy and disordered angiogenesis in selected rheumatic diseases: Rheumatoid arthritis and systemic sclerosis. Arthritis Res. Ther. 2007, 9 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Davis, P.J. Angiogenesis and anti-angiogenesis strategies in cancer. In Anti-Angiogenesis Strategies in Cancer Therapies, 1st ed.; Mousa, S.A., Davis, P.J., Eds.; Academic Press: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-a. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar] [PubMed]
- Javaherian, K.; Lee, T.-Y.; Tjin Tham Sjin, R.M.; Parris, G.E.; Hlatky, L. Two endogenous antiangiogenic inhibitors, endostatin and angiostatin, demonstrate biphasic curves in their antitumor profiles. Dose Response 2011, 9, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, S.Y. Angiopoietin-2: Development of inhibitors for cancer therapy. Curr. Oncol. Rep. 2009, 11, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A. Platelet factor 4: An inhibitor of angiogenesis. Semin. Thromb. Hemost. 2004, 30, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.M.; Zhou, B.; Han, Z.C. Roles of platelet factor 4 in hematopoiesis and angiogenesis. Growth Factors 2006, 24, 242–252. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Staton, C.A.; Lewis, C.E. Angiogenesis inhibitors found within the haemostasis pathway. J. Cell. Mol. Med. 2005, 9, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef] [PubMed]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.M.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. In Matrix Metalloproteinase Inhibitors; Springer Basel: Basel, Switzerland, 2012; pp. 209–279. [Google Scholar]
- McCrae, K.R.; Donate, F.; Merkulov, S.; Sun, D.; Qi, X.; Shaw, D.E. Inhibition of angiogenesis by cleaved high molecular weight kininogen (HKa) and HKa domain 5. Curr. Cancer Drug Targets 2005, 5, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Holroyd, E.W.; Chir, B.; Delacroix, S.; Larsen, K.; Harbuzariu, A.; Psaltis, P.J.; Wang, L.; Pan, S.; White, T.A.; Witt, T.A.; et al. Tissue factor pathway inhibitor blocks angiogenesis via its carboxyl terminus: Holroyd: TFPI regulates angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Huegel, R.; Velasco, P.; De la Luz Sierra, M.; Christophers, E.; Schroder, J.M.; Schwarz, T.; Tosato, G.; Lange-Asschenfeldt, B. Novel anti-inflammatory properties of the angiogenesis inhibitor vasostatin. J. Investig. Dermatol. 2007, 127, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lange-Asschenfeldt, B.; Velasco, P.; Streit, M.; Hawighorst, T.; Pike, S.E.; Tosato, G.; Detmar, M. The angiogenesis inhibitor vasostatin does not impair wound healing at tumor-inhibiting doses. J. Investig. Dermatol. 2001, 117, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Pike, S.E.; Yao, L.; Setsuda, J.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Atreya, C.D.; Teruya-Feldstein, J.; et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999, 94, 2461–2468. [Google Scholar] [PubMed]
- Takawale, A.; Zhang, P.; Azad, A.; Wang, W.; Wang, X.; Murray, A.G.; Kassiri, Z. Myocardial overexpression of TIMP3 following myocardial infarction exerts beneficial effects through promoting angiogenesis and suppressing early proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, Y.; Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Noguchi, R.; Tsujinoue, H.; Yanase, K.; Namisaki, T.; Imazu, H.; Masaki, T.; et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) inhibits tumor growth and angiogenesis in the TIMP-1 transgenic mouse model. Int. J. Cancer 2003, 105, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Fu, C.C.; Tsai, M.L.; Chung, W.J. Effect of U-995, a potent shark cartilage-derived angiogenesis inhibitor, on anti-angiogenesis and anti-tumor activities. Anticancer Res. 1998, 18, 4435–4441. [Google Scholar] [PubMed]
- Vazquez, F.; Hastings, G.; Ortega, M.A.; Lane, T.F.; Oikemus, S.; Lombardo, M.; Iruela-Arispe, M.L. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J. Biol. Chem. 1999, 274, 23349–23357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Volpert, O.; Shi, Y.H.; Bouck, N. Maspin is an angiogenesis inhibitor. Nat. Med. 2000, 6, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ponce, M.L.; Kleinman, H.K. Identification of redundant angiogenic sites in laminin alpha1 and gamma1 chains. Exp. Cell Res. 2003, 285, 189–195. [Google Scholar] [CrossRef]
- Pouliot, N.; Kusuma, N. Laminin-511: A multi-functional adhesion protein regulating cell migration, tumor invasion and metastasis. Cell Adhes. Migr. 2013, 7, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.G.; Leu, S.J.; Chen, N.; Tebeau, C.M.; Lin, S.X.; Yeung, C.Y.; Lau, L.F. CCN3 (nov) is a novel angiogenic regulator of the CCN protein family. J. Biol. Chem. 2003, 278, 24200–24208. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, E.; Colladel, R.; Andreuzzi, E.; Marastoni, S.; Todaro, F.; Schiappacassi, M.; Ligresti, G.; Colombatti, A.; Mongiat, M. MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF-a/VEGFR2 pathway. Oncogene 2012, 31, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. Hostile takeover: How tumors hijack pre-existing vascular environments to thrive. J. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A. Mechanisms of Angiogenesis: Potential Therapeutic Targets; Eurekah.com/Landes Bioscience: Georgetown, WA, USA, 2000. [Google Scholar]
- National Cancer Institute. Angiogenesis Inhibitors. Available online: http://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet (accessed on 12 April 2017).
- Ward, J.P. Oxygen sensors in context. Biochim. Biophys. Acta 2008, 1777, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Endo, M.; Ferrara, N.; Hlatky, L.; Jain, R.K. Formation of endothelial cell networks. Nature 2000, 405, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Algenstaedt, N.; Stoll, B.R.; Padera, T.P.; Dolmans, D.E.; Hicklin, D.J.; Fukumura, D.; Jain, R.K. Tumor oxygenation in hormone-dependent tumors during vascular endothelial growth factor receptor-2 blockade, hormone ablation, and chemotherapy. Cancer Res. 2000, 60, 4556–4560. [Google Scholar] [PubMed]
- Pavlakovic, H.; Havers, W.; Schweigerer, L. Multiple angiogenesis stimulators in a single malignancy: Implications for anti-angiogenic tumour therapy. Angiogenesis 2001, 4, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.H.; O’Donnell, A.L.; Balu, D.; Pohl, M.B.; Seyler, M.J.; Mohamed, S.; Mousa, S.; Dandona, P. Estrogen receptor-alpha in the inhibition of cancer growth and angiogenesis. Cancer Res. 2000, 60, 7094–7098. [Google Scholar] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Lin, H.Y.; Tang, H.Y.; Hercbergs, A.; Luidens, M.K.; Davis, P.J. Modulation of angiogenesis by thyroid hormone and hormone analogues: Implications for cancer management. Angiogenesis 2014, 17, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Srinivasan, M.; Mousa, S.A. Nanobiomaterials in drug delivery. In Nanobiomaterials in Drug Delivery: Applications of Nanobiomaterials; Grumezescu, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 9, pp. 1–39. [Google Scholar]
- Srinivasan, M.; Rajabi, M.; Mousa, S.A. Nanobiomaterials in cancer therapy. In Nanobiomaterials in Cancer Therapy: Applications of Nanobiomaterials; Grumezescu, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 7, pp. 57–89. [Google Scholar]
- Rajabi, M.; Sudha, T.; Darwish, N.H.E.; Davis, P.J.; Mousa, S.A. Synthesis of MR-49, a deiodinated analog of tetraiodothyroacetic acid (tetrac), as a novel pro-angiogenesis modulator. Bioorg. Med. Chem. Lett. 2016, 26, 4112–4116. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Solito, R.; Morbidelli, L.; Giachetti, A.; Ziche, M.; Donnini, S. Prostaglandin E2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J. Biol. Chem. 2008, 283, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci. Transl. Med. 2014, 6, 242ra284. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Natesan, V.; Shi, H.; Hamik, A.; Kawanami, D.; Hao, C.; Mahabaleshwar, G.H.; Wang, W.; Jin, Z.G.; Atkins, G.B.; et al. A novel role of CCN3 in regulating endothelial inflammation. J. Cell. Commun. Signal. 2010, 4, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; van der Voort, D.; Shi, H.; Zhang, R.; Qing, Y.; Hiraoka, S.; Takemoto, M.; Yokote, K.; Moxon, J.V.; Norman, P.; et al. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J. Clin. Investig. 2016, 126, 1282–1299. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Cheng, H.C.; Wang, J.; Wang, S.W.; Tai, H.C.; Lin, C.W.; Tang, C.H. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget 2014, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Connor, A.R.; Sounni, N.E.; Eckhard, U.; Morrison, C.J.; Noel, A.; Overall, C.M. Degradomic and yeast 2-hybrid inactive catalytic domain substrate trapping identifies new membrane-type 1 matrix metalloproteinase (MMP14) substrates: CCN3 (Nov) and CCN5 (WISP2). Matrix Biol. 2017, 59, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Andreuzzi, E.; Colladel, R.; Pellicani, R.; Tarticchio, G.; Cannizzaro, R.; Spessotto, P.; Bussolati, B.; Brossa, A.; De Paoli, P.; Canzonieri, V.; et al. The angiostatic molecule multimerin 2 is processed by MMP-9 to allow sprouting angiogenesis. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Colladel, R.; Pellicani, R.; Andreuzzi, E.; Paulitti, A.; Tarticchio, G.; Todaro, F.; Colombatti, A.; Mongiat, M. MULTIMERIN2 binds VEGF-a primarily via the carbohydrate chains exerting an angiostatic function and impairing tumor growth. Oncotarget 2016, 7, 2022–2037. [Google Scholar] [PubMed]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Belcik, J.T.; Qi, Y.; Kaufmann, B.A.; Xie, A.; Bullens, S.; Morgan, T.K.; Bagby, S.P.; Kolumam, G.; Kowalski, J.; Oyer, J.A.; et al. Cardiovascular and systemic microvascular effects of anti-vascular endothelial growth factor therapy for cancer. J. Am. Coll. Cardiol. 2012, 60, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, D.S.; Bernatchez, P.N.; Rollin, S.; Bazan, N.G.; Sirois, M.G. Immediate and delayed VEGF-mediated NO synthesis in endothelial cells: Role of PI3K, PKC and PLC pathways. Br. J. Pharmacol. 2002, 137, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H1054–H1058. [Google Scholar]
- Sane, D.C.; Anton, L.; Brosnihan, K.B. Angiogenic growth factors and hypertension. Angiogenesis 2004, 7, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Gressett, S.M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Emblem, K.E.; Mouridsen, K.; Bjornerud, A.; Farrar, C.T.; Jennings, D.; Borra, R.J.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; Rosen, B.R.; et al. Vessel architectural imaging identifies cancer patient responders to anti-angiogenic therapy. Nat. Med. 2013, 19, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Gerstner, E.R.; Emblem, K.E.; Duda, D.G.; Kalpathy-Cramer, J.; Snuderl, M.; Ancukiewicz, M.; Polaskova, P.; Pinho, M.C.; Jennings, D.; et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl. Acad. Sci. USA 2013, 110, 19059–19064. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Moimas, S.; Zacchigna, S.; Pattarini, L.; Zentilin, L.; Ruozi, G.; Mano, M.; Sinigaglia, M.; Maione, F.; Serini, G.; et al. Neuropilin-1 identifies a subset of bone marrow Gr1- monocytes that can induce tumor vessel normalization and inhibit tumor growth. Cancer Res. 2012, 72, 6371–6381. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.G.; Emblem, K.E.; Polaskova, P.; Jennings, D.; Kim, H.; Ancukiewicz, M.; Wang, M.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012, 72, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Debreli Coskun, M.; Keating, K.A.; Lin, H.Y.; Davis, P.J.; Mousa, S.A. Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int. J. Nanomed. 2017, 12, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Coskun, M.D.; Keating, K.A.; Lin, H.Y.; Davis, P.J.; Mousa, S.A. Targeted delivery of cisplatin to tumor xenografts via the nanoparticle component of nano-diamino-tetrac. Nanomedicine 2017, 12, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69 (Suppl. 3), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.E.; El-Remessy, A.B. Angiogenesis inhibitors in cancer therapy: Mechanistic perspective on classification and treatment rationales. Br. J. Pharmacol. 2013, 170, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, A.; Hahnfeldt, P.; Maercker, C.; Grone, H.J.; Debus, J.; Ansorge, W.; Folkman, J.; Hlatky, L.; Huber, P.E. Endostatin’s antiangiogenic signaling network. Mol. Cell 2004, 13, 649–663. [Google Scholar] [CrossRef]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Endogenous inhibitors of angiogenesis: A historical review. Leuk. Res. 2009, 33, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Sugimoto, H.; Birrane, G.; Maeshima, Y.; Cooke, V.G.; Kieran, M.; Kalluri, R. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc. Natl. Acad. Sci. USA 2008, 105, 15040–15045. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Fontanini, G.; Cuccato, S.; De Placido, S.; Bianco, A.R.; Tortora, G. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin. Cancer Res. 2001, 7, 1459–1465. [Google Scholar] [PubMed]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Wakelee, H.A. Monoclonal antibodies targeting vascular endothelial growth factor: Current status and future challenges in cancer therapy. BioDrugs 2009, 23, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Giantonio, B.J.; Levy, D.E.; O’Dwyer, P.J.; Meropol, N.J.; Catalano, P.J.; Benson, A.B. A phase II study of high-dose bevacizumab in combination with irinotecan, 5-fluorouracil, leucovorin, as initial therapy for advanced colorectal cancer: Results from the Eastern Cooperative Oncology Group study E2200. Ann. Oncol. 2006, 17, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Kabbinavar, F.; Hurwitz, H.I.; Fehrenbacher, L.; Meropol, N.J.; Novotny, W.F.; Lieberman, G.; Griffing, S.; Bergsland, E. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU) leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J. Clin. Oncol. 2003, 21, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Fehrenbacher, L.; Novotny, W.F.; Herbst, R.S.; Nemunaitis, J.J.; Jablons, D.M.; Langer, C.J.; DeVore, R.F., 3rd; Gaudreault, J.; Damico, L.A.; et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Y.; Yu, P.; Qu, X.J.; Liu, Y.P.; Zhang, J.D. Phase III trials of standard chemotherapy with or without bevacizumab for ovarian cancer: A meta-analysis. PLoS ONE 2013, 8, e81858. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Witzig, T.E. A review of angiogenesis and antiangiogenic therapy with thalidomide in multiple myeloma. Cancer Treat. Rev. 2000, 26, 351–362. [Google Scholar] [CrossRef] [PubMed]
- List, A.; Kurtin, S.; Roe, D.J.; Buresh, A.; Mahadevan, D.; Fuchs, D.; Rimsza, L.; Heaton, R.; Knight, R.; Zeldis, J.B. Efficacy of lenalidomide in myelodysplastic syndromes. N. Engl. J. Med. 2005, 352, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.; Lenz, H.J.; Meropol, N.J.; Posey, J.; Ryan, D.P.; Picus, J.; Bergsland, E.; Stuart, K.; Baum, C.M.; Fuchs, C.S. A phase 2 study to evaluate the efficacy and safety of SU 11248 in patients (pts) with unresectable neuroendocrine tumors (NETs). J. Clin. Oncol. 2005, 23, 310S–310S. [Google Scholar] [CrossRef]
- Socinski, M.A.; Novello, S.; Sanchez, J.M.; Brahmer, J.A.; Govindan, R.; Belani, C.P.; Atkins, J.N.; Gillenwater, H.H.; Palleres, C.; Chao, R.C. Efficacy and safety of sunitinib in previously treated, advanced non-small cell lung cancer (NSCLC): Preliminary results of a multicenter phase II trial. J. Clin. Oncol. 2006, 24, 364S–364S. [Google Scholar]
- Miller, K.D.; Burstein, H.J.; Elias, A.D.; Rugo, H.; Cobleigh, M.A.; Pegram, M.D.; Eisenberg, P.D.; Collier, M.; Adams, B.J.; Baum, C.M. Phase II study of SU11248, a multitargeted receptor tyrosine kinase inhibitor (TKI), in patients (pts) with previously treated metastatic breast cancer (MBC). In Proceedings of the San Antonio Breast Cancer Symposium 28th Annual Meeting, San Antonio, TX, USA, 8–11 December 2005. [Google Scholar]
- Lenz, H.; Marshall, J.; Rosen, L.; Belt, R.; Hurwitz, H.; Eckhardt, S.; Bergsland, E.; Haller, D.; Chao, R.; Saltz, L. Phase II trial of SU11248 in patients with metastatic colorectal cancer (MCRC) after failure of standard chemotherapy. In Proceedings of the American Society of Clinical Oncology Gastrointestinal 4th Annual Meeting, San Francisco, CA, USA, 27–29 January 2006. [Google Scholar]
- Keating, G.M.; Santoro, A. Sorafenib a review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Tao, L.; Wang, X.F.; Liu, Q.Y.; Zhang, W.; Li, Q.Y.; He, C.F.; Xue, D.Y.; Zhang, J.; Liu, C. Sorafenib inhibits renal fibrosis induced by unilateral ureteral obstruction via inhibition of macrophage infiltration. Cell. Physiol. Biochem. 2016, 39, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; Gokare, P.; Gallant, J.N.; Dicker, D.; Whitcomb, T.; Cooper, T.; Liao, J.G.; Derr, J.; Liu, J.; Goldenberg, D.; et al. Sorafenib and quinacrine target anti-apoptotic protein MCL1: A poor prognostic marker in anaplastic thyroid cancer (ATC). Clin. Cancer Res. 2016, 22, 6192–6203. [Google Scholar] [CrossRef] [PubMed]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Lee, C.W.; Goel, H.L.; Bosari, S.; Languino, L.R.; Altieri, D.C. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 2007, 26, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Heng, D.Y.; Bukowski, R.M. Anti-angiogenic targets in the treatment of advanced renal cell carcinoma. Curr. Cancer Drug Targets 2008, 8, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Medina, E.C.; Esquivel, J.A., 2nd; Espitia, C.M.; Smith, S.; Oberheu, K.; Swords, R.; Kelly, K.R.; Mita, M.M.; Mita, A.C.; et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin. Cancer Res. 2010, 16, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Puzanov, I.; Atkins, M.B. Opportunities and obstacles to combination targeted therapy in renal cell cancer. Clin. Cancer Res. 2007, 13, 764s–769s. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 658–671. [Google Scholar] [PubMed]
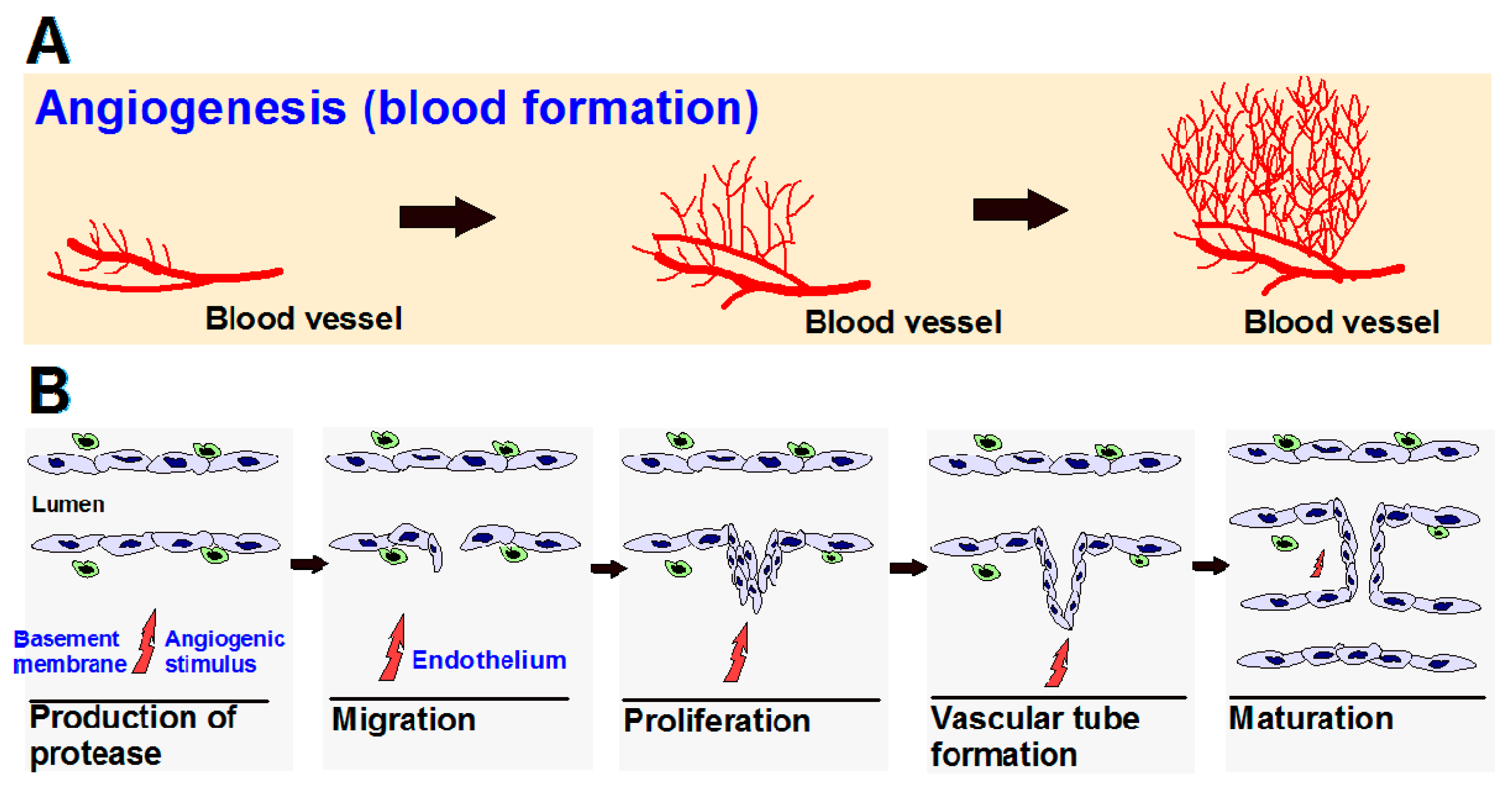



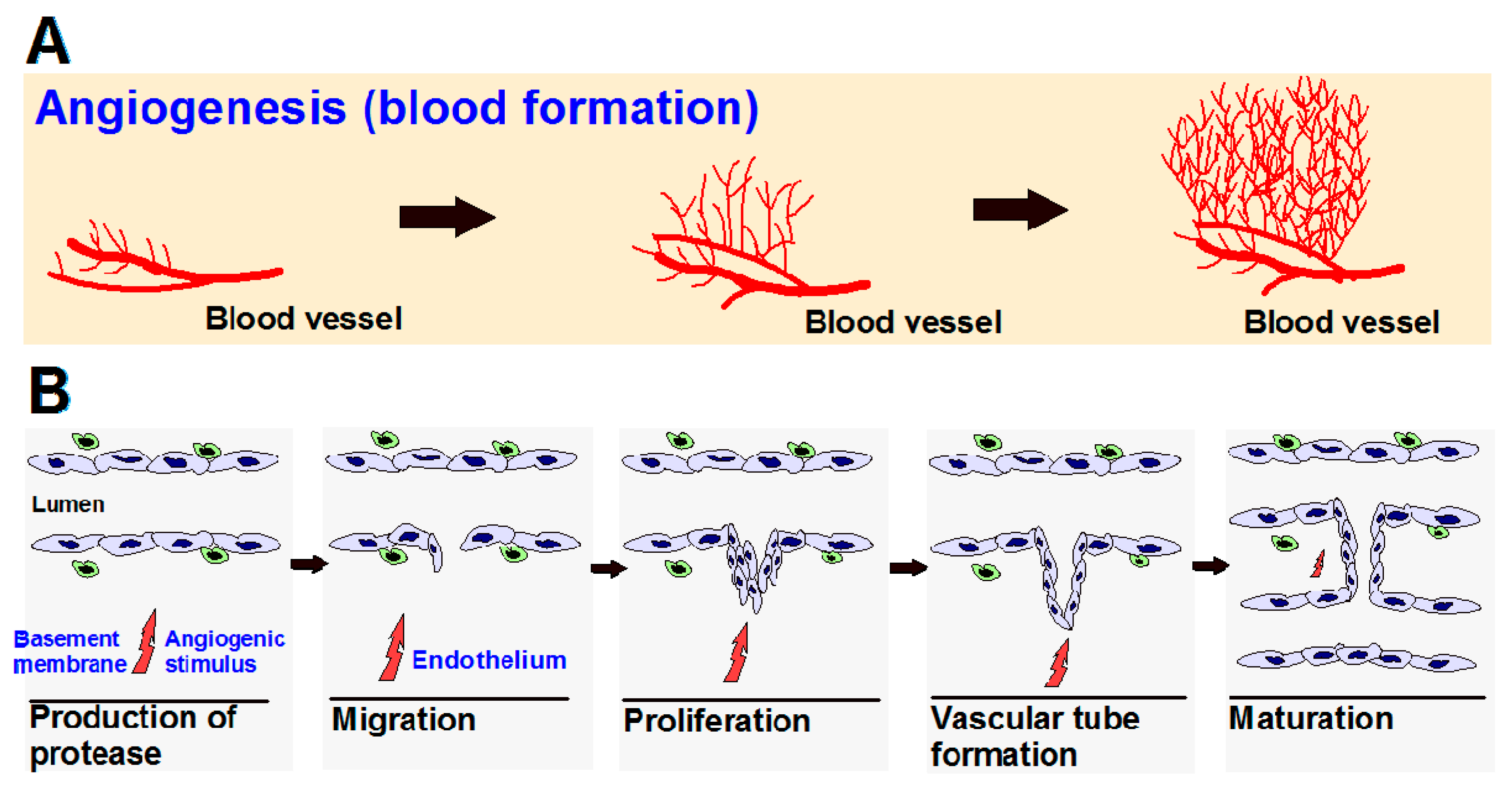
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endogenous Angiogenesis Inhibitors | Mechanisms | Reference |
|---|---|---|
| Soluble VEGF-1 | Decoy receptors for VEGF-B | [5] |
| Angiostatin | Suppress EC adhesion, migration, proliferation | [6] |
| Thrombospondin-1 and -2 | Suppress EC adhesion, migration, proliferation | [7] |
| Angiopoietin-2 | Oppose Angiopoietin 1 | [8] |
| Platelet Factor-4 | Inhibit bFGF (FGF2) and VEGF binding | [9,10] |
| Endostatin | Suppress EC adhesion, migration, proliferation | [6,11] |
| Anti-thrombin III Fragment | Suppress EC adhesion, migration, proliferation | [12] |
| Osteopontin | Serve as ligand for integrin binding | [13] |
| Collagen | Substrate for MMPs | [14,15] |
| Kininogen Domains | Suppress EC adhesion, migration, proliferation | [16] |
| Tissue Factor Pathways Inhibitor | Antagonist for Tissue Factor | [17] |
| Vasostatin | Suppress EC adhesion | [18,19] |
| Calreticulin | Suppress EC adhesion | [20] |
| TIMPs | Suppress EC adhesion | [21,22] |
| A cartilage-derived angiogenesis inhibitor | Suppress EC adhesion | [23] |
| Meth-1 and Meth-2 | Suppress EC adhesion | [24] |
| Maspin | Inhibits proteases | [25] |
| Laminin 511 | Suppresses metastases | [26,27] |
| CCN3 | Suppresses EC adhesion | [28] |
| Endorepellin | Suppresses EC adhesion | [29] |
| MULTIMERIN2 (Endoglyx-1) | Suppresses EC migration | [30] |
| Generic Name | FDA-Approved Indication |
|---|---|
| Bevacizumab | Colorectal, non-small-cell lung, and glioblastoma multiforme |
| Thalidomide | Myeloma |
| Lenalidomide | Myeloma (myelodysplastic syndrome (MDS)) |
| Sorafenib | Renal cell and hepatocellular carcinoma |
| Sunitinib | Renal cell and gastrointestinal carcinoma |
| Temsirolimus | Renal cell carcinoma |
| Axitinib | Renal cell carcinoma |
| Pazopanib | Renal cell carcinoma, kidney cancer, and advanced soft tissue sarcoma |
| Cabozantinib | Thyroid cancer |
| Everolimus | Kidney cancer, advanced breast cancer, pancreatic neuroendocrine tumors (PNETs), and subependymal giant cell astrocytoma |
| Ramucirumab | Stomach cancer and gastroesophageal junction adenocarcinoma |
| Regorafenib | Colorectal cancer and gastrointestinal stromal tumor |
| Vandetanib | Thyroid cancer |
| Ziv-aflibercept | Colorectal cancer |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. https://doi.org/10.3390/biomedicines5020034
Rajabi M, Mousa SA. The Role of Angiogenesis in Cancer Treatment. Biomedicines. 2017; 5(2):34. https://doi.org/10.3390/biomedicines5020034
Chicago/Turabian StyleRajabi, Mehdi, and Shaker A. Mousa. 2017. "The Role of Angiogenesis in Cancer Treatment" Biomedicines 5, no. 2: 34. https://doi.org/10.3390/biomedicines5020034
APA StyleRajabi, M., & Mousa, S. A. (2017). The Role of Angiogenesis in Cancer Treatment. Biomedicines, 5(2), 34. https://doi.org/10.3390/biomedicines5020034