Abstract
Approximately 30–35% of people with epilepsy experience seizures despite taking antiseizure medications. Recurrent seizures that are independent of status epilepticus can be associated with neuronal injury and structural changes to the brain, as well as diminished cognitive function, mood, and quality of life. A treatment that alters the underlying biology of epilepsy, thereby reducing the seizure burden and its attendant consequences, would be of great value in preventing these detrimental effects. In this review, we summarize preclinical and clinical research on pharmacological treatments that may favorably alter the underlying biology of epilepsy (i.e., disease modification or antiepileptogenesis). A reduction in seizures over time (e.g., increase in responder rates) or prevention of epilepsy in susceptible individuals has been observed with therapies that target neurotransmission (cenobamate, cannabidiol, vigabatrin, and diazepam nasal spray) and inflammation (everolimus), though evidence is limited and in preliminary stages. Pharmacological treatments that target neuroinflammation and oxidative stress have the potential to modify seizure phenotype and 1 or more comorbidities in preclinical studies (e.g., stress/anxiety and depression). Gene therapies and stem-cell-derived treatments also hold promise in reducing seizure burden in preclinical models, with several therapeutic candidates having advanced to phase 1/2 and 3 clinical trials. Effective disease-modifying strategies in epilepsy might include seizure control with novel antiseizure medications in combination with therapeutic targeting of key pathophysiological mechanisms. Standard criteria and a definition of disease modification should be established. Importantly, given the heterogeneity of the epilepsies, syndrome- or seizure-specific methods and trial design would likely be required.
1. Introduction
Epilepsy is a common, heterogeneous neurological disorder affecting approximately 50 million people worldwide [1,2]. Some epilepsies and their associated symptoms are progressive, whereas others may remain static or even improve over time [3,4]. The most common epilepsy treatments consist of chronic, daily antiseizure medications (ASMs) and intermittent rescue therapies (immediate-use seizure medications [ISMs]) for acute, as-needed treatment of seizure emergencies such as seizure clusters. As 30–35% of people with epilepsy continue to experience seizures despite the use of appropriate ASMs (i.e., drug-resistant epilepsy [DRE]) [5,6], other modalities such as neuromodulation and surgical therapy are also used [7].
Glutamatergic neurotransmission primarily mediates the increase in neuronal excitability in most epileptic seizures, along with a concurrent decrease in γ-aminobutyric (GABA)-ergic-mediated inhibitory neurotransmission [8]. Recurrent or continuous (i.e., status epilepticus [SE]) seizure activity can further alter the expression and localization of glutamate and GABA receptors as well as modify ionic gradients (e.g., chloride), reducing the seizure threshold and affecting pharmacoresponsiveness [9,10]. Glutamatergic-mediated increases in calcium signaling as well as production of reactive oxygen species may lead to neuronal injury and death [11]. On a larger scale, neural networks comprising functionally connected cortical and subcortical structures underlie epileptic seizures [12]. Secondary networks, where initiation, amplification, and expression of seizures occur at different sites, may explain how patients with Lennox–Gastaut syndrome can have similar electroclinical features despite differing etiologies [13].
Pharmacological treatments that produce long-term, persistent effects on disease course have been characterized in multiple neurological disorders, such as multiple sclerosis (MS), amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and migraine headaches [14,15,16,17,18,19,20]. For example, in MS, interferon β-1b, a modulator of the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway, has been used for approximately 30 years in the US and EU [19,21,22,23]. Subsequent disease-modifying therapies have been introduced since then that use monoclonal antibodies to other targets, treating both relapses and disease progression [24,25].
Similarly, it may be possible to modify the disease course by altering the underlying biology of epilepsy. This may prevent worsening of seizure frequency, severity, or phenotype; improve pharmacosensitivity; or reduce the need for ASMs [26]. Secondary comorbidities (e.g., declining memory, depression, and anxiety) might also be improved and considered as a positive effect on the disease course [26,27]. A related concept, antiepileptogenesis, refers to the prevention of epilepsy in a person who has a risk factor for developing epilepsy, such as traumatic brain injury (TBI), brain tumor, infection, surgery, or stroke. In addition to targeting underlying physiological mechanisms to alter the disease biology, improving seizure control with medical management might alter the underlying biology of epilepsy. Currently, there are no ASMs available with a proven disease modification or antiepileptogenesis benefit in humans.
In this article, we discuss the negative effects of recurrent seizures and examine the potential long-term, persistent effects of pharmacological treatments on the course of epilepsy. Additionally, we discuss the possibility of altering the biology and natural history of epilepsy through the treatment of seizures and by modifying the underlying pathophysiological processes that lead to epilepsy.
2. Potential Harmful Effects of Recurrent Seizures on the Underlying Biology of Epilepsy
Recurrent seizures may produce a variety of negative effects, including neuronal loss and structural changes, for example, hippocampal and temporal sclerosis, cortical dysplasia and thinning, brain atrophy, and gliosis [4,28]. Progressive impairment of cognition may also be associated with recurrent seizures [28]. In a longitudinal, case–control study, cortical thinning was detected in 77% of patients with temporal lobe epilepsy (TLE), independent of age, with the most pronounced changes occurring within 5 years of the onset of seizures [29]. In a study that examined the hippocampi in patients with TLE, which is often drug-resistant, hippocampal volumes were 14% to 18% smaller in patients with drug-resistant TLE compared with healthy controls; there were no differences between newly diagnosed patients, those with chronic TLE and well-controlled seizures, and healthy controls [30]. Moreover, the magnitude of the reduction in hippocampal volume was associated with total seizure number [30]. In a systematic review and meta-analysis of patients with drug-resistant TLE, marked ipsilateral hippocampal atrophy was correlated with epilepsy duration (r = −0.42; p < 0.0001) and seizure frequency (r = −0.35; p < 0.0001) [31]. In a single-center study of patients with drug-resistant TLE, neuronal loss or dysfunction was associated with frequent generalized tonic–clonic seizures but not complex partial seizures (focal impaired consciousness seizures) [32], suggesting that seizure type influences neuronal or hippocampal volume loss. Lesions or atrophy of various brain structures have also been characterized in progressive myoclonic disorders [33] and SE [34]. In a prospective study of patients with a history of moderate to severe TBI and nonconvulsive electrographic seizures, hippocampal atrophy was greater in patients with TBI and seizures than in patients with TBI but no seizures (21% vs. 12%; p = 0.017) [35]. Whether this represents a causal effect is not known, as volume loss may be correlated with seizure propensity.
A review of cognition in patients with epilepsy identified 19 controlled studies, a subset of which investigated the association of seizure frequency with cognitive endpoints [36]. Associations were noted between seizure frequency and adverse effects on memory, executive function, and intellectual function in adults; improved seizure control was associated with a gain in IQ. While data are limited, there are reported associations between seizure frequency or poorly controlled seizures and adverse effects on intelligence [36]. In a recent study of children aged 36 months with tuberous sclerosis complex (TSC), cumulative seizure burden was the most outstanding predictor of neurodevelopmental outcome [37]. A cross-sectional study in adults found a learning peak at age 16 to 17 years in patients with TLE compared with a peak at age 23 to 24 years in healthy controls. A decline in performance then ran in parallel, and people with TLE had persistently worse learning. This underscores the importance of epilepsy control at an early age to prevent development impairment and injury [38]. Additionally, other factors/characteristics (e.g., medication and seizure type) in people with epilepsy have been associated with cognitive deficits [36]. Uncontrolled seizures, particularly tonic–clonic seizures, are associated with increased mortality rates from all causes and from sudden unexpected death in epilepsy (SUDEP) [39,40]. Frequency of tonic–clonic seizures relates to SUDEP risk, with a ceiling effect at 3 or more seizures per year [41]. Uncontrolled seizures are also associated with increased medical morbidity, risk of injury, hospitalization, and emergency department visits [42,43,44]. Epilepsy, regardless of etiology, can negatively influence the quality of life, for which seizure control is a considerable factor [45,46,47]. Persistent seizures are associated with increased rates of suicidality and post-ictal psychosis [42]. Additionally, depression, stigma, and fear of recurrent seizures, as well as diminished social well-being (less social engagement and fewer relationships) and ASM-induced adverse reactions, can all contribute to decreased quality of life [45,48,49].
3. Evidence for Potential Therapeutic Alteration of the Underlying Biology of Epilepsy
3.1. Preclinical Evidence
Preclinical studies are often employed to identify and characterize molecular mechanisms that might underlie human disease and relate to clinical outcomes. Small molecules, such as traditional ASMs, are the predominant therapeutic modality in the drug development landscape of epilepsy; however, emerging approaches such as gene therapies for genetic epilepsies, cell therapies for acquired epilepsies, and brain stimulation may transform underlying disease processes. In the following section, antiepileptogenesis was defined as the prevention, delay, or reduction in severity of the onset of epilepsy (spontaneous recurring seizures [SRSs]). Disease-modifying was defined as a modification of the disease course with established SRSs. Evidence of disease modification includes any positive change in SRSs, behavior, and/or pathology. A change in any of these domains qualifies as disease modification.
Results summarized in Table 1 suggest that a number of modalities (i.e., small molecules, cell-based therapies, and gene therapies) have been found to modify the seizure phenotype, behavioral comorbidities, and pathophysiology in animal models of genetic epilepsy (e.g., absence, CLN2 disease, and Dravet syndrome) and acquired epilepsy induced by TBI, viral encephalitis, kindling, and SE. At a mechanistic level, therapies that decrease neuroinflammation, reduce oxidative stress, block DNA hypermethylation (e.g., adenosine sparing), increase neurotrophic potential, and reduce neuronal excitability, including therapies that modulate glutamatergic or GABAergic neurotransmission, may modify disease (Table 1). For example, cannabidiol [50] and lamotrigine [51] have been shown to have disease-modifying and/or antiepileptogenic effects in acquired epilepsy models, whereas carbamazepine [52], valproic acid [53], phenobarbital [54], and lacosamide [55,56] either had no effect or minor effects in the post–SE-induced models. In animal models of absence epilepsy, ethosuximide [57] and perampanel [58] have been reported to be disease-modifying or antiepileptogenic. Unfortunately, none of these agents have been demonstrated to modify or prevent epilepsy in humans. The fact that ASMs are largely ineffective as disease-modifying therapies in post-SE models (despite their ability to reduce neuronal excitability and attenuate symptomatic seizures in animals and patients with epilepsy) is consistent with clinical studies in which phenobarbital, phenytoin, carbamazepine, valproate, and magnesium failed to prevent the development of posttraumatic epilepsy [59]. It is possible that other agents could yield different results, and the study of disease modification after SE and other brain injuries should not be abandoned.

Table 1.
Summary of Published Preclinical Results.
The approaches with the greatest promise as disease-modifying therapies in preclinical studies include those therapies that target neuroinflammation and oxidative stress. These insults contribute to the development of epilepsy (Table 1). Many anti-inflammatory and antioxidant therapies modify both seizure phenotype and one or more comorbidities, including stress/anxiety [77,79], depression [61,62,79], and cognitive decline [65,76,77] associated with brain insult in animal models. However, soticlestat, an inhibitor of cholesterol 24-hydroxylase that reduces inflammatory markers, failed to meet primary clinical endpoints for seizure reduction in Dravet syndrome and Lennox–Gastaut syndrome [102], despite favorable results in a mouse model of Dravet syndrome [103]. Studies of anti-inflammatory agents are still worthwhile, as this agent used differently or other agents may prove beneficial. Disease-modifying results have also been observed in animal models with therapies that reduce neuronal excitability by replacing inhibitory interneurons within the epileptic circuit using targeted implantation of neuronal stem cells [87,91,92,93] or neurotrophic approaches using adeno-associated virus (AAV) vector-driven enhancement of inhibitory neuronal function [63,64,99]. These therapies will be discussed in the next section, as they have entered human trials, with preclinical studies guiding clinical development.
Target-driven therapeutic discovery offers hope for identifying specific disease-modifying treatments that may go beyond seizure control. Preclinical models provide platforms to evaluate these mechanisms in a controlled and systematic manner and allow scientists to explore the processes underlying epileptogenesis, such as neuroinflammation, oxidative stress, and disruptions in neuronal-glial interactions. This targeted approach opens the door to personalized therapies tailored to the pathophysiology of individual patients, potentially offering opportunities for disease modification and improved long-term outcomes.
3.2. Clinical Evidence
3.2.1. Nonpharmacological Therapies
Surgical resection and neurostimulation can alter the trajectory of epilepsy and its comorbidities for patients with epilepsy. Surgical resection can result in seizure freedom, but surgery is appropriate for only a subset of patients with refractory epilepsy, and approximately half of the patients continue to experience seizures postresection [104,105]. However, even when seizures persist, surgery can abolish tonic–clonic seizures in many patients [106], and surgery is associated with reduced mortality [107]. In children with TSC, surgery has been associated with neurodevelopmental improvement [108], providing strong evidence that seizure control improves developmental outcomes.
Long-term neurostimulation is also appropriate for a subset of patients with refractory epilepsy and includes responsive neurostimulation (RNS), deep brain stimulation (DBS), and vagus nerve stimulation (VNS). Neurostimulation appears to alter neural (seizure) networks over time, reduce the likelihood of seizures [109,110], yield extended periods of seizure freedom with progressively reduced frequency over time [111], and reduce mortality risk [112]. In three open-label extension (OLE) studies, the median reduction in seizure frequency in patients with RNS at 2, 5, and 9 years of observation were 53%, 66%, and 75%, respectively [113,114,115]. In patients with DBS who participated in a randomized controlled trial, the median reduction in seizure frequency at 2, 5, and 7 years of follow-up were 56%, 69%, and 75%, respectively [116,117,118]. In a meta-analysis of patients treated with VNS, mean seizure reduction at 1, 2, and 3 years was 32.9%, 44.4%, and 53.5%, respectively [119]. While surgical and some neurostimulation strategies reduce seizures over time, suggesting a change in the natural history of epilepsy, there remains an unmet need for noninvasive, less restrictive options for which a heterogeneous patient population would be eligible.
3.2.2. Pharmacological Therapies
There is evidence to suggest that some ASMs may have long-term, persistent effects on the course of epilepsy (e.g., progressive reduction in seizure counts, increases in responder rates, or other measures of seizure control over time). Pharmacological treatments that might alter the underlying biology of epilepsy include ASMs (daily and rescue treatments) as well as other treatments. The ability of an ASM to alter the underlying biology of epilepsy might result from direct action on the pathophysiology of epilepsy, indirect action as a product of enhanced seizure control (i.e., prevent negative outcomes associated with recurrent seizures), or some combination of the two. We have included recent clinical trials of ASMs that demonstrated favorable long-term improvements in seizure control, as these treatments might alter the biology of epilepsy as a result of fewer and/or less severe seizures (Table 2).
Table 2.
Summary of Published Clinical Results.
Daily Treatments
(1) Cenobamate
Cenobamate, an inhibitor of voltage-gated sodium currents and a positive allosteric modulator of GABA-A receptors, has exhibited long-term efficacy in controlling seizures [120,132,133]. In a randomized, placebo-controlled trial of adult patients with focal seizures, the median percent change in seizure frequency during the 18-week treatment period was 55% in the 200 mg and 400 mg cenobamate groups compared with 24% in the placebo group (p < 0.001) [120]. In a long-term, open-label study, rates of participants who achieved seizure freedom during consecutive 6-month intervals increased over time, from 25% during months 3 to 9 to 44% during months 27 to 33 (median dose at month 33, 300 mg; Figure 1) [121].
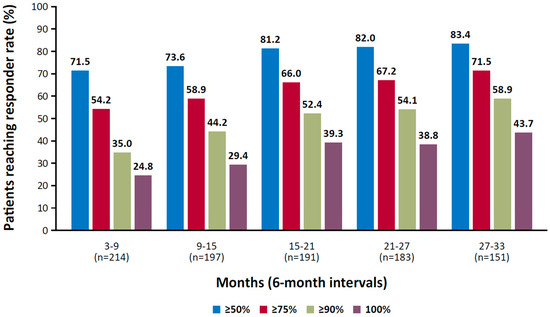
Figure 1.
Responder rate at consecutive 6-month intervals in a phase 3, open-label study of cenobamate. Reproduced with permission from Sperling et al., Efficacy of cenobamate for uncontrolled focal seizures: Post hoc analysis of a Phase 3, multicenter, open-label study; published by Wiley, 2021.
(2) Cannabidiol
There is a wide variety of molecular targets for cannabidiol, including nonselective ion channels (transient-receptor potential channels), voltage-gated sodium channels, potassium channels, G protein–coupled receptor 55, and 5-HT receptors [134]. In a long-term OLE study of cannabidiol oral solution in patients with Lennox–Gastaut syndrome, the median percent reduction in total seizures increased over time, from 48% at weeks 1 to 12 to 65% at weeks 145 to 156 (mean modal dose, 24 mg/kg/d; Figure 2A) [122]. In a long-term OLE in patients with Dravet syndrome, the median percent reduction in total seizures increased from 49% at weeks 1 to 12 to 78% at weeks 145 to 156 (mean modal dose, 22 mg/kg/d; Figure 2B) [123].
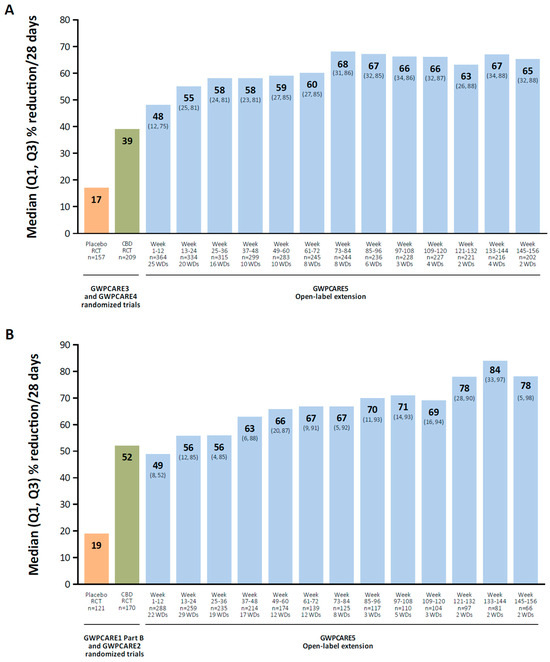
Figure 2.
Median reduction in total seizure frequency in OLEs of cannabidiol oral solution. Patients with Lennox–Gastaut (A) or Dravet syndrome (B). CBD, cannabidiol; OLE, open-label extension; RCT, randomized controlled trial; WD, withdrawal. Reproduced with permission from Patel et al., Long-term safety and efficacy of add-on cannabidiol in patients with Lennox–Gastaut syndrome: Results of a long-term open-label extension trial; published by Wiley, 2021 and Scheffer et al., Add-on cannabidiol in patients with Dravet syndrome: Results of a long-term open-label extension trial; published by Wiley, 2021.
(3) Vigabatrin
Vigabatrin is an inhibitor of GABA transaminase, a GABA-degrading enzyme; inhibition of this enzyme increases GABA availability and attenuates excitability associated with seizures [135]. It has been suggested that vigabatrin could alter the course or prevent the development of epilepsy in TSC [124,125,126]. In a retrospective study, 65% of children with TSC who received early treatment with vigabatrin achieved seizure freedom; in those who received vigabatrin later, 24% achieved seizure freedom (p < 0.01) [124]. In a prospective study of vigabatrin that compared early treatment (within a week of the appearance of epileptiform discharges in the EEG) with standard treatment (within a week of onset of seizures), infants with TSC and early treatment had lower rates of intellectual disability (14% vs. 48%; p = 0.031), fewer polytherapy requirements (21% vs. 55%; p = 0.039), and a lower incidence of DRE (7% vs. 42%; p = 0.004) [125]. This study was limited by its design—an open-label, single-center study that prospectively compared a preventative treatment group with a prior group of infants who had received standard treatment based on conventional practice [125].
In a pooled analysis from a randomized controlled trial (EPISTOP), children at 24 months of age who received prophylactic vigabatrin that was administered after epileptiform activity was detected but before onset of seizures had a lower likelihood of developing clinical seizures (OR, 0.21; 95% CI, 0.04–0.9), infantile spasms (OR, 0; 95% CI, 0–0.33), and DRE (OR, 0.23; 95% CI, 0.06–0.83) compared with children receiving conventional treatment (ASMs after the development of electrographic or clinical seizures) [126]. There were no differences in cognitive scores (Bayley Scales of Infant and Toddler Development, 3rd ed [Bayley-III]) between groups, and the incidence of neurodevelopmental delay was also similar [126]. Limitations of the trial were small sample size (only 27 children randomized) and the absence of a placebo-treated control group. In a larger randomized, double-blind, placebo-controlled trial (PREVeNT), no differences were observed between vigabatrin and placebo treatments on cognitive (Bayley-III) and behavioral (Vineland-II adaptive behavior) outcomes in infants with TSC, consistent with the EPISTOP study [126,127]. In both studies, infants in the comparator groups received open-label vigabatrin for seizure control once seizures developed. In the PREVeNT study, most infants in the placebo group experienced seizures soon after randomization, so the duration of vigabatrin exposure between placebo and vigabatrin groups was similar, potentially affecting the analysis of cognitive measures [127]. Moreover, there were no differences between vigabatrin and placebo in the number of infants who experienced seizures or had DRE at 24 months of age, which was not consistent with EPISTOP, although vigabatrin delayed the onset and reduced the prevalence of infantile spasms. Despite randomization, the TSC genotype was unbalanced between placebo and vigabatrin groups; infants with TSC1 mutations or variants of unknown significance/no mutation genotypes were more common in the placebo arm. Therefore, the potentially milder form of TSC in the placebo group might have masked any between-treatment separation. Future trials should control for multiple TSC-associated factors that affect seizure risk, DRE, and cognitive outcomes [127].
Intermittent Treatments
- Diazepam Nasal Spray
Diazepam is a positive allosteric modulator of the GABA-A receptor, and diazepam nasal spray is an aqueous spray that utilizes dodecyl maltoside (Intravail®) to facilitate intranasal absorption and vitamin E to enhance the solubility of diazepam [136,137]. The safety and effectiveness of diazepam nasal spray were examined in a phase 3, long-term, open-label, repeat-dose safety study conducted in patients with seizure clusters aged 6 to 65 years [138] and a phase 1/2a, open-label, single-dose, PK study with a 180-day, open-label safety period and optional extension enrolling patients with epilepsy aged 2 to 5 years [139]. Diazepam nasal spray had a comparable safety profile to diazepam rectal gel, and <20% of seizure clusters were treated with a second dose, supporting its effectiveness [138,139].
A post hoc analysis of the phase 3 study in patients aged 6 to 65 years was conducted to examine the duration (in days) between treated seizure clusters (seizure interval [SEIVAL]) over time [128]. SEIVAL was assessed using 90-day periods, as this period length would allow for the inclusion of a sufficient number of patients with an assessable SEIVAL for each period; four 90-day periods approximated the study’s 365-day treatment period. In the analysis of patients with SEIVALs in each of the four 90-day periods (second doses excluded [i.e., excluding retreatment of a seizure cluster within 24 h]), mean SEIVAL increased from 13.9 days in Period 1 to 26.8 days in Period 4 (p ≤ 0.001; Figure 3). The increase in SEIVAL was independent of age, duration of exposure, and change in concomitant medications. This was consistent with results for the overall study population, which included variable cohorts for each period [128]. Similar results were observed regardless of sex, whether the medication was self-administered or administered by a caregiver, or the presence of developmental epileptic encephalopathies [140,141].
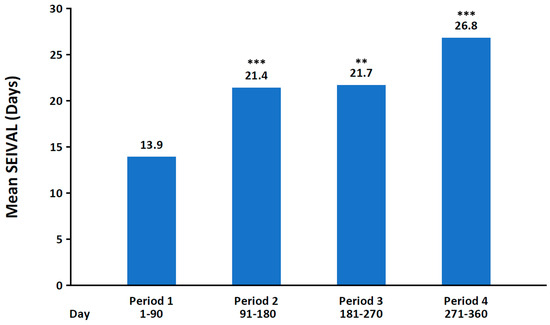
Figure 3.
Mean interval between seizure clusters for Periods 1–4 of phase 3, open-label study of diazepam nasal spray (n = 76). SEIVAL, seizure interval. ** p < 0.01, *** p ≤ 0.001 compared with Period 1. Reproduced with permission from Misra et al., Significant improvements in seizure interval (time between seizure clusters) across time in patients treated with diazepam nasal spray as intermittent rescue therapy for seizure clusters; published by Wiley, 2022.
A subsequent analysis of the open-label safety study evaluated the effect of treatment on the proportion of prolonged seizures recorded in the study population over time (Periods 1–4) [142]. In this analysis, prolonged seizures were defined as those treated 5 to 15 min after seizure onset. When anchored to the patient’s first treated seizure, the proportion of prolonged seizures decreased from 31% in Period 1 to 17% in Period 4. When anchoring the start of Period 1 to the patient’s first treated prolonged seizure, the proportion of prolonged seizures decreased from 44% in Period 1 to 15% in Period 4 [142].
The mechanism for the increase in SEIVAL should be elucidated. A preclinical, repeat-dosing method of diazepam has been developed in rats to create an experimental condition for intermittent use of diazepam similar to the rescue treatment of seizure clusters in patients [143]. If progressively increased SEIVAL is caused by rapid cessation of seizure activity or prevention of clusters, these preclinical studies will provide an opportunity to specifically evaluate the effects of intermittent diazepam administration on the underlying biology of seizures [143].
Other Pharmacological Treatments
Everolimus inhibits mammalian target of rapamycin (mTOR) signaling, which is involved in the pathophysiology of TSC [129]. In a randomized, placebo-controlled trial conducted in patients with focal onset seizures in TSC, the number of patients who achieved ≥50% reduction in seizure frequency (responders) from baseline was greater in low-dose (28.2%; p = 0.0077; 3–7 ng/mL) and high-dose (40.0%; p < 0.001; 9–15 ng/mL) everolimus groups compared with placebo (15.1%). Median percentage changes in seizure frequency from baseline were 14.9%, 29.3%, and 39.6% with placebo, low-dose, and high-dose everolimus, respectively [129]. In a separate study, the effect of everolimus on white matter in TSC was examined using diffusion tensor imaging [130], with changes in white matter having been associated with neurological comorbidity [144]. Everolimus treatment was associated with a decrease in mean diffusivity and radial diffusivity, while axial diffusivity increased in untreated patients, suggesting that everolimus modifies white matter changes that occur with increased mTOR activity in TSC [130]. Hence, treating underlying pathophysiology affects epilepsy outcomes.
A retrospective study conducted in pediatric patients with DRE who received pulsatile corticoid therapy (dexamethasone; up to 10 cycles) assessed whether treatment reduced the burden of interictal epileptiform activity [131]. Such burden was reduced after dexamethasone treatment compared with baseline; sleep physiology and quality of life (QOL) also improved following treatment. The authors concluded that pulsatile corticoid therapy may attenuate the pathophysiology of epilepsy and improve associated outcomes of sleep and cognitive function [131].
4. Discussion
Research is needed to develop therapies that produce long-term beneficial effects on the course of disease. The nature of epilepsy and epileptogenesis is variable and complex [145], and complex and innovative solutions will likely be required. Nevertheless, preliminary evidence suggests that pharmacological modification of the long-term disease course may be achievable. Agents that enhance inhibition, such as vigabatrin and diazepam, or address pathophysiological mechanisms associated with epilepsy, such as everolimus, hold promise for altering the natural history of epilepsy. Challenges that come with evaluating disease modification in clinical trials include establishing validated biomarkers for disease modification (which are likely syndrome-specific) that can demonstrate target engagement of the drug or intervention. Rigorous trial designs that use validated longitudinal endpoints, allow for variability of natural history, and have sufficient statistical power are necessary to identify successful disease-modifying therapies.
Focusing on the use of relevant animal models that display many parallels to the human condition should guide clinical trials. Data emerging from these studies can provide important insights into the pathophysiology of the disease and the potential for therapeutic interventions that target disease-specific defects. As described below, preclinical disease-modifying studies have the potential to inform clinical trials designed to demonstrate human disease modification and play an important role in providing proof of concept that the treatment, through engagement of its intended target, has the potential to modify the outcome in an appropriate animal model. Past experience suggests that animal models often best predict results in animal models, so no assumptions can be made about effects in human epilepsy without investigation in humans.
Ongoing Research
Targeted therapies acting on underlying anatomic and physiological derangements have now entered clinical trials. Stem– or progenitor–cell-based interventions to replace and/or repair altered networks and gene/RNA therapies (e.g., AAV vectors and antisense oligonucleotides) are in the early stages of clinical development. In preliminary findings from an ongoing phase 1/2 clinical trial (NCT05135091) [146], adult participants with drug-resistant, unilateral TLE (n = 5) experienced 82% seizure reduction following transplantation of medial ganglionic eminence GABAergic interneurons cells (NRTX-1001) in the hippocampus [147], and clinical observations are consistent with preclinical results reported in a mouse model of mesial TLE [93]. Multiple phase 1/2 studies are currently examining ETX101, an AAV serotype 9 (AAV9) gene therapy designed to express voltage-gated sodium channels (NaV1.1) in GABAergic inhibitory interneurons [64] in children with SCN1A-positive Dravet syndrome [148,149,150]. These studies rely on preclinical efficacy (disease-modifying) and safety reported in Scn1a+/− knockout mice and nonhuman primates, respectively [64]. Additionally, a phase 1/2 study examining the safety, tolerability, and efficacy of AMT-260, an AAV9 vector that reduces expression of GluK2 and GluK2-containing kainate receptors, is being conducted in adults with unilateral refractory mesial TLE [151]. In phase 1/2a trials of zorevunersen (STK-001), an antisense oligonucleotide that increases expression of NaV1.1 channels [152], median convulsive seizure frequency was reduced at 3 and 6 months after the last dose compared with baseline in children with SCN1A-positive Dravet syndrome [153,154]. The efficacy, safety, and tolerability of zorevunersen will be further examined in a phase 3, multicenter, randomized, double-blind, sham-controlled clinical trial [155].
5. Conclusions
Exciting steps are being taken to study disease modification in epilepsy. Major problems that remain are the tenuous link between animal models and human epilepsies and the huge variety of types of seizures, epilepsies, and variations in the pathophysiology of human epilepsy. For example, the methods typically used to induce SE in animals differ from the common causes of SE in humans (e.g., stroke and brain trauma) [156]. Some preclinical methods in animals that induce seizures and epilepsy in relatively rapid fashion are synthetically induced (e.g., chemical or electrical stimuli), and the methods themselves might influence study outcomes apart from epileptogenesis. Homogeneity in preclinical methods, including animal models and strains, is best for reducing variability and detecting statistically significant differences but may affect generalizability to human epilepsy. Moreover, human brains differ substantially from animal brains and may respond differently to identical insults. Commonalities in underlying neural mechanisms, however, provide hope that results from some animal models will transfer to humans. It is hoped that better characterization of small-molecule therapies and newer therapeutic techniques will provide insights that allow for modification of disease course and improvement in patient lives.
Author Contributions
All authors were involved in the conception, design, review of the literature, and drafting of the manuscript. All authors critically reviewed and revised the work. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was funded by Neurelis, Inc. (San Diego, CA, USA).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Medical writing support was provided by Kirk W. Evanson, from Citrus Health Group, Inc. (Chicago, IL, USA), and was funded by Neurelis, Inc. (San Diego, CA, USA).
Conflicts of Interest
Sperling has consulted for Medtronic and Neurelis, Inc. He has received research support from Medtronic; SK Life Science; Takeda; Xenon; Cerevel; UCB Pharma; Janssen; Equilibre; Epiwatch; Byteflies, Biohaven, and Cavion. He receives royalties from Oxford University Press and Cambridge University Press. Peters has served as a speaker and consultant for Greenwich Biosciences; Neurelis, Inc.; and Novartis. Wu and Guignet received research funding from Neurelis, Inc. White received grant funding from Neurelis, Inc., UCB Pharma, and Eisai Pharmaceuticals; consultant fees from Biogen, GW, Neurelis, Inc., Takeda, Inc., and JAZZ Pharmaceuticals; and speaker honoraria from SK Pharmaceuticals and UCB Pharma. He is also co-founder of NeuroAdjuvants, Inc. Shih was an employee of and had received stock options from Neurelis, Inc. Ngo is an employee of and has received stock options from Neurelis, Inc. Carrazana is an employee of and has received stock and stock options from Neurelis, Inc. Rabinowicz is an employee of and has received stock options from Neurelis, Inc.
References
- World Health Organization. Epilepsy: A Public Health Imperative. 2019. Available online: https://www.who.int/publications/i/item/epilepsy-a-public-health-imperative (accessed on 10 September 2024).
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Kalviainen, R. Progressive myoclonus epilepsies. Semin. Neurol. 2015, 35, 293–299. [Google Scholar] [CrossRef]
- Jiruska, P.; Freestone, D.; Gnatkovsky, V.; Wang, Y. An update on the seizures beget seizures theory. Epilepsia 2023, 64 (Suppl. S3), S13–S24. [Google Scholar] [CrossRef]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef]
- Stern, J.M.; Sankar, R.; Sperling, M. (Eds.) Medication-Resistant Epilepsy Diagnosis and Treatment; Cambridge University Press: Cambridge, UK, 2020. [Google Scholar]
- Rao, V.R. Chronic electroencephalography in epilepsy with a responsive neurostimulation device: Current status and future prospects. Expert Rev. Med. Devices 2021, 18, 1093–1105. [Google Scholar] [CrossRef]
- Bromfield, E.B.; Cavazos, J.E.; Sirven, J.I. (Eds.) An Introduction to Epilepsy; American Epilepsy Society: West Hartford, CT, USA, 2006. [Google Scholar]
- Naylor, D.E. In the fast lane: Receptor trafficking during status epilepticus. Epilepsia Open 2023, 8 (Suppl. S1), S35–S65. [Google Scholar] [CrossRef]
- Kapur, J.; Long, L.; Dixon-Salazar, T. Consequences: Bench to home. Epilepsia 2022, 63 (Suppl. S1), S14–S24. [Google Scholar] [CrossRef]
- Walker, M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef]
- Lehnertz, K.; Brohl, T.; Wrede, R.V. Epileptic-network-based prediction and control of seizures in humans. Neurobiol. Dis. 2023, 181, 106098. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.S.; Warren, A.E.; Jackson, G.D.; Abbott, D.F. Conceptualizing Lennox-Gastaut syndrome as a secondary network epilepsy. Front. Neurol. 2014, 5, 225. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Lalli, S.; Monsurro, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.A.; Isaranuwatchai, W.; Grossman, L.; Straus, S.E. Disease-modifying drugs for Alzheimer disease: Implications for people in Canada. CMAJ 2023, 195, E1446–E1448. [Google Scholar] [CrossRef]
- Cummings, J.L.; Osse, A.M.L.; Kinney, J.W. Alzheimer’s disease: Novel targets and investigational drugs for disease modification. Drugs 2023, 83, 1387–1408. [Google Scholar] [CrossRef]
- Olejnik, P.; Roszkowska, Z.; Adamus, S.; Kasarello, K. Multiple sclerosis: A narrative overview of current pharmacotherapies and emerging treatment prospects. Pharmacol. Rep. 2024, 76, 926–943. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Pitzer, K.; Fiske, B.; Navangul, A.; Beissert, K.; Pilcicka, A.; Fuest, R.; Wyse, R.K.; et al. Parkinson’s disease drug therapies in the clinical trial pipeline: 2024 update. J. Park. Dis. 2024, 14, 899–912. [Google Scholar] [CrossRef]
- Spain, R.I.; Cameron, M.H.; Bourdette, D. Recent developments in multiple sclerosis therapeutics. BMC Med. 2009, 7, 74. [Google Scholar] [CrossRef]
- Bayer AG. BETAFERON (Interferon beta-1b); Summary of Product Characteristics; Bayer AG: Berlin, Germany, 2006. [Google Scholar]
- Bayer HealthCare Pharmaceuticals Inc. BETASERON (Interferon beta-1b); Full Prescribing Information; Bayer HealthCare Pharmaceuticals Inc.: Whippany, NJ, USA, 2023. [Google Scholar]
- Rae-Grant, A.; Day, G.S.; Marrie, R.A.; Rabinstein, A.; Cree, B.A.C.; Gronseth, G.S.; Haboubi, M.; Halper, J.; Hosey, J.P.; Jones, D.E.; et al. Comprehensive systematic review summary: Disease-modifying therapies for adults with multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018, 90, 789–800. [Google Scholar] [CrossRef]
- Fox, R.J.; Bar-Or, A.; Traboulsee, A.; Oreja-Guevara, C.; Giovannoni, G.; Vermersch, P.; Syed, S.; Li, Y.; Vargas, W.S.; Turner, T.J.; et al. Tolebrutinib in nonrelapsing secondary progressive multiple sclerosis. N. Engl. J. Med. 2025, 392, 1883–1892. [Google Scholar] [CrossRef]
- French, J.A.; Bebin, M.; Dichter, M.A.; Engel, J., Jr.; Hartman, A.L.; Jozwiak, S.; Klein, P.; McNamara, J., Sr.; Twyman, R.; Vespa, P. Antiepileptogenesis and disease modification: Clinical and regulatory issues. Epilepsia Open 2021, 6, 483–492. [Google Scholar] [CrossRef]
- Rauh, R.; Schulze-Bonhage, A.; Metternich, B. Assessment of anxiety in patients with epilepsy: A literature review. Front. Neurol. 2022, 13, 836321. [Google Scholar] [CrossRef]
- Klein, P.; Carrazana, E.; Glauser, T.; Herman, B.P.; Penovich, P.; Rabinowicz, A.L.; Sutula, T.P. Do seizures damage the brain?-Cumulative effects of seizures and epilepsy: A 2025 perspective. Epilepsy Curr. 2025. [Google Scholar] [CrossRef]
- Galovic, M.; van Dooren, V.Q.H.; Postma, T.S.; Vos, S.B.; Caciagli, L.; Borzi, G.; Cueva Rosillo, J.; Vuong, K.A.; de Tisi, J.; Nachev, P.; et al. Progressive cortical thinning in patients with focal epilepsy. JAMA Neurol. 2019, 76, 1230–1239. [Google Scholar] [CrossRef]
- Kalviainen, R.; Salmenpera, T.; Partanen, K.; Vainio, P.; Riekkinen, P.; Pitkanen, A. Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy. Neurology 1998, 50, 1377–1382. [Google Scholar] [CrossRef]
- Caciagli, L.; Bernasconi, A.; Wiebe, S.; Koepp, M.J.; Bernasconi, N.; Bernhardt, B.C. A meta-analysis on progressive atrophy in intractable temporal lobe epilepsy: Time is brain? Neurology 2017, 89, 506–516. [Google Scholar] [CrossRef]
- Tasch, E.; Cendes, F.; Li, L.M.; Dubeau, F.; Andermann, F.; Arnold, D.L. Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann. Neurol. 1999, 45, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Valetto, A.; Bertini, V.; Esposito, M.; Carli, N.; Minassian, B.A.; Bonuccelli, A.; Peroni, D.; Michelucci, R.; Striano, P. The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure 2019, 71, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Mendes, A.; Sampaio, L. Brain magnetic resonance in status epilepticus: A focused review. Seizure 2016, 38, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.M.; McArthur, D.L.; Xu, Y.; Eliseo, M.; Etchepare, M.; Dinov, I.; Alger, J.; Glenn, T.P.; Hovda, D. Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology 2010, 75, 792–798. [Google Scholar] [CrossRef]
- Kesselmayer, R.F.; Morel, G.M.; Bordenavem, J.M.; Jones, J.; Hermann, B. Epilepsy and cognition. In The Comorbidities of Epilepsy; Mula, M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 245–272. [Google Scholar]
- Ihnen, S.K.Z.; Alperin, S.; Capal, J.K.; Cohen, A.L.; Peters, J.M.; Bebin, E.M.; Northrup, H.A.; Sahin, M.; Krueger, D.A.; Group, T.S. Accumulated seizure burden predicts neurodevelopmental outcome at 36 months of age in patients with tuberous sclerosis complex. Epilepsia 2025, 66, 117–133. [Google Scholar] [CrossRef]
- Helmstaedter, C.; Elger, C.E. Chronic temporal lobe epilepsy: A neurodevelopmental or progressively dementing disease? Brain 2009, 132, 2822–2830. [Google Scholar] [CrossRef]
- Trinka, E.; Rainer, L.J.; Granbichler, C.A.; Zimmermann, G.; Leitinger, M. Mortality, and life expectancy in epilepsy and status epilepticus—Current trends and future aspects. Front Epidemiol 2023, 3, 1081757. [Google Scholar] [CrossRef]
- Ryvlin, P.; Nashef, L.; Lhatoo, S.D.; Bateman, L.M.; Bird, J.; Bleasel, A.; Boon, P.; Crespel, A.; Dworetzky, B.A.; Hogenhaven, H.; et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): A retrospective study. Lancet Neurol. 2013, 12, 966–977. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Tomson, T.; Benn, E.; Sander, J.W.; Nilsson, L.; Langan, Y.; Walczak, T.S.; Beghi, E.; Brodie, M.J.; Hauser, A.; et al. Combined analysis of risk factors for SUDEP. Epilepsia 2011, 52, 1150–1159. [Google Scholar] [CrossRef]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.A.; Wang, Z.J.; Chang, E.; Powers, A.; Copher, R.; Cherepanov, D.; Broder, M.S. Healthcare utilization and costs in adults with stable and uncontrolled epilepsy. Epilepsy Behav. 2014, 31, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug resistance in epilepsy: Clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Siebenbrodt, K.; Willems, L.M.; von Podewils, F.; Mross, P.M.; Struber, M.; Langenbruch, L.; Bierhansl, L.; Gorny, I.; Schulz, J.; Gaida, B.; et al. Determinants of quality of life in adults with epilepsy: A multicenter, cross-sectional study from Germany. Neurol Res Pr. 2023, 5, 41. [Google Scholar] [CrossRef]
- Conway, L.; Smith, M.L.; Ferro, M.A.; Speechley, K.N.; Connoly, M.B.; Snead, O.C.; Widjaja, E.; Team, P.S. Correlates of health-related quality of life in children with drug resistant epilepsy. Epilepsia 2016, 57, 1256–1264. [Google Scholar] [CrossRef]
- Ridsdale, L.; Wojewodka, G.; Robinson, E.; Landau, S.; Noble, A.; Taylor, S.; Richardson, M.; Baker, G.; Goldstein, L.H.; Team, S. Characteristics associated with quality of life among people with drug-resistant epilepsy. J. Neurol. 2017, 264, 1174–1184. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Aledo-Serrano, A.; Coppola, A.; Didelot, A.; Bates, E.; Sainz-Fuertes, R.; Lawthom, C. The impact of epilepsy on quality of life: Findings from a European survey. Epilepsy Behav. 2023, 142, 109179. [Google Scholar] [CrossRef]
- Yogarajah, M.; Mula, M. Social cognition, psychiatric comorbidities, and quality of life in adults with epilepsy. Epilepsy Behav. 2019, 100, 106321. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Mbilinyi, R.H.; Ramakrishnan, S. Efficacy of the FDA-approved cannabidiol on the development and persistence of temporal lobe epilepsy and complex focal onset seizures. Exp. Neurol. 2023, 359, 114240. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Wang, Q.; van Luijtelaar, G.; Sun, M. The effects of lamotrigine and ethosuximide on seizure frequency, neuronal loss, and astrogliosis in a model of temporal-lobe epilepsy. Brain Res. 2019, 1712, 1–6. [Google Scholar] [CrossRef]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef]
- Langer, M.; Brandt, C.; Zellinger, C.; Loscher, W. Therapeutic window of opportunity for the neuroprotective effect of valproate versus the competitive AMPA receptor antagonist NS1209 following status epilepticus in rats. Neuropharmacology 2011, 61, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.; Knox, K.; Zierath, D.; Koneval, Z.; Metcalf, C.; Wilcox, K.S.; White, H.S. Development of an antiepileptogenesis drug screening platform: Effects of everolimus and phenobarbital. Epilepsia 2021, 62, 1677–1688. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Stohr, T.; Matagne, A. The acute and chronic effects of the novel anticonvulsant lacosamide in an experimental model of status epilepticus. Epilepsy Res. 2011, 94, 10–17. [Google Scholar] [CrossRef]
- Licko, T.; Seeger, N.; Zellinger, C.; Russmann, V.; Matagne, A.; Potschka, H. Lacosamide treatment following status epilepticus attenuates neuronal cell loss and alterations in hippocampal neurogenesis in a rat electrical status epilepticus model. Epilepsia 2013, 54, 1176–1185. [Google Scholar] [CrossRef]
- Dezsi, G.; Ozturk, E.; Stanic, D.; Powell, K.L.; Blumenfeld, H.; O’Brien, T.J.; Jones, N.C. Ethosuximide reduces epileptogenesis and behavioral comorbidity in the GAERS model of genetic generalized epilepsy. Epilepsia 2013, 54, 635–643. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Franco, V.; Marchiselli, R.; Perucca, E.; De Sarro, G.; Russo, E. Perampanel effects in the WAG/Rij rat model of epileptogenesis, absence epilepsy, and comorbid depressive-like behavior. Epilepsia 2017, 58, 231–238. [Google Scholar] [CrossRef]
- Temkin, N.R. Preventing and treating posttraumatic seizures: The human experience. Epilepsia 2009, 50 (Suppl. S2), 10–13. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; De Fazio, P.; De Sarro, G.; Russo, E. Antidepressants but not antipsychotics have antiepileptogenic effects with limited effects on comorbid depressive-like behaviour in the WAG/Rij rat model of absence epilepsy. Br. J. Pharmacol. 2015, 172, 3177–3188. [Google Scholar] [CrossRef]
- Leo, A.; Citraro, R.; Amodio, N.; De Sarro, C.; Gallo Cantafio, M.E.; Constanti, A.; De Sarro, G.; Russo, E. Fingolimod exerts only temporary antiepileptogenic effects but longer-lasting positive effects on behavior in the WAG/Rij rat absence epilepsy model. Neurotherapeutics 2017, 14, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Nesci, V.; Tallarico, M.; Amodio, N.; Gallo Cantafio, E.M.; De Sarro, G.; Constanti, A.; Russo, E.; Citraro, R. IL-6 receptor blockade by tocilizumab has anti-absence and anti-epileptogenic effects in the WAG/Rij rat model of absence epilepsy. Neurotherapeutics 2020, 17, 2004–2014. [Google Scholar] [CrossRef]
- Takahashi, K.; Eultgen, E.M.; Wang, S.H.; Rensing, N.R.; Nelvagal, H.R.; Dearborn, J.T.; Danos, O.; Buss, N.; Sands, M.S.; Wong, M.; et al. Gene therapy ameliorates spontaneous seizures associated with cortical neuron loss in a Cln2R207X mouse model. J. Clin. Investig. 2023, 133, e165908. [Google Scholar] [CrossRef]
- Tanenhaus, A.; Stowe, T.; Young, A.; McLaughlin, J.; Aeran, R.; Lin, I.W.; Li, J.; Hosur, R.; Chen, M.; Leedy, J.; et al. Cell-selective adeno-associated virus-mediated SCN1A gene regulation therapy rescues mortality and seizure phenotypes in a Dravet syndrome mouse model and is well tolerated in nonhuman primates. Hum. Gene Ther. 2022, 33, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Dyomina, A.V.; Zubareva, O.E.; Smolensky, I.V.; Vasilev, D.S.; Zakharova, M.V.; Kovalenko, A.A.; Schwarz, A.P.; Ischenko, A.M.; Zaitsev, A.V. Anakinra reduces epileptogenesis, provides neuroprotection, and attenuates behavioral impairments in rats in the lithium-pilocarpine model of epilepsy. Pharmaceuticals 2020, 13, 340. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Y.; Xu, C.; Liu, K.; Wang, Y.; Chen, L.; Wu, X.; Gao, F.; Guo, Y.; Zhu, J.; et al. Therapeutic potential of an anti-high mobility group box-1 monoclonal antibody in epilepsy. Brain. Behav. Immun. 2017, 64, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Pitsch, J.; Kuehn, J.C.; Gnatkovsky, V.; Muller, J.A.; van Loo, K.M.J.; de Curtis, M.; Vatter, H.; Schoch, S.; Elger, C.E.; Becker, A.J. Anti-epileptogenic and anti-convulsive effects of fingolimod in experimental temporal lobe epilepsy. Mol. Neurobiol. 2019, 56, 1825–1840. [Google Scholar] [CrossRef]
- Chen, M.; Arumugam, T.V.; Leanage, G.; Tieng, Q.M.; Yadav, A.; Ullmann, J.F.; She, D.T.; Truong, V.; Ruitenberg, M.J.; Reutens, D.C. Disease-modifying effect of intravenous immunoglobulin in an experimental model of epilepsy. Sci. Rep. 2017, 7, 40528. [Google Scholar] [CrossRef]
- Jimenez-Pacheco, A.; Diaz-Hernandez, M.; Arribas-Blazquez, M.; Sanz-Rodriguez, A.; Olivos-Ore, L.A.; Artalejo, A.R.; Alves, M.; Letavic, M.; Miras-Portugal, M.T.; Conroy, R.M.; et al. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J. Neurosci. 2016, 36, 5920–5932. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Heck, T.D.; Dahle, E.J.; Vanegas, F.; Pruess, T.H.; Wilcox, K.S.; White, H.S. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 2016, 57, 1958–1967. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef]
- Polascheck, N.; Bankstahl, M.; Loscher, W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2010, 224, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Gericke, B.; Brandt, C.; Theilmann, W.; Welzel, L.; Schidlitzki, A.; Twele, F.; Kaczmarek, E.; Anjum, M.; Hillmann, P.; Loscher, W. Selective inhibition of mTORC1/2 or PI3K/mTORC1/2 signaling does not prevent or modify epilepsy in the intrahippocampal kainate mouse model. Neuropharmacology 2020, 162, 107817. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Carlson, S.; Gregory-Flores, A.; Hinojo-Perez, A.; Olson, A.; Thippeswamy, T. Mechanisms of disease-modifying effect of saracatinib (AZD0530), a Src/Fyn tyrosine kinase inhibitor, in the rat kainate model of temporal lobe epilepsy. Neurobiol. Dis. 2021, 156, 105410. [Google Scholar] [CrossRef] [PubMed]
- Puttachary, S.; Sharma, S.; Verma, S.; Yang, Y.; Putra, M.; Thippeswamy, A.; Luo, D.; Thippeswamy, T. 1400W, a highly selective inducible nitric oxide synthase inhibitor is a potential disease modifier in the rat kainate model of temporal lobe epilepsy. Neurobiol. Dis. 2016, 93, 184–200. [Google Scholar] [CrossRef]
- Jiang, Z.; Guo, M.; Shi, C.; Wang, H.; Yao, L.; Liu, L.; Xie, C.; Pu, S.; LaChaud, G.; Shen, J.; et al. Protection against cognitive impairment and modification of epileptogenesis with curcumin in a post-status epilepticus model of temporal lobe epilepsy. Neuroscience 2015, 310, 362–371. [Google Scholar] [CrossRef]
- Sandouka, S.; Singh, P.K.; Saadi, A.; Taiwo, R.O.; Sheeni, Y.; Zhang, T.; Deeb, L.; Guignet, M.; White, S.H.; Shekh-Ahmad, T. Repurposing dimethyl fumarate as an antiepileptogenic and disease-modifying treatment for drug-resistant epilepsy. J. Transl. Med. 2023, 21, 796. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Tchekalarova, J.D.; Ivanova, N.M.; Pechlivanova, D.M.; Atanasova, D.; Lazarov, N.; Kortenska, L.; Mitreva, R.; Lozanov, V.; Stoynev, A. Antiepileptogenic and neuroprotective effects of losartan in kainate model of temporal lobe epilepsy. Pharmacol. Biochem. Behav. 2014, 127, 27–36. [Google Scholar] [CrossRef]
- Gong, L.; Zhu, T.; Chen, C.; Xia, N.; Yao, Y.; Ding, J.; Xu, P.; Li, S.; Sun, Z.; Dong, X.; et al. Miconazole exerts disease-modifying effects during epilepsy by suppressing neuroinflammation via NF-kappaB pathway and iNOS production. Neurobiol. Dis. 2022, 172, 105823. [Google Scholar] [CrossRef]
- Shekh-Ahmad, T.; Eckel, R.; Dayalan Naidu, S.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 141, 1390–1403. [Google Scholar] [CrossRef]
- Brandt, C.; Glien, M.; Gastens, A.M.; Fedrowitz, M.; Bethmann, K.; Volk, H.A.; Potschka, H.; Loscher, W. Prophylactic treatment with levetiracetam after status epilepticus: Lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacology 2007, 53, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Casillas-Espinosa, P.M.; Anderson, A.; Harutyunyan, A.; Li, C.; Lee, J.; Braine, E.L.; Brady, R.D.; Sun, M.; Huang, C.; Barlow, C.K.; et al. Disease-modifying effects of sodium selenate in a model of drug-resistant, temporal lobe epilepsy. Elife 2023, 12, e78877. [Google Scholar] [CrossRef] [PubMed]
- Sandau, U.S.; Yahya, M.; Bigej, R.; Friedman, J.L.; Saleumvong, B.; Boison, D. Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice. Epilepsia 2019, 60, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Reschke, C.R.; Silva, L.F.A.; Norwood, B.A.; Senthilkumar, K.; Morris, G.; Sanz-Rodriguez, A.; Conroy, R.M.; Costard, L.; Neubert, V.; Bauer, S.; et al. Potent anti-seizure effects of locked nucleic acid antagomirs targeting miR-134 in multiple mouse and rat models of epilepsy. Mol. Ther. Nucleic Acids 2017, 6, 45–56. [Google Scholar] [CrossRef]
- Reschke, C.R.; Silva, L.F.A.; Vangoor, V.R.; Rosso, M.; David, B.; Cavanagh, B.L.; Connolly, N.M.C.; Brennan, G.P.; Sanz-Rodriguez, A.; Mooney, C.; et al. Systemic delivery of antagomirs during blood-brain barrier disruption is disease-modifying in experimental epilepsy. Mol. Ther. 2021, 29, 2041–2052. [Google Scholar] [CrossRef]
- Lentini, C.; d’Orange, M.; Marichal, N.; Trottmann, M.M.; Vignoles, R.; Foucault, L.; Verrier, C.; Massera, C.; Raineteau, O.; Conzelmann, K.K.; et al. Reprogramming reactive glia into interneurons reduces chronic seizure activity in a mouse model of mesial temporal lobe epilepsy. Cell Stem Cell 2021, 28, 2104–2121.e2110. [Google Scholar] [CrossRef]
- Bittencourt, S.; Ferrazoli, E.; Valente, M.F.; Romariz, S.; Janisset, N.; Macedo, C.E.; Antonio, B.B.; Barros, V.; Mundim, M.; Porcionatto, M.; et al. Modification of the natural progression of epileptogenesis by means of biperiden in the pilocarpine model of epilepsy. Epilepsy Res. 2017, 138, 88–97. [Google Scholar] [CrossRef]
- Brandt, C.; Nozadze, M.; Heuchert, N.; Rattka, M.; Loscher, W. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J. Neurosci. 2010, 30, 8602–8612. [Google Scholar] [CrossRef]
- Costard, L.S.; Neubert, V.; Veno, M.T.; Su, J.; Kjems, J.; Connolly, N.M.C.; Prehn, J.H.M.; Schratt, G.; Henshall, D.C.; Rosenow, F.; et al. Electrical stimulation of the ventral hippocampal commissure delays experimental epilepsy and is associated with altered microRNA expression. Brain Stimul. 2019, 12, 1390–1401. [Google Scholar] [CrossRef]
- Casalia, M.L.; Howard, M.A.; Baraban, S.C. Persistent seizure control in epileptic mice transplanted with gamma-aminobutyric acid progenitors. Ann. Neurol. 2017, 82, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.W.; Gupta, J.; Tagliatela, S.; Litvina, E.; Zheng, X.; Van Zandt, M.A.; Woods, N.; Grund, E.; Lin, D.; Royston, S.; et al. Long-term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J. Neurosci. 2014, 34, 13492–13504. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Broer, S.; Parekh, M.; Maury, Y.; Havlicek, S.; Kriks, S.; Fuentealba, L.; Lee, S.; Zhou, R.; Subramanyam, G.; et al. Human pallial MGE-type GABAergic interneuron cell therapy for chronic focal epilepsy. Cell Stem Cell 2023, 30, 1331–1350.e1311. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, L.; Yang, F.; Meng, X.D.; Wu, C.; Ma, H.; Jiang, W. Disease-modifying effects of RHC80267 and JZL184 in a pilocarpine mouse model of temporal lobe epilepsy. CNS Neurosci. Ther. 2014, 20, 905–915. [Google Scholar] [CrossRef]
- Meller, S.; Kaufer, C.; Gailus, B.; Brandt, C.; Loscher, W. Scopolamine prevents aberrant mossy fiber sprouting and facilitates remission of epilepsy after brain injury. Neurobiol. Dis. 2021, 158, 105446. [Google Scholar] [CrossRef]
- Pereira, H.A.; Benassi, S.K.; Mello, L.E. Plastic changes and disease-modifying effects of scopolamine in the pilocarpine model of epilepsy in rats. Epilepsia 2005, 46 (Suppl. S5), 118–124. [Google Scholar] [CrossRef]
- Gol, M.; Costa, A.M.; Biagini, G.; Lucchi, C. Seizure progression is slowed by enhancing neurosteroid availability in the brain of epileptic rats. Epilepsia 2024, 65, e41–e46. [Google Scholar] [CrossRef]
- Casillas-Espinosa, P.M.; Shultz, S.R.; Braine, E.L.; Jones, N.C.; Snutch, T.P.; Powell, K.L.; O’Brien, T.J. Disease-modifying effects of a novel T-type calcium channel antagonist, Z944, in a model of temporal lobe epilepsy. Prog. Neurobiol. 2019, 182, 101677. [Google Scholar] [CrossRef]
- Agostinho, A.S.; Mietzsch, M.; Zangrandi, L.; Kmiec, I.; Mutti, A.; Kraus, L.; Fidzinski, P.; Schneider, U.C.; Holtkamp, M.; Heilbronn, R.; et al. Dynorphin-based “release on demand” gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol. Med. 2019, 11, e9963. [Google Scholar] [CrossRef]
- Russmann, V.; Seeger, N.; Zellinger, C.; Hadamitzky, M.; Pankratova, S.; Wendt, H.; Bock, E.; Berezin, V.; Potschka, H. The CNTF-derived peptide mimetic Cintrofin attenuates spatial-learning deficits in a rat post-status epilepticus model. Neurosci. Lett. 2013, 556, 170–175. [Google Scholar] [CrossRef]
- Paolone, G.; Falcicchia, C.; Lovisari, F.; Kokaia, M.; Bell, W.J.; Fradet, T.; Barbieri, M.; Wahlberg, L.U.; Emerich, D.F.; Simonato, M. Long-term, targeted delivery of GDNF from encapsulated cells is neuroprotective and reduces seizures in the pilocarpine model of epilepsy. J. Neurosci. 2019, 39, 2144–2156. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of Soticlestat as an Add-on Therapy in Children and Young Adults With Dravet Syndrome. 2021. Available online: https://clinicaltrials.gov/study/NCT04940624 (accessed on 30 January 2025).
- Hawkins, N.A.; Jurado, M.; Thaxton, T.T.; Duarte, S.E.; Barse, L.; Tatsukawa, T.; Yamakawa, K.; Nishi, T.; Kondo, S.; Miyamoto, M.; et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia 2021, 62, 2845–2857. [Google Scholar] [CrossRef]
- de Tisi, J.; Bell, G.S.; Peacock, J.L.; McEvoy, A.W.; Harkness, W.F.; Sander, J.W.; Duncan, J.S. The long-term outcome of adult epilepsy surgery, patterns of seizure remission, and relapse: A cohort study. Lancet 2011, 378, 1388–1395. [Google Scholar] [CrossRef]
- Bell, G.S.; de Tisi, J.; Gonzalez-Fraile, J.C.; Peacock, J.L.; McEvoy, A.W.; Harkness, W.F.J.; Foong, J.; Pope, R.A.; Diehl, B.; Sander, J.W.; et al. Factors affecting seizure outcome after epilepsy surgery: An observational series. J. Neurol. Neurosurg. Psychiatry 2017, 88, 933–940. [Google Scholar] [CrossRef]
- Alcala-Zermeno, J.L.; Romozzi, M.; Sperling, M.R. The effect of epilepsy surgery on tonic-clonic seizures. Epilepsia 2025, 66, 1048–1058. [Google Scholar] [CrossRef]
- Sperling, M.R.; Barshow, S.; Nei, M.; Asadi-Pooya, A.A. A reappraisal of mortality after epilepsy surgery. Neurology 2016, 86, 1938–1944. [Google Scholar] [CrossRef]
- Grayson, L.E.; Peters, J.M.; McPherson, T.; Krueger, D.A.; Sahin, M.; Wu, J.Y.; Northrup, H.A.; Porter, B.; Cutter, G.R.; O’Kelley, S.E.; et al. Pilot study of neurodevelopmental impact of early epilepsy surgery in tuberous sclerosis complex. Pediatr. Neurol. 2020, 109, 39–46. [Google Scholar] [CrossRef]
- Khambhati, A.N.; Shafi, A.; Rao, V.R.; Chang, E.F. Long-term brain network reorganization predicts responsive neurostimulation outcomes for focal epilepsy. Sci. Transl. Med. 2021, 13, eabf6588. [Google Scholar] [CrossRef] [PubMed]
- Kokkinos, V.; Sisterson, N.D.; Wozny, T.A.; Richardson, R.M. Association of closed-loop brain stimulation neurophysiological features with seizure control among patients with focal epilepsy. JAMA Neurol. 2019, 76, 800–808. [Google Scholar] [CrossRef]
- Touma, L.; Dansereau, B.; Chan, A.Y.; Jette, N.; Kwon, C.S.; Braun, K.P.J.; Friedman, D.; Jehi, L.; Rolston, J.D.; Vadera, S.; et al. Neurostimulation in people with drug-resistant epilepsy: Systematic review and meta-analysis from the ILAE Surgical Therapies Commission. Epilepsia 2022, 63, 1314–1329. [Google Scholar] [CrossRef]
- Devinsky, O.; Friedman, D.; Duckrow, R.B.; Fountain, N.B.; Gwinn, R.P.; Leiphart, J.W.; Murro, A.M.; Van Ness, P.C. Sudden unexpected death in epilepsy in patients treated with brain-responsive neurostimulation. Epilepsia 2018, 59, 555–561. [Google Scholar] [CrossRef]
- Nair, D.R.; Laxer, K.D.; Weber, P.B.; Murro, A.M.; Park, Y.D.; Barkley, G.L.; Smith, B.J.; Gwinn, R.P.; Doherty, M.J.; Noe, K.H.; et al. Nine-year prospective efficacy and safety of brain-responsive neurostimulation for focal epilepsy. Neurology 2020, 95, e1244–e1256. [Google Scholar] [CrossRef]
- Bergey, G.K.; Morrell, M.J.; Mizrahi, E.M.; Goldman, A.; King-Stephens, D.; Nair, D.; Srinivasan, S.; Jobst, B.; Gross, R.E.; Shields, D.C.; et al. Long-term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology 2015, 84, 810–817. [Google Scholar] [CrossRef]
- Heck, C.N.; King-Stephens, D.; Massey, A.D.; Nair, D.R.; Jobst, B.C.; Barkley, G.L.; Salanova, V.; Cole, A.J.; Smith, M.C.; Gwinn, R.P.; et al. Two-year seizure reduction in adults with medically intractable partial onset epilepsy treated with responsive neurostimulation: Final results of the RNS System pivotal trial. Epilepsia 2014, 55, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Salanova, V.; Witt, T.; Worth, R.; Henry, T.; Gross, R.; Oommen, K.; Osorio, I.; Nazzaro, J.; Labar, D.; et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 2010, 51, 899–908. [Google Scholar] [CrossRef]
- Salanova, V.; Witt, T.; Worth, R.; Henry, T.R.; Gross, R.E.; Nazzaro, J.M.; Labar, D.; Sperling, M.R.; Sharan, A.; Sandok, E.; et al. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology 2015, 84, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Salanova, V.; Sperling, M.R.; Gross, R.E.; Irwin, C.P.; Vollhaber, J.A.; Giftakis, J.E.; Fisher, R.S.; SANTE Study Group. The SANTE study at 10 years of follow-up: Effectiveness, safety, and sudden unexpected death in epilepsy. Epilepsia 2021, 62, 1306–1317. [Google Scholar] [CrossRef]
- Skrehot, H.C.; Englot, D.J.; Haneef, Z. Neuro-stimulation in focal epilepsy: A systematic review and meta-analysis. Epilepsy Behav. 2023, 142, 109182. [Google Scholar] [CrossRef]
- Krauss, G.L.; Klein, P.; Brandt, C.; Lee, S.K.; Milanov, I.; Milovanovic, M.; Steinhoff, B.J.; Kamin, M. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: A multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020, 19, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.R.; Abou-Khalil, B.; Aboumatar, S.; Bhatia, P.; Biton, V.; Klein, P.; Krauss, G.L.; Vossler, D.G.; Wechsler, R.; Ferrari, L.; et al. Efficacy of cenobamate for uncontrolled focal seizures: Post hoc analysis of a phase 3, multicenter, open-label study. Epilepsia 2021, 62, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.D.; Mazurkiewicz-Beldzinska, M.; Chin, R.F.; Gil-Nagel, A.; Gunning, B.; Halford, J.J.; Mitchell, W.; Scott Perry, M.; Thiele, E.A.; Weinstock, A.; et al. Long-term safety and efficacy of add-on cannabidiol in patients with Lennox-Gastaut syndrome: Results of a long-term open-label extension trial. Epilepsia 2021, 62, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Halford, J.J.; Miller, I.; Nabbout, R.; Sanchez-Carpintero, R.; Shiloh-Malawsky, Y.; Wong, M.; Zolnowska, M.; Checketts, D.; Dunayevich, E.; et al. Add-on cannabidiol in patients with Dravet syndrome: Results of a long-term open-label extension trial. Epilepsia 2021, 62, 2505–2517. [Google Scholar] [CrossRef]
- Cusmai, R.; Moavero, R.; Bombardieri, R.; Vigevano, F.; Curatolo, P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. 2011, 22, 735–739. [Google Scholar] [CrossRef]
- Jozwiak, S.; Kotulska, K.; Domanska-Pakiela, D.; Lojszczyk, B.; Syczewska, M.; Chmielewski, D.; Dunin-Wasowicz, D.; Kmiec, T.; Szymkiewicz-Dangel, J.; Kornacka, M.; et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2011, 15, 424–431. [Google Scholar] [CrossRef]
- Kotulska, K.; Kwiatkowski, D.J.; Curatolo, P.; Weschke, B.; Riney, K.; Jansen, F.; Feucht, M.; Krsek, P.; Nabbout, R.; Jansen, A.C.; et al. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann. Neurol. 2021, 89, 304–314. [Google Scholar] [CrossRef]
- Bebin, E.M.; Peters, J.M.; Porter, B.E.; McPherson, T.O.; O’Kelley, S.; Sahin, M.; Taub, K.S.; Rajaraman, R.; Randle, S.C.; McClintock, W.M.; et al. Early treatment with vigabatrin does not decrease focal seizures or improve cognition in tuberous sclerosis complex: The PREVeNT trial. Ann. Neurol. 2023, 95, 15–26. [Google Scholar] [CrossRef]
- Misra, S.N.; Sperling, M.R.; Rao, V.R.; Peters, J.M.; Davis, C.; Carrazana, E.; Rabinowicz, A.L. Significant improvements in SEIzure interVAL (time between seizure clusters) across time in patients treated with diazepam nasal spray as intermittent rescue therapy for seizure clusters. Epilepsia 2022, 63, 2684–2693. [Google Scholar] [CrossRef]
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N.; et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 2153–2163. [Google Scholar] [CrossRef]
- Peters, J.M.; Prohl, A.; Kapur, K.; Nath, A.; Scherrer, B.; Clancy, S.; Prabhu, S.P.; Sahin, M.; Franz, D.N.; Warfield, S.K.; et al. Longitudinal effects of everolimus on white matter diffusion in tuberous sclerosis complex. Pediatr. Neurol. 2019, 90, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Schiller, K.; Thomas, J.; Avigdor, T.; Mansilla, D.; Kortas, A.; Unterholzner, G.; Rauchenzauner, M.; Frauscher, B. Pulsatile corticoid therapy reduces interictal epileptic activity burden in children with genetic drug-resistant epilepsy. Epilepsia Open 2024, 9, 1265–1276. [Google Scholar] [CrossRef]
- SK Life Science, Inc. XCOPRI (Cenobamate Tablets); Full Prescribing Information; SK Life Science, Inc.: Paramus, NJ, USA, 2022. [Google Scholar]
- Klein, P.; Aboumatar, S.; Brandt, C.; Dong, F.; Krauss, G.L.; Mizne, S.; Sanchez-Alvarez, J.C.; Steinhoff, B.J.; Villanueva, V. Long-term efficacy and safety from an open-label extension of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology 2022, 99, e989–e998. [Google Scholar] [CrossRef]
- Gilmartin, C.G.S.; Dowd, Z.; Parker, A.P.J.; Harijan, P. Interaction of cannabidiol with other antiseizure medications: A narrative review. Seizure 2021, 86, 189–196. [Google Scholar] [CrossRef]
- Ben-Menachem, E. Mechanism of action of vigabatrin: Correcting misperceptions. Acta Neurol. Scand. Suppl. 2011, 124, 5–15. [Google Scholar] [CrossRef]
- Becker, D.A.; Wheless, J.W.; Sirven, J.; Tatum, W.O.; Rabinowicz, A.L.; Carrazana, E. Treatment of seizure clusters in epilepsy: A narrative review on rescue therapies. Neurol. Ther. 2023, 12, 1439–1455. [Google Scholar] [CrossRef]
- Gidal, B.; Detyniecki, K. Rescue therapies for seizure clusters: Pharmacology and target of treatments. Epilepsia 2022, 63 (Suppl. S1), S34–S44. [Google Scholar] [CrossRef]
- Wheless, J.W.; Miller, I.; Hogan, R.E.; Dlugos, D.; Biton, V.; Cascino, G.D.; Sperling, M.R.; Liow, K.; Vazquez, B.; Segal, E.B.; et al. Final results from a phase 3, long-term, open-label, repeat-dose safety study of diazepam nasal spray for seizure clusters in patients with epilepsy. Epilepsia 2021, 62, 2485–2495. [Google Scholar] [CrossRef]
- Segal, E.B.; Wheless, J.W.; Zafar, M.; Shih, E.K.; Ngo, L.Y.; Carrazana, E.; Rabinowicz, A.L.; DIAZ Study Group. Pharmacokinetics and 180-day safety of diazepam nasal spray in pediatric patients with epilepsy aged 2-5 years. Epilepsia 2025. [Google Scholar] [CrossRef]
- Wheless, J.W.; Hogan, R.E.; Davis, C.S.; Carrazana, E.; Rabinowicz, A.L. Safety and effectiveness of diazepam nasal spray in male and female patients: Post hoc analysis of data from a phase 3 safety study. Epilepsia Open 2024, 9, 793–799. [Google Scholar] [CrossRef]
- Misra, S.N.; Sperling, M.R.; Rao, V.R.; Peters, J.M.; Penovich, P.; Wheless, J.; Hogan, R.E.; Davis, C.S.; Carrazana, E.; Rabinowicz, A.L. Analyses of patients who self-administered diazepam nasal spray for acute treatment of seizure clusters. Epilepsy Behav Rep 2024, 25, 100644. [Google Scholar] [CrossRef]
- Jarrar, R.; Stern, J.M.; Becker, D.A.; Davis, C.; Rabinowicz, A.L.; Carrazana, E. Treatment of prolonged seizure with diazepam nasal spray: A post hoc cohort analysis. In Proceedings of the American Epilepsy Society 2024 Annual Meeting, Los Angeles, CA, USA, 5–9 December 2024. [Google Scholar]
- Guignet, M.; White, H.S.; Misra, S.N.; Carrazana, E.; Rabinowicz, A.L. Development of a novel dosing paradigm to model diazepam rescue therapy in preclinical seizure and epilepsy models. Epilepsia Open 2024, 9, 1575–1581. [Google Scholar] [CrossRef]
- Baumer, F.M.; Peters, J.M.; Clancy, S.; Prohl, A.K.; Prabhu, S.P.; Scherrer, B.; Jansen, F.E.; Braun, K.P.J.; Sahin, M.; Stamm, A.; et al. Corpus callosum white matter diffusivity reflects cumulative neurological comorbidity in tuberous sclerosis complex. Cereb. Cortex 2018, 28, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S.; Bumanglag, A.V. Defining “epileptogenesis” and identifying “antiepileptogenic targets” in animal models of acquired temporal lobe epilepsy is not as simple as it might seem. Neuropharmacology 2013, 69, 3–15. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A First-in-Human (FIH) Study of Inhibitory Interneurons (NRTX- 1001) in Drug-Resistant Unilateral Mesial Temporal Lobe Epilepsy (MTLE). 2022. Available online: https://www.clinicaltrials.gov/study/NCT05135091 (accessed on 9 January 2025).
- Hixson, J.; Blum, D.; Banik, G.; Bershteyn, M.; Bulfone, A.; Feld, B.; Finefrock, H.; Fuenteabla, L.; Jung, J.H.; Kowal, T.; et al. First-in-human trial of NRTX-1001 gabaergic interneuron cell therapy for drug-resistant focal epilepsy—Updated Clinical Trial Results (P8-9.003). Neurology 2025, 104. [Google Scholar] [CrossRef]
- ClinicalTrials. WAYFINDER: A Clinical Study to Evaluate the Safety and Efficacy of ETX101, an AAV9-Delivered Gene Therapy in Children with SCN1A-Positive Dravet Syndrome. 2024. Available online: https://clinicaltrials.gov/study/NCT06112275 (accessed on 10 January 2025).
- ClinicalTrials. ENDEAVOR: A Clinical Study to Evaluate the Safety and Efficacy of ETX101, an AAV9-Delivered Gene Therapy in Infants and Children with SCN1A-Positive Dravet Syndrome. 2024. Available online: https://clinicaltrials.gov/study/NCT05419492 (accessed on 10 January 2025).
- ClinicalTrials. EXPEDITION: A Clinical Study to Evaluate the Safety and Efficacy of ETX101, an AAV9-Delivered Gene Therapy in Children with SCN1A-Positive Dravet Syndrome. 2024. Available online: https://clinicaltrials.gov/study/NCT06283212 (accessed on 10 January 2025).
- ClinicalTrials. A Multi-Center, Phase 1/2a, First-in-Human (Fih) Study Investigating the Safety, Tolerability, and Efficacy of Amt-260 in Adults with Unilateral Refractory Mesial Temporal Lobe Epilepsy (Mtle) Administered via Magnetic Resonance Imaging (MRI)-Guided Convection-Enhanced De-Livery (Ced). 2024. Available online: https://clinicaltrials.gov/study/NCT06063850 (accessed on 10 January 2025).
- Bialer, M.; Johannessen, S.I.; Koepp, M.J.; Levy, R.H.; Perucca, E.; Perucca, P.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Sixteenth EILAT Conference on New Antiepileptic Drugs and Devices (EILAT XVI): II. Drugs in more advanced clinical development. Epilepsia 2022, 63, 2883–2910. [Google Scholar] [CrossRef]
- Parkerson, K.A.; Laux, L.; Cross, J.H.; Brunklaus, A.; Desurkar, A.; Schreiber, J.; Sullivan, J.; Phillips, S.; Lallas, M.; Devinsky, O.; et al. MONARCH and ADMIRAL interim analyses: Ongoing, open-label, phase 1/2a studies in US and UK investigating safety and drug exposure of STK-001, an antisense oligonucleotide (ASO), in children and adolescents with dravet syndrome (DS). Neurotherapeutics 2023, 20, 1244. [Google Scholar]
- Stoke Therapeutics. Stoke Therapeutics Announces Landmark New Data That Support the Potential for Stk-001 to Be the First Disease-Modifying Medicine for the Treatment of Patients with Dravet Syndrome. 2024. Available online: https://investor.stoketherapeutics.com/news-releases/news-release-details/stoke-therapeutics-announces-landmark-new-data-support-potential (accessed on 17 May 2025).
- ClinicalTrials.gov. A Double-Blind Study Evaluating the Efficacy, Safety, and Tolerability of Zorevunersen in Patients with Dravet Syndrome. 2025. Available online: https://clinicaltrials.gov/study/NCT06872125 (accessed on 17 May 2025).
- Loscher, W. Mammalian models of status epilepticus—Their value and limitations. Epilepsy Behav. 2024, 158, 109923. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).