
Antibody–Drug Conjugates for Cancer Therapy

 ,
, 
Abstract
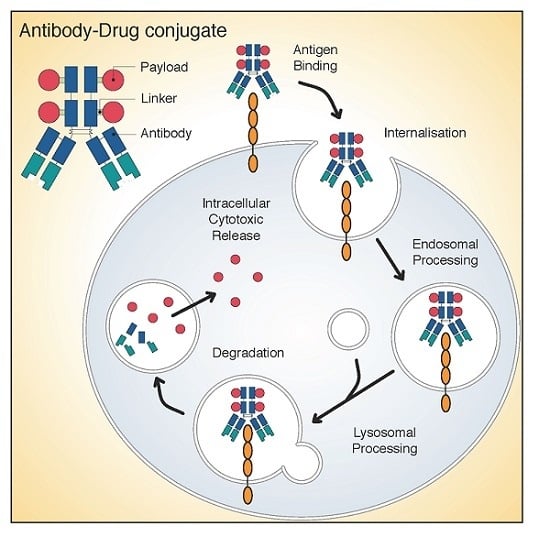
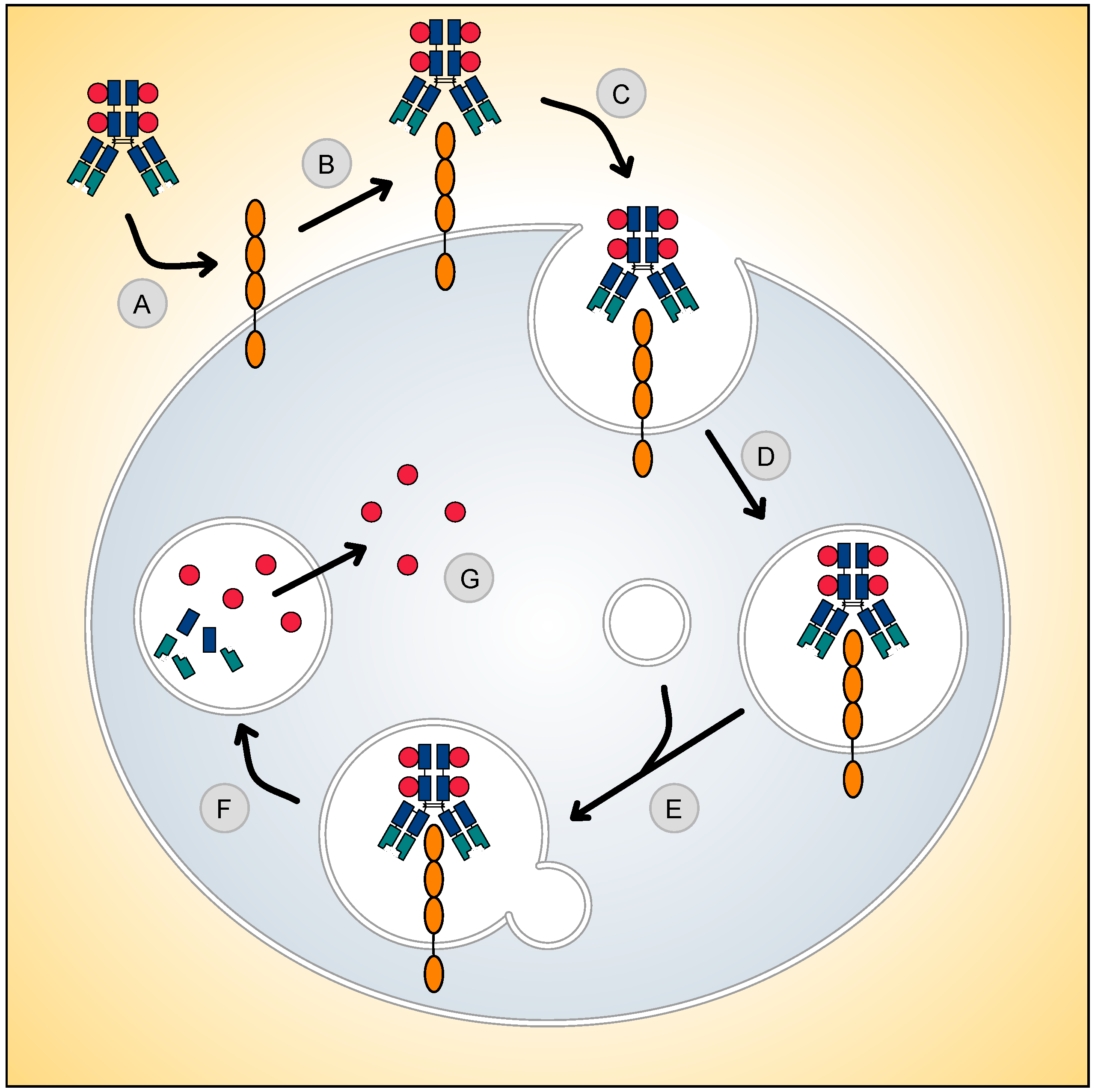
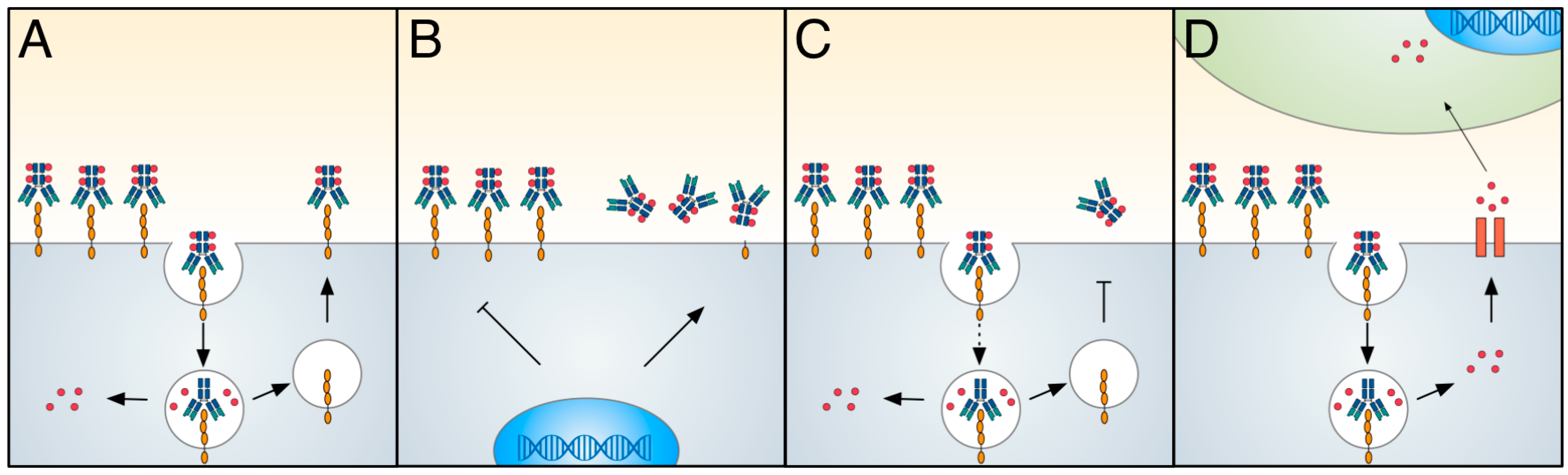
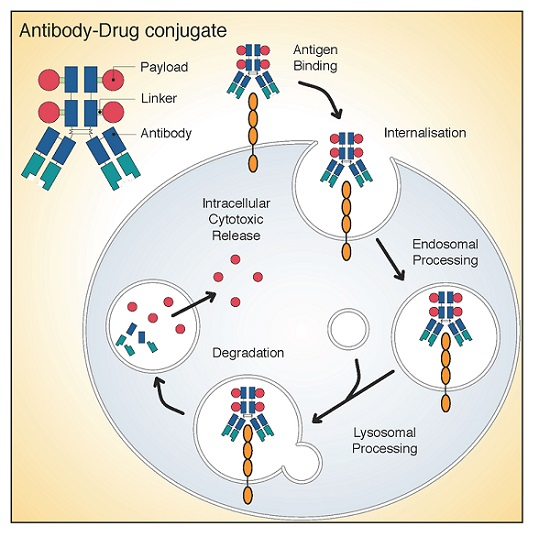
:
1. Introduction
2. Selecting an Appropriate Target
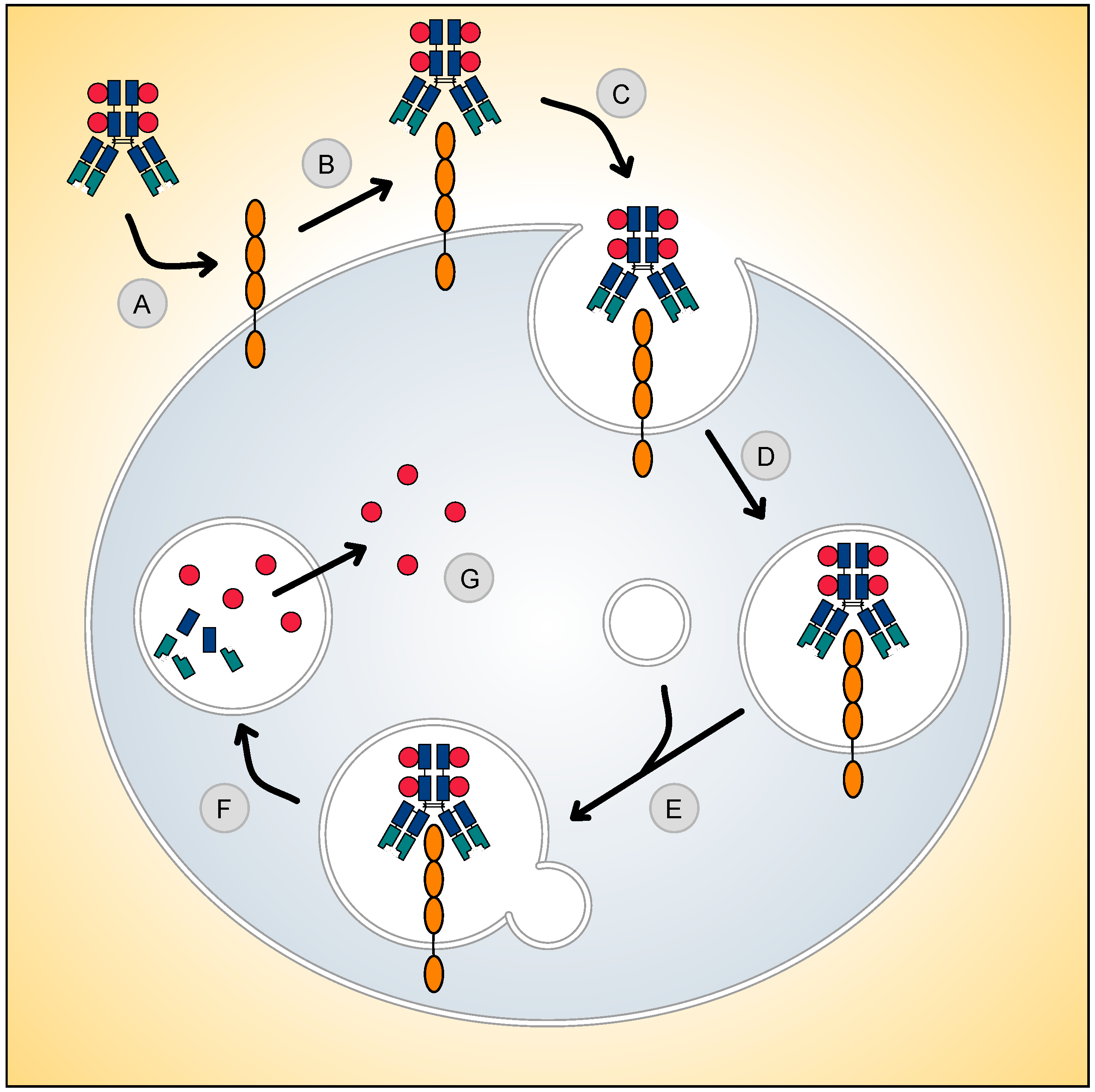
3. Antibody–Drug Conjugation
4. Antibody–Drug Conjugate Payloads
5. Clinically Approved Antibody–Drug Conjugates
6. Antibody–Drug Conjugate Toxicities
7. Recent Antibody–Drug Conjugate Developments
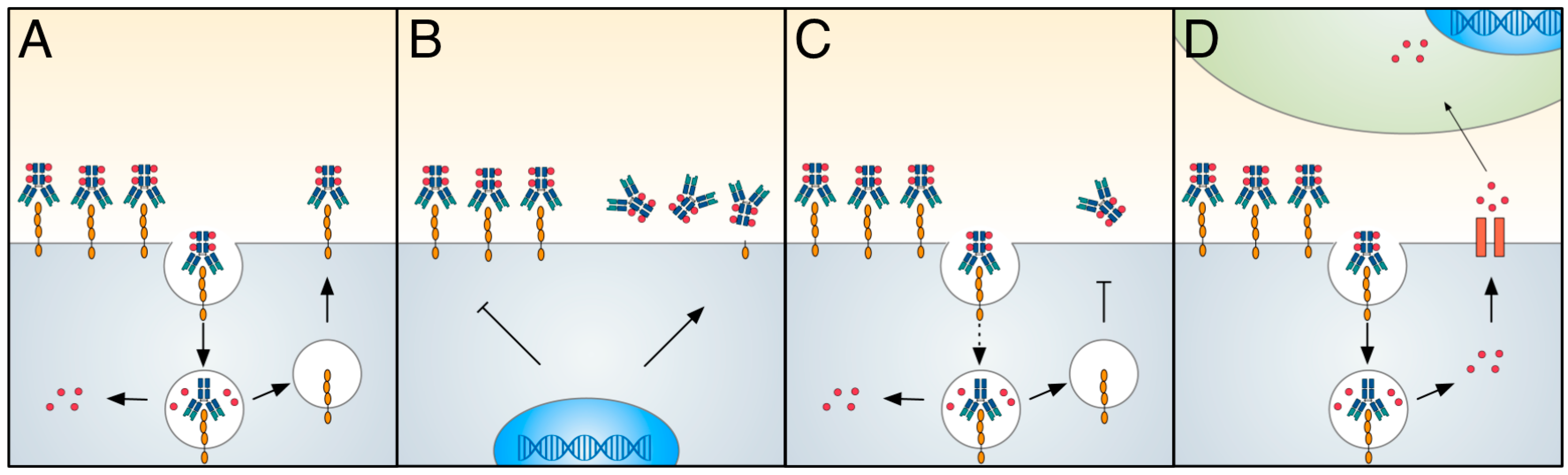
8. Resistance to Antibody–Drug Conjugate Therapy
9. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | adenosine triphosphate-binding cassette |
| ADC | antibody–drug conjugate |
| ADCC | antibody-dependent cytotoxicity |
| ALCL | anaplastic large-cell lymphoma |
| ALL | acute lymphocytic leukaemia |
| AML | acute myeloid leukaemia |
| ASCT | autologous stem cell transplantation |
| CDC | complement-dependent cytotoxicity |
| CR | complete response |
| CTLA-4 | cytotoxic T lymphocyte-associated protein-4 |
| DAR | drug to antibody ratio |
| DLBCL | diffuse large B-cell lymphoma |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor mutant (exon deletion 2–7) |
| FDA | Food and Drug Administration |
| FL | follicular lymphoma |
| GBM | glioblastoma multiforme |
| GPNMB | glycoprotein-NMB |
| HER2 | epidermal growth factor receptor 2 |
| HL | Hodgkin lymphoma |
| iNHL | indolent Non-Hodgkin lymphoma |
| mAb | monoclonal antibody |
| MBC | metastatic breast cancer |
| mc | Maleimidocaproyl |
| mcc | Maleimidomethyl cyclohexane-1-carboxylate, linked to cysteine of mAb |
| MCC | Maleimidomethyl cyclohexane-1-carboxylate, linked to thiol of cytotoxin |
| MDR | multidrug resistance |
| MMAE | monomethylauristatin E |
| MMAF | monomethylauristatin F |
| MMTV | mouse mammary tumour virus |
| NHL | non-Hodgkin lymphoma |
| NSCLC | non-small cell lung cancer |
| ORR | overall response rate |
| P-gp | P-glycoprotein |
| PABC | Para-aminobenzyloxycarbonyl |
| PBD | pyrrolobenzodiazepine |
| PD-1 | programmed cell death protein-1 |
| RCC | renal cell cancer |
| sALCL | systemic anaplastic large-cell lymphoma |
| SCLC | small cell lung cancer |
| SPDB | N-succinimidyl-4-(2-pyridyldithio) butanoate |
| T-DM1 | Ado-trastuzumab emtansine |
| TM-ADC | trastuzumab-maytansinoid ADC |
| TICs | tumour initiating cells |
| TILs | infiltrating lymphocytes |
| vc | valine-citrulline |
| vc-seco-DUBA | valine-citrulline-seco duocarmycin hydroxybenzamide azaindole |
References
- Schwartz, R.S. Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Shuptrine, C.W.; Surana, R.; Weiner, L.M. Monoclonal antibodies for the treatment of cancer. Semin. Cancer Biol. 2012, 22, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Niwa, R.; Satoh, M.; Akinaga, S.; Shitara, K.; Hanai, N. Engineered therapeutic antibodies with improved effector functions. Cancer Sci. 2009, 100, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody–Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Panowksi, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J. Targeted Cancer Therapy: Conferring Specificity to Cytotoxic Drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Shefet-Carasso, L.; Benhar, I. Antibody-targeted drugs and drug resistance—Challenges and solutions. Drug Resist. Updates 2015, 18, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Parslow, A.C.; Gan, H.K.; Scott, A.M. Antibody-mediated delivery of therapeutics for cancer therapy. Expert Opin. Drug Deliv. 2016, 13, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V.J. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody–Drug Conjugates for the Treatment of Cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Sochaj, A.M.; Świderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody–drug conjugates. Biotechnol. Adv. 2015, 33, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody–drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-Drug Conjugates for the Treatment of Non-Hodgkin's Lymphoma: Target and Linker-Drug Selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Kigawa, J.; Minagawa, Y.; Kanamori, Y.; Itamochi, H.; Cheng, X.; Okada, M.; Oishi, T.; Terakawa, N. Glutathione concentration may be a useful predictor of response to second-line chemotherapy in patients with ovarian cancer. Cancer 1998, 82, 697–702. [Google Scholar] [CrossRef]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Saito, G.; Swanson, J.A.; Lee, K.-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- Tothill, R.; Estall, V.; Rischin, D. Merkel Cell Carcinoma: Emerging Biology, Current Approaches, and Future Directions. Am. Soc. Clin. Oncol. 2015, 35, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K. Antibody-Maytansinoid Conjugates Are Activated in Targeted Cancer Cells by Lysosomal Degradation and Linker-Dependent Intracellular Processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Girish, S.; Gupta, M.; Wang, B.; Lu, D.; Krop, I.E.; Vogel, C.L.; Burris, H.A., III; LoRusso, P.M.; Yi, J.-H.; Saad, O.; et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother. Pharmacol. 2012, 69, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.K.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P. SGN-CD33A: A novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody-Drug Conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, M.M.C.; Groothuis, P.G.; Ubink, R.; van der Vleuten, M.A.J.; van Achterberg, T.A.; Loosveld, E.M.; Damming, D.; Jacobs, D.C.H.; Rouwette, M.; Egging, D.F.; et al. The Preclinical Profile of the Duocarmycin-Based HER2-Targeting ADC SYD985 Predicts for Clinical Benefit in Low HER2-Expressing Breast Cancers. Mol. Cancer Ther. 2015, 14, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, G.; Salnikov, A.V.; Luttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic Potential of Amanitin-Conjugated Anti-Epithelial Cell Adhesion Molecule Monoclonal Antibody Against Pancreatic Carcinoma. J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, J.; Edelman, M.; Harb, W.; Matei, D.; Nguyen, B.; Burgess, S. P1.08. A phase 1 study of 3 different schedules of the folic acid-tubulysin small-molecule drug conjugate EC1456 in pts with advanced solid tumors. Ann. Oncol. 2015, 26. [Google Scholar] [CrossRef]
- Sahin, U.; Hartmann, F.; Senter, P.; Pohl, C.; Engert, A.; Diehl, V.; Pfreundschuh, M. Specific activation of the prodrug mitomycin phosphate by a bispecific anti-CD30/anti-alkaline phosphatase monoclonal antibody. Cancer Res. 1990, 50, 6944–6948. [Google Scholar] [PubMed]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentuximab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Fromm, J.R.; McEarchern, J.A.; Kennedy, D.; Thomas, A.; Shustov, A.R.; Gopal, A.K. Clinical binding properties, internalization kinetics, and clinicopathologic activity of brentuximab vedotin: An antibody-drug conjugate for CD30-positive lymphoid neoplasms. Clin. Lymphoma Myeloma Leuk. 2012, 12, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Mao, W.; Wang, X.; Wang, B.-E.; Pham, T.; Flygare, J.; Yu, S.-F.; Yee, S.; Goldenberg, D.; Fields, C.; et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.L.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of Gemtuzumab Ozogamicin to Induction Chemotherapy Improves Survival in Older Patients With Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.; Recher, C.; Pigneux, A.; Witz, F.; Vey, N.; Blanchet, O.; Lefebvre, P.; Luquet, I.; Guillerme, I.; Volteau, C. Addition of gemtuzumab ozogamycin to chemotherapy improves event-free survival but not overall survival of AML patients with intermediate cytogenetics not eligible for allogeneic transplantation. Results of the GOELAMS AML 2006 IR study. Blood 2011, 118, 79. [Google Scholar]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Ravandi, F.; Estey, E.H.; Appelbaum, F.R.; Lo-Coco, F.; Schiffer, C.A.; Larson, R.A.; Burnett, A.K.; Kantarjian, H.M. Gemtuzumab Ozogamicin: Time to Resurrect? J. Clin. Oncol. 2012, 30, 3921–3923. [Google Scholar] [CrossRef] [PubMed]
- Estey, E. Treatment of AML: Resurrection for gemtuzumab ozogamicin? Lancet 2012, 379, 1468–1469. [Google Scholar] [CrossRef]
- Dang, N.H.; Ogura, M.; Castaigne, S.; Fayad, L.; Jerkeman, M.; Radford, J.A.; Pezzutto, A.; Bondarenko, I.; Stewart, D.A.; Shnaidman, M. Randomized, phase 3 trial of inotuzumab ozogamicin plus rituximab (R-InO) versus chemotherapy for relapsed/refractory aggressive B-cell non-Hodgkin lymphoma (B-NHL). J. Clin. Oncol. 2014, 32, 8529. [Google Scholar]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Hernandez-Ilizaliturri, F.; Chanan-Khan, A.; Patel, M.; Kelly, K.R.; Running, K.L.; Murphy, M.; Guild, R.; Carrigan, C.; Ladd, S. Phase I study of lorvotuzumab mertansine (LM, IMGN901) in combination with lenalidomide (Len) and dexamethasone (Dex) in patients with CD56-positive relapsed or relapsed/refractory multiple myeloma (MM). Blood 2012, 120, 728. [Google Scholar]
- Kelly, K.R.; Chanan-Khan, A.; Somlo, G.; Heffner, L.T. Indatuximab Ravtansine (BT062) in Combination with Lenalidomide and Low-Dose Dexamethasone in Patients with Relapsed and/or Refractory Multiple. Blood 2014, 124, 4736. [Google Scholar]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Boni, V.; Rixe, O.; Rasco, D.; Gomez-Roca, C.; Calvo, E.; Morris, J.C.; Tolcher, A.W.; Assadourian, S.; Guillemin, H.; Delord, J.-P. Abstract A73: A Phase I first-in-human (FIH) study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs) (NCT01156870). Mol. Cancer Ther. 2013, 12. [Google Scholar] [CrossRef]
- Morschhauser, F.; Flinn, I. Preliminary results of a phase II randomized study (ROMULUS) of polatuzumab vedotin (PoV) or pinatuzumab vedotin (PiV) plus rituximab (RTX) in patients (Pts) with relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL). In Proceedings of the 50th Annual Meeting of American Society Of Clinical Oncology, Chicago, IL, USA, 30 May–3 June 2014.
- Ott, P.A.; Hamid, O.; Pavlick, A.C.; Kluger, H.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Crowley, E.; Green, J.A.; Hawthorne, T.; et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients With Advanced Melanoma. J. Clin. Oncol. 2014, 32, 3659–3666. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Smith, D.C.; Appleman, L.J.; Fleming, M.T.; Hussain, A.; Dreicer, R.; Sartor, A.O.; Shore, N.D.; Vogelzang, N.J.; Youssoufian, H. A phase 2 trial of prostate-specific membrane antigen antibody drug conjugate (PSMA ADC) in taxane-refractory metastatic castration-resistant prostate cancer (mCRPC). Blood 2014, 32, 5023. [Google Scholar]
- Gan, H.K.; Papadopoulos, K.P.; Fichtel, L.; Lassman, A.B.; Merrell, R.; Van Den Bent, M.J.; Kumthekar, P.; Scott, A.M.; Pedersen, M.; Gomez, E.J.; et al. Phase I study of ABT-414 mono- or combination therapy with temozolomide (TMZ) in recurrent glioblastoma (GBM). In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Starodub, A.; Ocean, A.J.; Messersmith, W.A.; Picozzi, V.J.; Guarino, M.J.; Thomas, S.S.; Bardia, A.; Shah, M.A.; Govindan, S.V.; Maliakal, P.P. Phase I/II trial of IMMU-132 (isactuzumab govitecan), an anti-Trop-2-SN-38 antibody drug conjugate (ADC): Results in patients with metastatic gastrointestinal (GI) cancers. Blood 2015, 33, 703–747. [Google Scholar]
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. U.S. Food and Drug Administration Approval Summary: Brentuximab Vedotin for the Treatment of Relapsed Hodgkin Lymphoma or Relapsed Systemic Anaplastic Large-Cell Lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody-Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Tanner, M.; Koninki, K.; Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011, 13, R46. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Dirix, L.; Kocsis, J.; Bianchi, G.V.; Lu, J.; Vinholes, J.; Guardino, E.; Song, C.; Tong, B.; Ng, V.; et al. Phase II Randomized Study of Trastuzumab Emtansine Versus Trastuzumab Plus Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2013, 31, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Dieras, V.; Harbeck, N.; Albain, K.; Burris, H.; Awada, A.; Crivellari, D.; Andre, F.; Choi, Y. J.; Huang, J.; Miller, K.D. Abstract P3-14-01: A phase Ib/II trial of trastuzumab-DM1 with pertuzumab for patients with HER2-positive, locally advanced or metastatic breast cancer: interim efficacy and safety results. Cancer Res. 2010, 70. [Google Scholar] [CrossRef]
- Burris, H.A.; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B. Antibody–drug conjugates for cancer: poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; LoRusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial, S.D.C.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I Multidose-Escalation Study of the Anti-CD19 Maytansinoid Immunoconjugate SAR3419 Administered by Intravenous Infusion Every 3 Weeks to Patients with Relapsed/Refractory B-Cell Lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Motzer, R. Phase I studies of anti-ENPP3 antibody drug conjugates (ADCs) in advanced refractory renal cell carcinomas (RRCC). In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Goff, L.W.; Papadopoulos, K.; Posey, J.A.; Phan, A.T.; Patnaik, A.; Miller, J.G.; Zildjian, S.; O’Leary, J.J.; Qin, A.; Tolcher, A. A phase II study of IMGN242 (huC242-DM4) in patients with CanAg-positive gastric or gastroesophageal (GE) junction cancer. In Proceedings of the 45th ASCO Annual Meeting, Orlando, IL, USA, 29 May–2 June 2009.
- Bendell, J.; Blumenschein, G.; Zinner, R.; Hong, D.; Jones, S.; Infante, J.; Burris, H.; Rajagopalan, P.; Kornacker, M.; Henderson, D.; et al. Abstract LB-291: First-in-human phase I dose escalation study of a novel anti-mesothelin antibody drug conjugate (ADC), BAY 94-9343, in patients with advanced solid tumors. Cancer Res. 2014, 73. [Google Scholar] [CrossRef]
- Moore, K.; Ponte, J.; LoRusso, P.; Birrer, M.; Bauer, T.M.; Borghei, H.; O’Malley, D.; Ruiz-Soto, R.; Lutz, R.J.; Malik, L. Relationship of pharmacokinetics (PK), toxicity and initial evidence of clinical activity with IMGN853, a folate receptor alpha (FRα)-targeting antibody drug conjugate in patients with epithelial ovarian cancer and other FRα-positive solid tumors. J. Clin. Oncol. 2014, 32, 5571. [Google Scholar]
- Oak, E.; Bartlett, N.L. A safety evaluation of brentuximab vedotin for the treatment of Hodgkin lymphoma. Expert Opin. Drug Saf. 2016, 15, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Goy, A.; Forero, A.; Wagner-Johnston, N.; Christopher Ehmann, W.; Tsai, M.; Hatake, K.; Ananthakrishnan, R.; Volkert, A.; Vandendries, E.; Ogura, M. A phase 2 study of inotuzumab ozogamicin in patients with indolent B-cell non-Hodgkin lymphoma refractory to rituximab alone, rituximab and chemotherapy, or radioimmunotherapy. Br. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Stankowski, R.V. Cardiovascular toxicity associated with adjuvant trastuzumab therapy: Prevalence, patient characteristics, and risk factors. Ther. Adv. Drug Saf. 2014, 5, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Martin, K.; Theurich, S.; Schreiner, J.; Savic, S.; Terszowski, G.; Lardinois, D.; Heinzelmann-Schwarz, V.A.; Schlaak, M.; Kvasnicka, H.-M.; et al. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol. Res. 2014, 2, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Kreuzaler, M.; Khan, T.; Thommen, D.S.; Martin, K.; Glatz, K.; Savic, S.; Harbeck, N.; Nitz, U.; Gluz, O.; et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2014, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Helman, E.; Wick, M.J.; Clark, M.J.; Gamez, L.; Boyle, S.; Papadopoulos, K.P.; Luo, S.; Tolcher, A.W.; Sripakdeevong, P.; Karbelashvili, M.; et al. Abstract 1457: Genomic characterization of a PDX model of T-DM1-resistant HER2+ invasive ductal carcinoma using augmented exome sequencing. Cancer Res. 2015, 75, 1457. [Google Scholar] [CrossRef]
- O’Brien, C.; Cavet, G.; Pandita, A.; Hu, X.; Haydu, L.; Mohan, S.; Toy, K.; Rivers, C.S.; Modrusan, Z.; Amler, L.C.; et al. Functional genomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res. 2008, 68, 5380–5389. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Sammet, B.; Steinkühler, C.; Sewald, N. Antibody-drug conjugates in tumor therapy. Pharm. Pat. Anal. 2012, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J. Membr. Boil. 2005, 206, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-Maytansinoid Conjugates Designed to Bypass Multidrug Resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Payload | Target Antigen | Antibody–Drug Conjugate | Lead Indication | Phase | Reference |
|---|---|---|---|---|---|
| Calicheamicin | CD22 | Inotuzumab Ozogamicin | B-cell malignancy | FDA Breakthrough Therapy Designation | [53] |
| CD33 | Gemtuzumab Ozogamicin (GO) | AML | FDA approved but withdrawn | [46] | |
| DM1 | CD22 | Brentuximab Vedotin | Hodgkin’s Lymphoma, Systemic ALCL | FDA approved | [54] |
| CD56 | Lorvotuzumab mertansine | Multiple myeloma | I/II | [55] | |
| CD138 | BT062 | Multiple myeloma | I/IIa | [56] | |
| HER2 | Trastuzumab emtansine (T-DM1) | Breast cancer | FDA approved | [57] | |
| MUC1 | SAR-566658 | Solid tumours | I/II | [58] | |
| DM4 | CD22 | Pinatuzumab vedotin + Rituximab | DLBCL, FL | II | [59] |
| CD79b | Polatuzumab vedotin + Rituximab | DLBCL, FL | II | [59] | |
| GPNMB | Glembatumumab vedotin | Melanoma | II | [60] | |
| MMAE | PSMA | PSMA ADC | Prostate cancer | II | [61] |
| MMAF | EGFR | ABT-414 | GBM | IIb/III | NCT02573324 [62] |
| SN-38 | CEACAM | IMMU-130 | Colorectal cancer | II | NCT01915472 |
| Trop2 | IMMU-132 | Epithelial cancers | I/II | [63] | |
| Liposomal doxorubicin | HER2 | MM-302 | HER2 positive metastatic breast cancer | II | NCT02213744 |
| Payload | Target Antigen | Antibody–Drug Conjugate | Lead Indication | Phase |
|---|---|---|---|---|
| Auristatin microtubule inhibitor | PTK7 | PF-06647020 | Solid tumours | Phase I |
| NOTCH-3 | PF-06650808 | Solid tumours | Preclinical | |
| DM1 | CD70 | AMG-172 | Renal cell carcinoma | Phase I |
| CD22 | Anti-CD22-MCC-DM1 | Non-Hodgkin lymphoma | Preclinical | |
| Mesothelin | BAY 94-9343 | Mesothelioma, pancreatic, ovarian, NSCLC | Phase I | |
| CD37 | IMGN-529 | NHL | Phase I | |
| Folate receptor 1 | IMGN853 | Ovarian cancer NSCLC | Phase I | |
| CD56 | Lorvotuzumab mertansine | SCLC, Merkel cell, ovarian | Phase I | |
| CD19 | SAR-3419 | NHL | Phase I | |
| DM4 | Nectin-4 | ASG-22ME | Solid tumours | Phase I |
| Carbonic anhydrase | BAY 79-4620 | Solid tumours | Phase I | |
| MMAE | SLC44A4 | ASG-5ME | Pancreatic cancer | Phase I |
| SLTRK6 | ASG-15ME | Urothelial tumours | Phase I | |
| CD22 | DCDT2980S | Non-Hodgkin lymphoma | Preclinical | |
| Sodium-dependent phosphate transporter | DNIB0600A | NSCLC, Ovarian cancer | Phase I | |
| Axl | HuMax-Axl-ADC | Solid, haematological malignancies | Preclinical | |
| CD19 | SGN CD19A | NHL | Phase I | |
| CD70 | SGN-75 | RCC | Phase I | |
| MMAF | ENPP3 | AGS-16M8F | Renal cell carcinoma | Phase I |
| 5T4 | PF 06263507 | Solid tumours | Phase I | |
| PBD | CD19 | ADCT-402 | NHL | Phase I |
| CD70 | SGN-CD70A | NHL | Preclinical |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parslow, A.C.; Parakh, S.; Lee, F.-T.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Biomedicines 2016, 4, 14. https://doi.org/10.3390/biomedicines4030014
Parslow AC, Parakh S, Lee F-T, Gan HK, Scott AM. Antibody–Drug Conjugates for Cancer Therapy. Biomedicines. 2016; 4(3):14. https://doi.org/10.3390/biomedicines4030014
Chicago/Turabian StyleParslow, Adam C., Sagun Parakh, Fook-Thean Lee, Hui K. Gan, and Andrew M. Scott. 2016. "Antibody–Drug Conjugates for Cancer Therapy" Biomedicines 4, no. 3: 14. https://doi.org/10.3390/biomedicines4030014
APA StyleParslow, A. C., Parakh, S., Lee, F.-T., Gan, H. K., & Scott, A. M. (2016). Antibody–Drug Conjugates for Cancer Therapy. Biomedicines, 4(3), 14. https://doi.org/10.3390/biomedicines4030014