
Genetic Modification of T Cells


Abstract
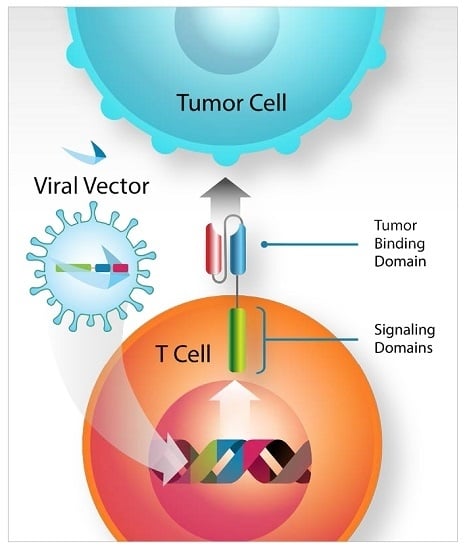
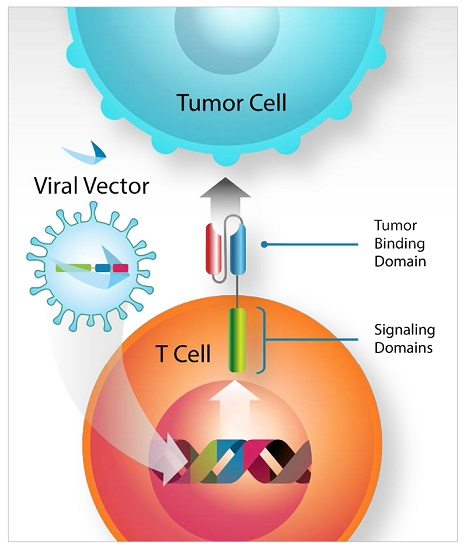
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
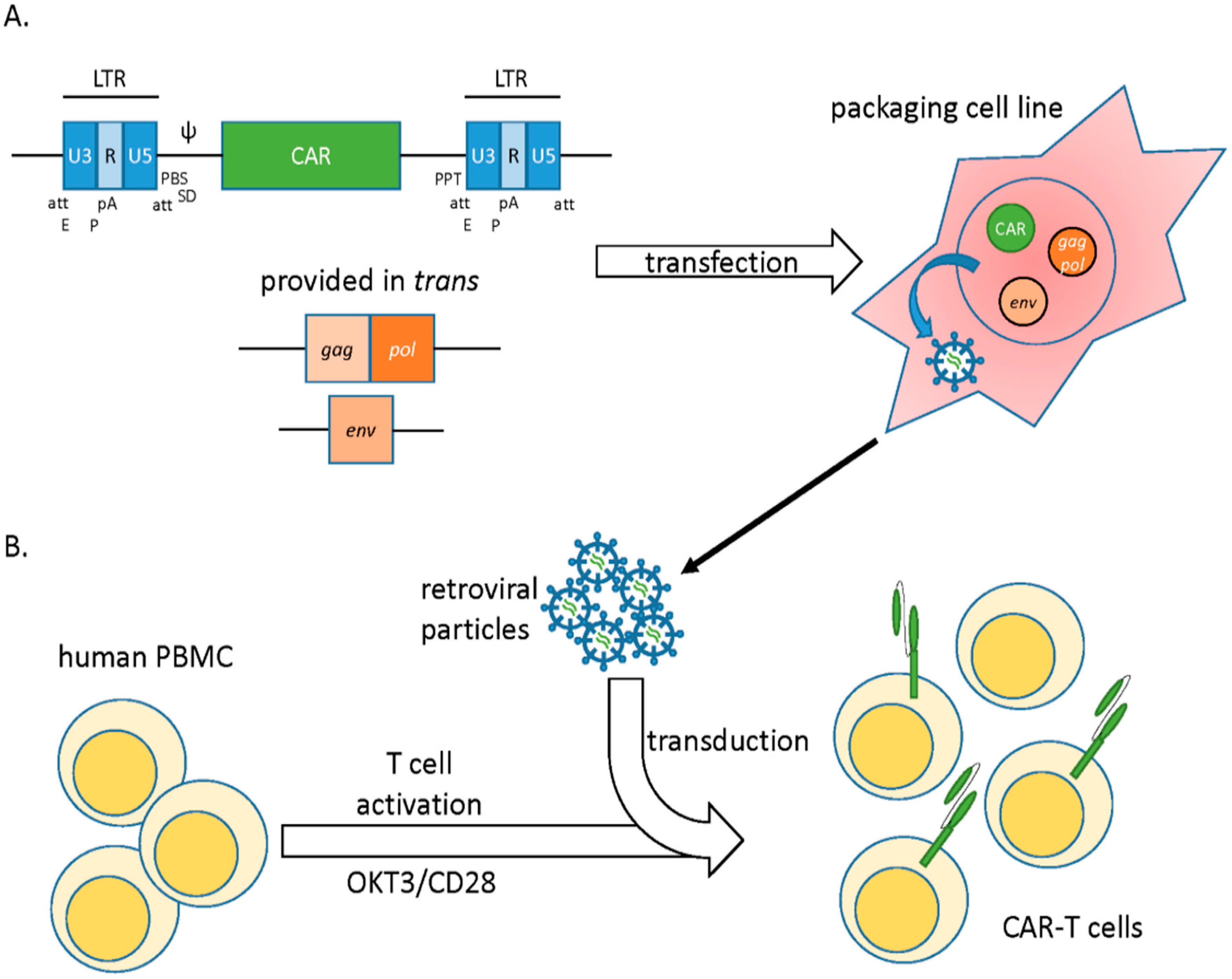
2. Viral Vectors
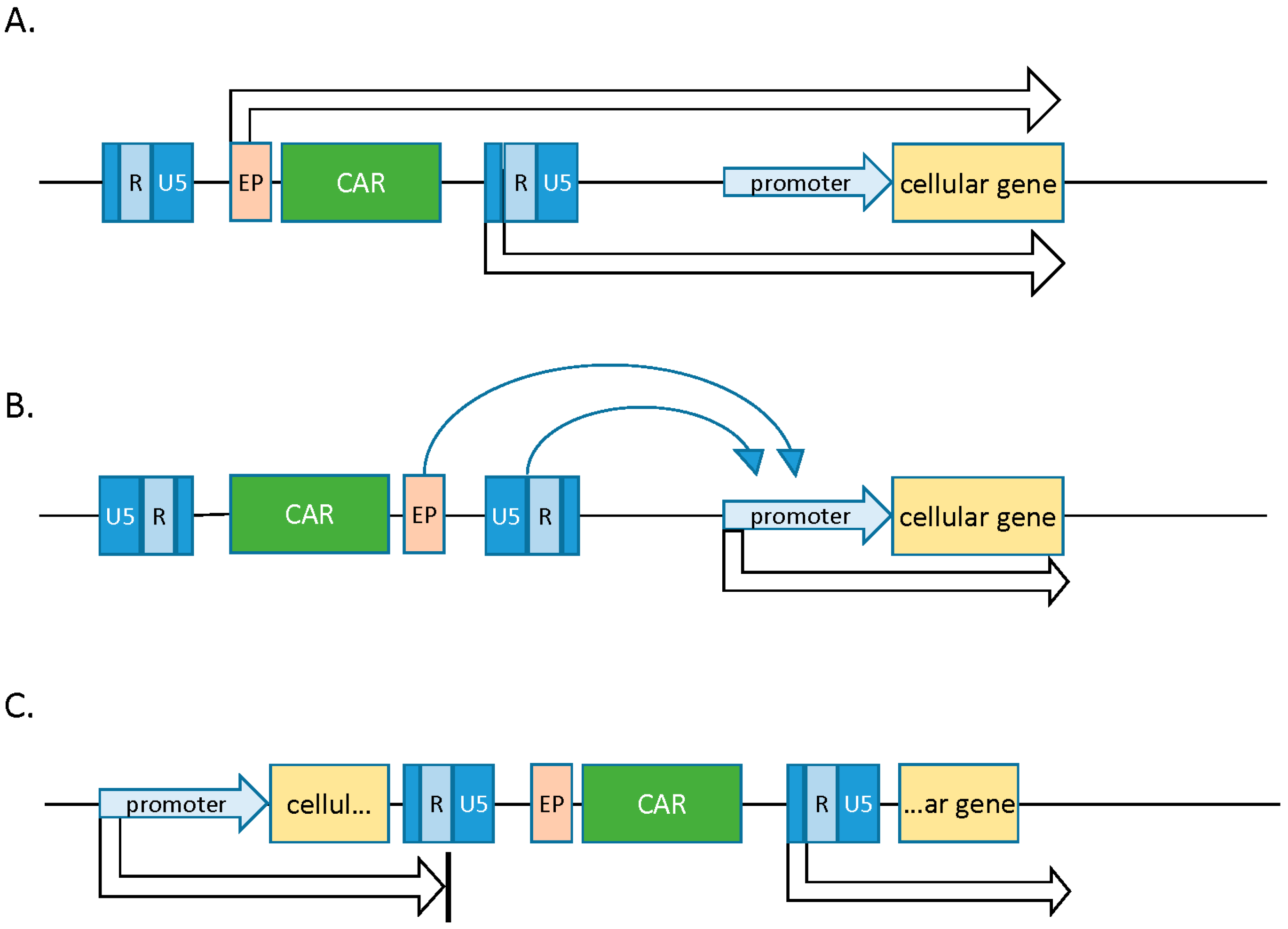
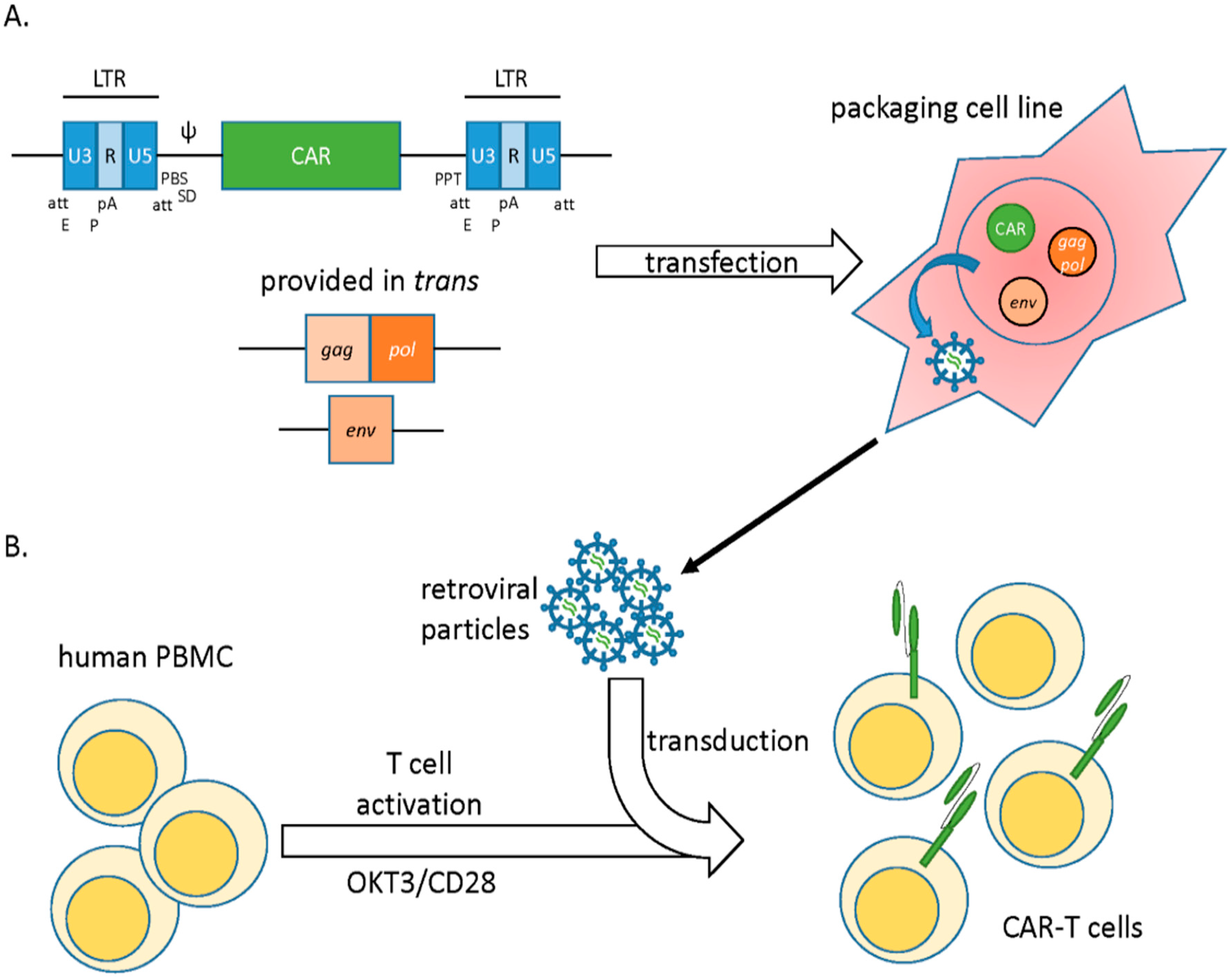
2.1. Gamma Retroviral Vectors
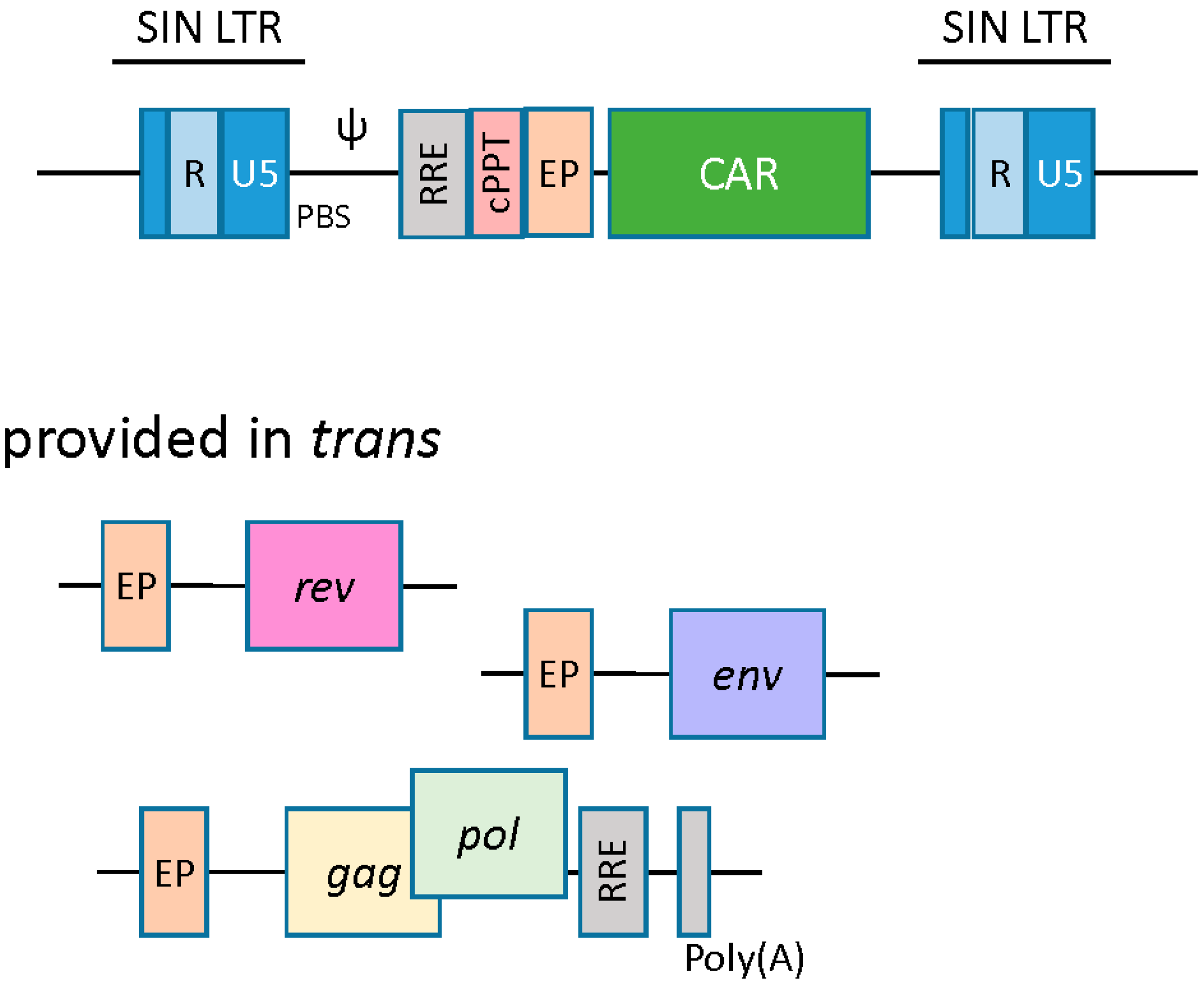
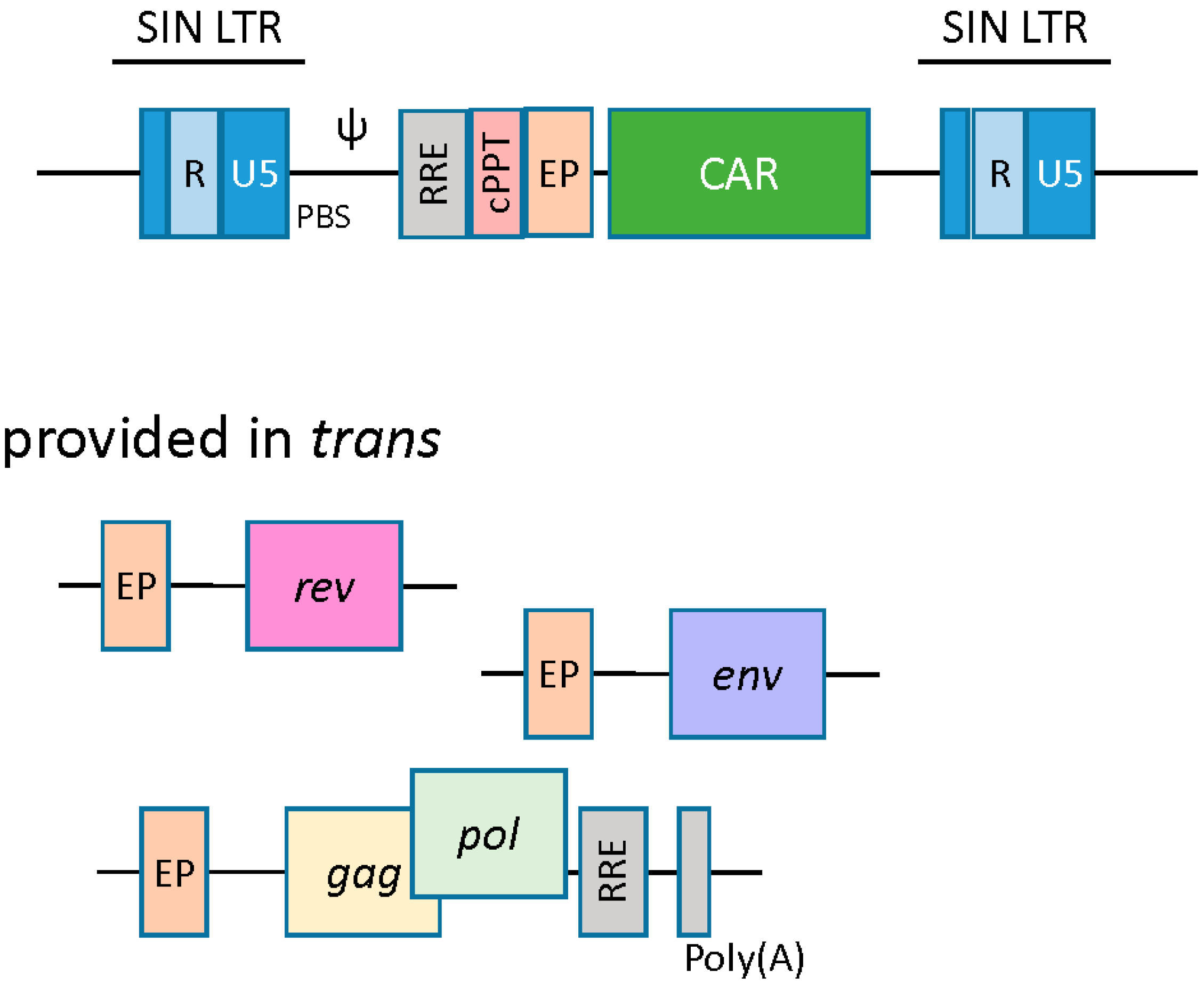
2.2. Lentiviral Vectors
2.3. Alpha Retroviral Vectors
3. Non-Viral Vector Systems
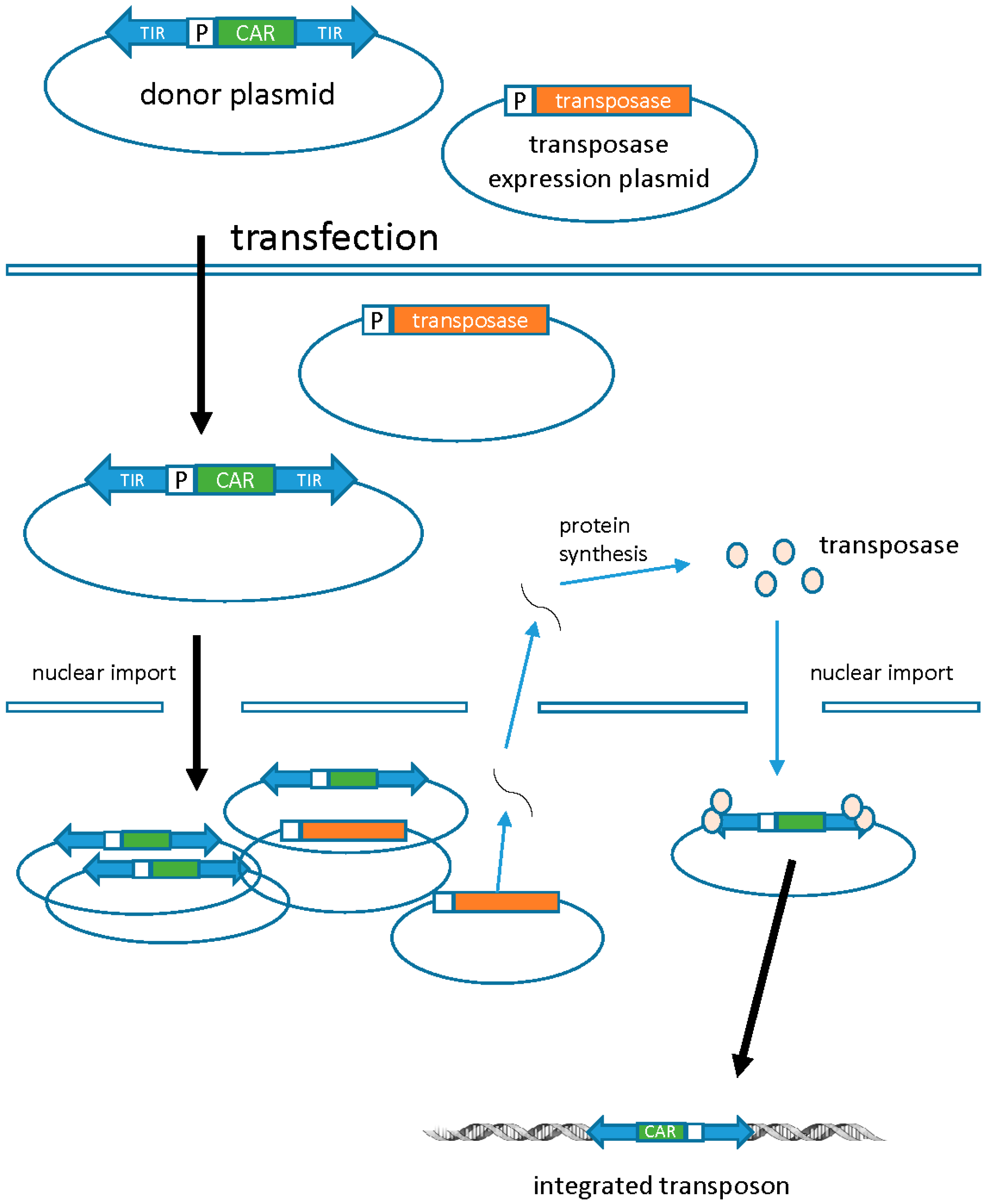
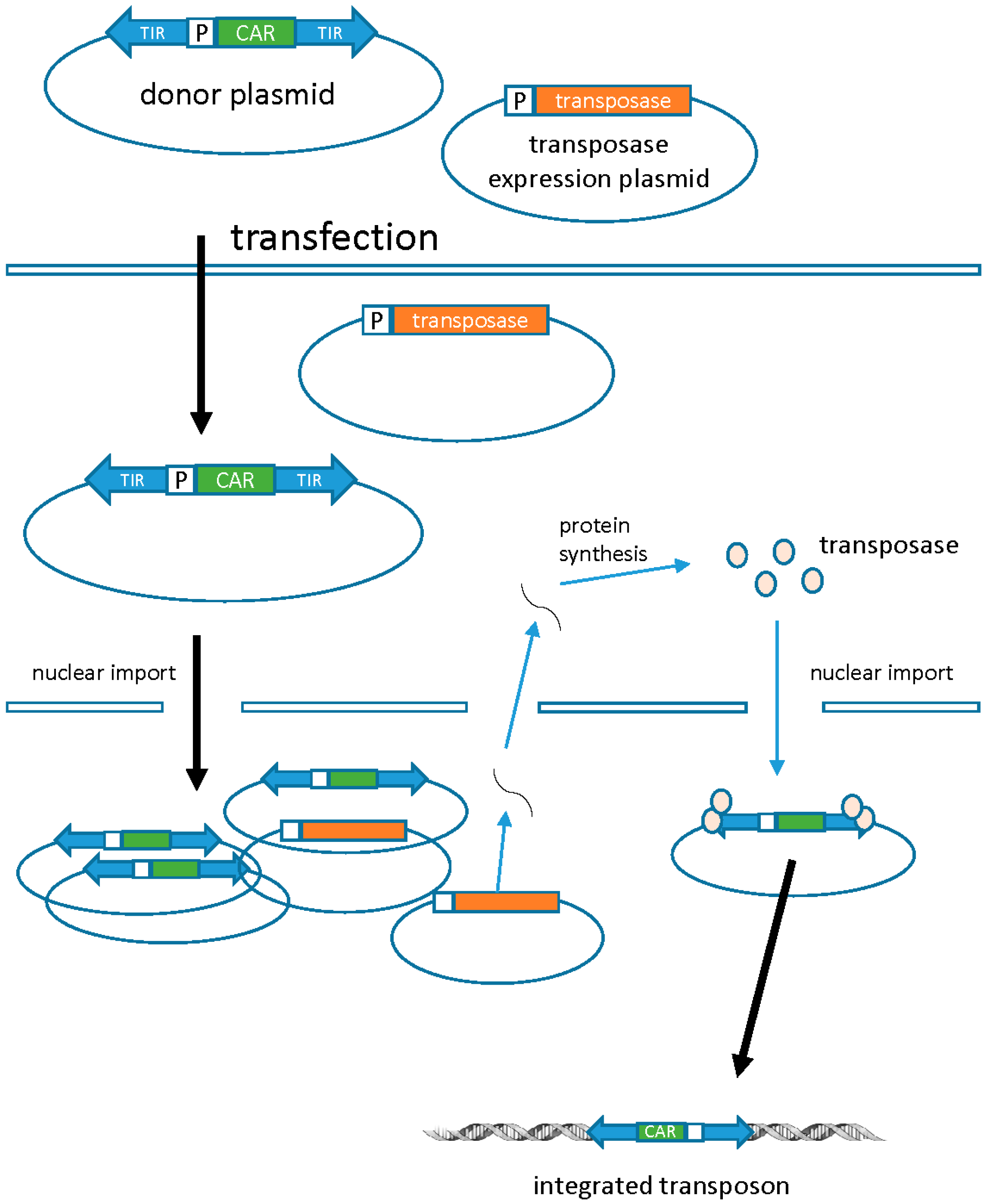
3.1. Transposons
3.2. mRNA Electroporation
4. Summary
Conflicts of Interest
References
- Suerth, J.; Labenski, V.; Schambach, A. Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy. Viruses 2014, 6, 4811–4838. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 28, 669–672. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Schambach, A.; Bohne, J.; Galla, M. Retrovirus vectors: Toward the plentivirus? Mol. Ther. 2006, 13, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.M.; Miller, A.D. Production of high-titer helper virus-free retroviral vectors by cocultivation of packaging cells with different host ranges. J. Virol. 1991, 65, 3887–3890. [Google Scholar] [PubMed]
- Suerth, J.D.; Schambach, A.; Baum, C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr. Opin. Immunol. 2012, 24, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Cam, H.; Schüler, T.; Indraccolo, S.; Gladow, M.; Baum, C.; Blankenstein, T. Uckert, Retroviral vectors for high-level transgene expression in T lymphocytes. Hum. Gene Ther. 2003, 14, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [PubMed]
- Lamers, C.; Willemsen, R.; van Elzakker, P. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
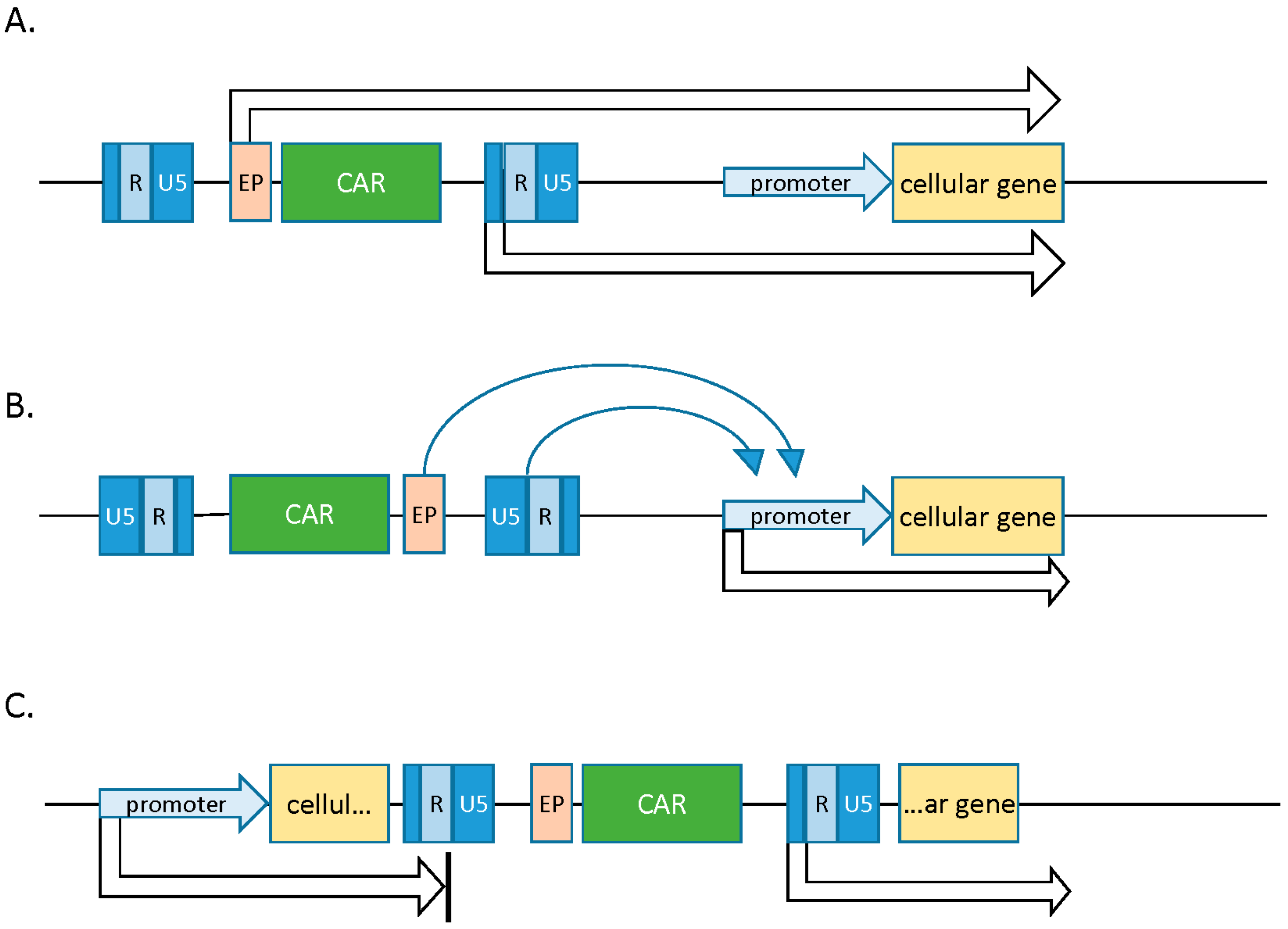
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Bushman, F.D. Retroviral DNA integration—Mechanism and consequences. Adv. Genet. 2005, 555, 147–181. [Google Scholar]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Nat. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, S.; Swapna, G.V.T.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, S.; De Rijck, J.; Demeulemeester, J.; Vets, S.; Madlala, P.; Cermakova, K.; Debyser, Z.; Gijsbers, R. BET-independent MLV-based vectors target away from promoters and regulatory elements. Mol. Ther. Nucleic Acids 2014, 3, e179. [Google Scholar] [CrossRef] [PubMed]
- Newrzela, S.; Cornils, K.; Li, Z.; Baum, C.; Brugman, M.H. Resistance of mature T cells to oncogene transformation. Blood 2008, 112, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Loew, R.; Meyer, Y.; Kuehlcke, K.; Gama-Norton, L.; Wirth, D. A new PG13-based packaging cell line for stable production of clinical-grade self-inactivating γ-retroviral vectors using targeted integration. Gene Ther. 2009, 17, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Mátrai, J.; Chuah, M.K.; VandenDriessche, T. Recent advances in lentiviral vector development and applications. Mol. Ther. 2010, 18, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Amirache, F.; Lévy, C.; Costa, C.; Mangeot, P.-E.; Torbett, B.E.; Wang, C.X.; Nègre, D.; Cosset, F.-L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef] [PubMed]
- Saukkonen, K.; Hemminki, A. Tissue-specific promoters for cancer Gene Therapy. Exp. Opin. Biol. Ther 2004, 4, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Yang, L.; Joo, K.; Yang, H. Targeting lentiviral vectors to antigen-specific immunoglobulins. Hum. Gene Ther. 2008, 19, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Maurice, M.; Verhoeyen, E.; Salmon, P.; Trono, D. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood 2002, 99, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Frecha, C.; Costa, C.; Negre, D.; Gauthier, E.; Russell, S.J. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 2008, 112, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Pistello, M.; Vannucci, L.; Ravani, A.; Bonci, F. Streamlined design of a self-inactivating feline immunodeficiency virus vector for transducing ex vivo dendritic cells and T lymphocytes. Genet. Vaccines Ther. 2007, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Korin, Y.D.; Zack, J.A. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 1998, 72, 3161–3168. [Google Scholar] [PubMed]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A. Mechanisms governing lentivirus integration site selection. Curr. Gene Ther. 2008, 8, 419–429. [Google Scholar] [CrossRef]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi, L.S.; Sanvito, F.; et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Cornetta, K. Design and Potential of Non-Integrating Lentiviral Vectors. Biomedicines 2014, 2, 14–35. [Google Scholar] [CrossRef]
- Verghese, S.; Goloviznina, N.; Skinner, A.; Lipps, H.; Kurre, P. S/MAR sequence confers long-term mitotic stability on non-integrating lentiviral vector episomes without selection. Nucleic Acids Res. 2014, 42, e53. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.-L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, S.; Gregory, P.D.; et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.; Hollis, R.P.; Kuftinec, G.; Senadheera, S.; Chan, R.; Kohn, D.B. Integrase-defective Lentiviral Vectors as a Delivery Platform for Targeted Modification of Adenosine Deaminase Locus. Mol. Ther. 2013, 21, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.; Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Self-inactivating alpharetroviral vectors with a split-packaging design. J. Virol. 2010, 84, 6626–6635. [Google Scholar] [PubMed]
- Suerth, J.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral Self-inactivating Vectors: Long-term Transgene Expression in Murine Hematopoietic Cells and Low Genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Izsvák, Z.; Hackett, P.B.; Cooper, L.; Ivics, Z. Translating Sleeping Beauty transposition into cellular therapies: Victories and challenges. Bioessays 2010, 32, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Izsvák, Z. Nonviral gene delivery with the sleeping beauty transposon system. Hum. Gene Ther. 2011, 22, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Moyes, J.S.; Huls, M.H.; Cooper, L.J. Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015, 22, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Hackett, P.B.; Largaespada, D.A.; Cooper, L. A transposon and transposase system for human application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Manuri, P.; Wilson, M.H.; Maiti, S.N.; Mi, T. Piggybac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Human Gene Ther. 2010, 21, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Huye, L.E.; Salsman, V.S.; Leen, A.M. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Iwase, N.; Kawakami, K.; Iwasaki, M.; Yamamoto, C.; Ohmine, K.; Uchibori, R.; Teruya, T.; Ido, H.; Saga, Y.; et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015, 22, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Bak, R.O.; Krogh, L.B.; Staunstrup, N.H.; Moldt, B.; Corydon, T.J.; Schrøder, L.D.; Mikkelsen, J.G. DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic Acids Res. 2014, 42, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Mikkelsen, J. Driving DNA transposition by lentiviral protein transduction. Mob. Genet. Elem. 2014, 4, e29591. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, G.; Andries, O.; Demeester, J. mRNA as gene therapeutic: How to control protein expression. J. Control. Release 2011, 150, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Lee, J.M.; Cho, H.I.; Kim, E.K.; Kim, H.S. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009, 16, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.; Zhao, Y.; Liu, X.; Jiang, S.; Carpenito, C.; Kalos, M.; Carroll, R.G.; June, C.H.; Grupp, S.A. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum. Gene Ther. 2011, 22, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.; Liu, X.; Jiang, S.; June, C.H.; Grupp, S.A.; Zhao, Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 2013, 24, 717–727. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, R.A.; Boyerinas, B. Genetic Modification of T Cells. Biomedicines 2016, 4, 9. https://doi.org/10.3390/biomedicines4020009
Morgan RA, Boyerinas B. Genetic Modification of T Cells. Biomedicines. 2016; 4(2):9. https://doi.org/10.3390/biomedicines4020009
Chicago/Turabian StyleMorgan, Richard A., and Benjamin Boyerinas. 2016. "Genetic Modification of T Cells" Biomedicines 4, no. 2: 9. https://doi.org/10.3390/biomedicines4020009
APA StyleMorgan, R. A., & Boyerinas, B. (2016). Genetic Modification of T Cells. Biomedicines, 4(2), 9. https://doi.org/10.3390/biomedicines4020009