Abstract
One major challenge in cellular neuroscience is to elucidate how the accurate alignment of presynaptic release sites with postsynaptic densely clustered ligand-gated ion channels at chemical synapses is achieved upon synapse assembly. The clustering of neurotransmitter receptors at postsynaptic sites is a key moment of synaptogenesis and determinant for effective synaptic transmission. The number of the ionotropic neurotransmitter receptors at these postsynaptic sites of both excitatory and inhibitory synapses is variable and is regulated by different mechanisms, thus allowing the modulation of synaptic strength, which is essential to tune neuronal network activity. Several well-regulated processes seem to be involved, including lateral diffusion within the plasma membrane and local anchoring as well as receptor endocytosis and recycling. The molecular mechanisms implicated are numerous and were reviewed recently in great detail. The role of pre-synaptically released neurotransmitters within the complex regulatory apparatus organizing the postsynaptic site underneath presynaptic terminals is not completely understood, even less for inhibitory synapses. In this mini review article, we focus on this aspect of synapse formation, summarizing and contrasting findings on the functional role of the neurotransmitters glycine and γ-aminobutyric acid (GABA) for initiation of postsynaptic receptor clustering and regulation of Cl− channel receptor numbers at inhibitory synapses gathered over the last two decades.
1. Introduction
One important goal of cellular neuroscience is to disclose the cellular and molecular mechanisms involved in regulating the assembly of chemical synapses, ensuring rapid synaptic transmission. Most synapses are established during development following genetically determined intrinsic mechanisms, but synapses continue to be formed and modulated in strength in the mature nervous system throughout life. Adjustments in the strength of neuronal connections occur in part postsynaptically by changes in the number of receptors at the postsynaptic sites [1,2,3]. The number of receptors that cluster postsynaptically depends first on lateral diffusion events from the extrasynaptic plasma membrane, where the membrane insertion of receptors carrying intracellular vesicle pools might occur (Figure 1). Receptor subunits are initially translated, folded, and assembled into complete receptor complexes within the rough endoplasmic reticulum (RER). Interestingly, in addition to the somatic region, a local de novo synthesis of γ-aminobutyric acid A-receptors (GABAARs) and even of gephyrin, a main scaffold protein at the inhibitory postsynapse, was also demonstrated in dendrites as driven by a reduced expression of two miRNAs and regulated by N-methyl-D-aspartate receptor (NMDAR) signaling [4,5]. The delivery of receptor-containing vesicles to the plasma membrane results from active cytoskeletal transport after segregation from the trans-Golgi compartment or previous endocytosis from the extrasynaptic plasma membrane. Indeed, a part of extrasynaptically localized membrane neurotransmitter receptors are internalized with the potential for recycling or lysosomal degradation [6,7]. Accordingly, a number of highly regulated processes implicating multiple intracellular signaling cascades are likely to affect receptor localization, stabilization, and maintenance at the synapse [8,9]. However, the specific mechanisms that target receptors from intracellular or extrasynaptic compartments and keep them at synapses are still not clear and even less well understood for inhibitory synapses compared to glutamatergic synapses. Cell adhesion molecules in cooperation with synaptic scaffold proteins were proposed to play a key role in facilitating the accumulation of surface neurotransmitter receptors at postsynaptic sites and modulating receptor dynamics and were reviewed recently in great detail [9,10]. What is the contribution of neurotransmitter signals to the organization of the appropriate neurotransmitter receptor clusters at the synapse? Searching for answers to this seemingly simple question, here we proposed to review studies that have provided insights into the part ligand activation itself accomplishes in regulating the formation and maintenance of postsynaptic glycine receptor (GlyR) and GABAAR clusters at inhibitory synapses.
The neurotransmitters glycine and GABA mediate fast inhibitory neurotransmission in the mature nervous system through activation of GlyRs and GABAARs, respectively, which both belong to the superfamily of heteropentameric ligand-gated ion channels [11]. Under normal physiological conditions, activation of GlyRs and GABAARs by ligand binding opens a chloride-permeable ion channel, allowing chloride ions (Cl−) to enter the neuron, resulting in membrane hyperpolarization and neuronal inhibition. However, in immature neurons, due to the relatively high intracellular Cl− concentration, activation of GlyRs and GABAARs leads to chloride efflux and consequently to a depolarization of the plasma membrane. Therefore, GlyRs and GABAARs are mostly excitatory during development and, through the activation of voltage-gated Ca2+ channels, orchestrate numerous Ca2+ -dependent processes [12,13,14] that may underlie some type and developmental stage dependent differences in receptor clustering properties. Ca2+ as a second messenger influences the activation of kinases like Ca2+/calmodulin-dependent protein kinase type II (CaMKII) and phosphatases like calcineurin, which in turn, by modifications of receptors and other proteins, affect receptor trafficking and other intracellular signaling pathways that ultimately lead to changes in gene expression and protein synthesis. Altogether these will shape postsynaptic receptor cluster formation and contribute to inhibitory synaptic plasticity [8,15].

Figure 1.
Overview of GABAAR and GlyR trafficking and putative roles of ligand binding for postsynaptic receptor clustering (modified from [16]). Vesicles carrying pentameric GlyRs or GABAAR are transported after segregation from the trans-Golgi compartment to the plasma membrane. After fusion of receptor-carrying vesicles with the plasma membrane, receptors diffuse in the plane of the membrane and might be trapped by the gephyrin scaffold beneath the postsynaptic membrane specialization. Here, several receptors can aggregate together, forming clusters. Receptors can also diffuse out from the postsynaptic membrane and might be removed from the cell surface by endocytosis. From this state they can be exocytosed again or might fuse with the early/late endosome for lysosomal degradation. Both neurotransmitters are able to induce the formation of new GlyR or GABAAR clusters; however, neurotransmitter binding seems to be essential only for GlyR cluster formation but not for the initial postsynaptic clustering of GABAARs.
Figure 1.
Overview of GABAAR and GlyR trafficking and putative roles of ligand binding for postsynaptic receptor clustering (modified from [16]). Vesicles carrying pentameric GlyRs or GABAAR are transported after segregation from the trans-Golgi compartment to the plasma membrane. After fusion of receptor-carrying vesicles with the plasma membrane, receptors diffuse in the plane of the membrane and might be trapped by the gephyrin scaffold beneath the postsynaptic membrane specialization. Here, several receptors can aggregate together, forming clusters. Receptors can also diffuse out from the postsynaptic membrane and might be removed from the cell surface by endocytosis. From this state they can be exocytosed again or might fuse with the early/late endosome for lysosomal degradation. Both neurotransmitters are able to induce the formation of new GlyR or GABAAR clusters; however, neurotransmitter binding seems to be essential only for GlyR cluster formation but not for the initial postsynaptic clustering of GABAARs.

In this manuscript we review in a time-course manner publications and discuss knowledge converged over the last 25 years with special focus on the role glycine and GABA signals might have in both the immature state and mature nervous system on the two main aspects of postsynaptic receptor cluster formation: First, the initiation of the de novo formation of receptor clusters, thus related to the number of postsynaptic clusters in a given dendritic or somatic region, and second, the increase and decrease in cluster size as one structural correlate of synaptic plasticity. In this context, the clustering of gephyrin, thought to be the main postsynaptic scaffold and organizer of inhibitory synapses [17,18], will also be discussed.
2. Postsynaptic Clustering of the Strychnine-Sensitive GlyRs in Early Developmental Stages In Vitro
GlyR cluster formation at postsynaptic membranes in relation to neurotransmitter-dependent receptor activation was first analyzed in 1998 by two independent studies [19,20]. Both studies used the same experimental system of activity blockade by strychnine of cultured rat spinal cord neurons, which recapitulate the developmental sequence of GlyR expression. Strychnine, an alkaloid extracted from the Indian tree Strychnox nux vomica, is a potent competitive GlyR antagonist that blocks the binding of glycine to its Cl− channel receptor, particularly within the spinal cord. Electron cryo-microscopy studies have provided insights into how strychnine binding to the GlyR promotes receptor domain rearrangement leading to rotation of the transmembrane domain toward the pore axis, occluding the ion conduction route [21,22]. The high affinity and specificity of strychnine for GlyRs make strychnine an ideal antagonist ligand to study the dynamics of GlyR topography in response to activity in cell culture or in vivo conditions. The model primarily targets the α1 GlyRs; thus, the findings might not accurately reflect the roles of other α GlyR subtypes with more restricted expression patterns. Furthermore, prolonged strychnine exposure can lead to changes in GlyR mRNA and protein levels, affecting the overall expression levels of GlyRs, while at higher concentrations its activity extends beyond GlyRs, all representing limitations that have to be considered when interpreting results [23,24]. In the work of Levi et al., chronic inhibition of the GlyR with increasing strychnine concentrations for 7 or 11 days reduced in a group of strychnine-sensitive neurons the number of synaptic GlyR clusters, visualized as mAb4a (known to recognize all GlyR α subunits) immunoreactivity, in a dose-dependent manner. In addition, the size of the few GlyR clusters found on the surface of strychnine-responsive neurons was also significantly smaller than those seen on the membrane of untreated neurons, suggesting that GlyR clusters were first destabilized [19]. The second study reported similar results in the sense that strychnine treatment for 8 days resulted in a significantly lower number of mAb4a co-localizations with presynaptic terminals, indicating postsynaptic GlyR clusters [20] (Figure 2). Interestingly, in this study, a very similar effect could be measured upon inhibition of voltage-dependent Ca2+ channels (verapamil and nifedipine), suggesting that the depolarizing effect of the GlyR activation in immature neurons causing the opening of L-type Ca2+ channels with subsequent Ca2+ influx is needed for GlyR clustering at developing postsynaptic sites. The downstream signaling mechanisms leading to the receptor cluster formation were not further characterized.
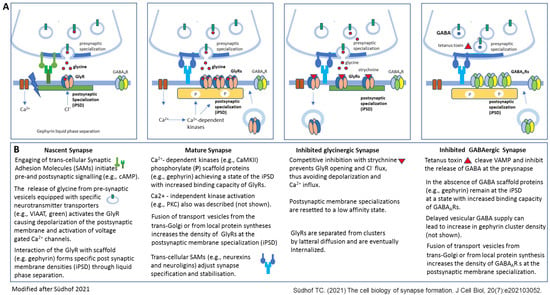
Figure 2.
Different stages of inhibitory synapse formation and effects of experimental activity blockade with focus on the postsynaptic membrane specializations (modified after [8]). Synapses initiate as nascent contacts that mature into plastic synaptic connections. (A) Schematic view of a nascent glycinergic synapse (first), a mature glycinergic synapse (second), a glycinergic synapse (third), and GABAergic synapse after experimental activity blockade (fourth). In nascent synapses Synaptic Adhesion Molecules (SAMs) mediate initial transneuronal interactions. For example, latrophilins, specifically latrophilin-1 at inhibitory synapses, are thought to initiate intracellular signaling cascades that organize inhibitory synaptic specializations. Binding of the postsynaptic neuroligins (specifically NLG2 at inhibitory synapses) to its presynaptic partners, neurexins, is proposed to shape subsequent synapse maturation and properties (second). Inhibition of synapses with the high-affinity competitive antagonist strychnine results in altered GlyR clustering due to elimination of GlyRs from the postsynaptic membrane specialization (third). Inhibition of GABA release due to VAMP cleavage through tetanus toxin does not affect GABAAR clustering at the postsynaptic membrane specialization. (B) Short description of the events presented in A. Further details are provided in the manuscript text. NLG—neurologin 2, VAMP—vesicle-associated membrane protein.
In both studies, in the subgroup of strychnine-responsive neurons, the reduced number of post-synaptic GlyR clusters was correlated with the intracytoplasmic accumulation of larger GlyR-containing structures next to the nucleus, which were not colocalized with either cis-Golgi apparatus or RER, arguing against neosynthesized GlyRs [19]. Based on these observations, the authors hypothesized that strychnine-mediated blocking of postsynaptic GlyRs ultimately led to receptor removal from the dendritic and somatic membrane surface via endocytosis, which was not compensated by new GlyR incorporation and lateral diffusion into the postsynaptic receptor cluster [19,20]. However, this hypothesis was refuted a few years later by the same group from the Triller lab. Evaluating GlyR clustering upon short-term exposure of neurons to strychnine (5 h, 1 h), they observed that the perinuclear accumulation of immunoreactive GlyRs precedes the disappearance of the receptors from postsynaptic sites [25]. By labeling the lumen or membrane of the endocytic compartments, they could show that the strychnine-induced redistribution and accumulation of GlyR is not the result of the endocytosis of plasma membrane receptor molecules but rather affects the activity blockade by strychnine, the receptors postsynaptic anchoring, and/or the reloading of postsynaptic clusters due to altered protein exocytic trafficking [25]. Anyway, these studies assign transmitter-induced activity a pivotal role in GlyR clustering at early stages of development but less for the maintenance of differentiated postsynaptic sites since when strychnine was added after 8 Div, when GlyR clusters had already formed, the postsynaptic localization of GlyR remained the same for several days [20]. Furthermore, it has to be emphasized that these conclusions were restricted to a subpopulation of strychnine-responsive neurons within the spinal cord culture containing also neurons with many GlyR clusters even upon strychnine treatment. Besides, it has to be noticed that, in addition to the above-mentioned limitations of the chronic strychnine treatment model, the presence or absence of glycine in the cell culture medium or altered conditions of the energy metabolism might have also influenced the results of these experiments.
3. Postsynaptic Clustering of GlyRs in Early Developmental Stages In Vivo
More than 10 years after the above-described in vitro studies investigated Yamanaka et al., whether neurotransmitter signaling is required for the initial assembly of GlyRs throughout synapse formation in vivo, using chronic strychnine treatment during the early development of zebrafish [26]. Bath-applied strychnine to zebrafish embryos from 22 hpf (hours postfertilization), before GlyR clusters had formed, hindered almost completely the formation of postsynaptic GlyR clusters at Mauthner-cells (M-cells) surface, which are a single pair of reticulospinal neurons in the hindbrain, receiving many inhibitory inputs from glycinergic interneurons on their somatic and dendritic membrane. Importantly, and in concordance with the earlier in vitro observations [19,20], after strychnine removal, GlyR clusters appeared on the surface of M-cells and even increased in density over time. Furthermore, labeling GlyR clusters with mAb4a antibody after strychnine application for 24 h at later stages of zebrafish development, when GlyR clusters had already been formed, the previously assembled GlyR clusters disappeared from the surface of M-cells in the early larval stages (2–3 dpf) and were markedly diminished in late larvae and juveniles (10–11 dpf, 15–16 dpf, 30–31 dpf) (Figure 2). The authors proposed that the age-dependent decrease in the activity-dependent maintenance of GlyR clusters might be at least partly assigned to the reduction of intracellular Cl− concentration along neuron development, although this is questionable since in immature spinal cord neurons in vitro no effect of late strychnine application on receptor cluster maintenance was observed [20]. However, this study was the first and so far the only one demonstrating, by immunolabeling supported by electrophysiological recordings, that glycinergic transmission is indeed necessary for GlyR cluster formation in vivo and also for maintenance of previously formed GlyR clusters, at least until juvenile periods. Mechanistically this work confirmed the functional role of the GlyR activation in vivo causing depolarization and opening of L-type Ca2+ channels since the specific inhibition of these channels as well as of the calcium-/calmodulin-dependent protein kinase II (CaMKII) resulted in the same robust reduction of GlyR clusters as the strychnine treatment itself [26]. The very fast response to strychnine application, namely, eliminating GlyR clusters within one hour, strongly indicated as a cellular mechanism for the disappearance of postsynaptic GlyRs after strychnine treatment the increased lateral diffusion of receptors out of the postsynaptic membrane area due probably to altered anchoring at the gephyrin scaffold, known to be modulated by CaMKII-dependent phosphorylation [27]. CaMKIIα, the predominant CaMKII isoform in the hippocampus and neocortex, is activated by Ca2+ influx and subsequent calmodulin binding, leading to its autophosphorylation and sustained activity [15]. Activated CaMKII likely promotes gephyrin phosphorylation, which accelerates the GlyR cluster formation at the postsynaptic sites. Indeed, in a later study, it could be shown that the phosphorylation of gephyrin at Ser325 by CaMKIIα promotes the synaptic accumulation of GlyR on zebrafish M-cells through enhancement of the gephyrin-GlyR binding, increasing inhibitory glycinergic inputs [28].
Whether the long-term strychnine treatment effects might also involve alterations in the expression and/or intracellular transport of GlyRs was not clarified within this study [26].
4. Postsynaptic GlyR Clustering in Mature Neurons In Vitro
Similar conclusions were drawn by a study published four years later [29] analyzing the consequences of chronic GlyR inhibition on postsynaptic glycine receptor clustering in mature-state spinal cord neuron cultures (Div13–15), in which, as is well known, the receptor activation is more likely to cause hyperpolarization of the postsynaptic membrane than depolarization, as in the immature state. In this study, the chronic inhibition by strychnine for 14 days resulted in a decrease in the size of the postsynaptic GlyR clusters detected as GlyRα1 puncta apposed to vesicular inhibitory amino acid transporter (VIAAT) signals. Applying a combination of fluorescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) techniques, it was convincingly presented that the smaller clusters emanated rather from the increased diffusive property of the receptors in the plane of the membrane after inhibition than from a decline in exocytosis of new intracellular GlyRs into the plasma membrane. Furthermore, the phenomenon could be reversed since wash-out of strychnine and intrinsic activation of GlyRs by glycine for one hour led to enlargement of the synaptic GlyR clusters coincident with a decrease in receptors lateral diffusion at synaptic sites. Thus, these results evidenced the important role of glycinergic transmission for the formation of synaptic GlyR clusters also in mature state neurons. However, the experiments testing the effect of strychnine blocking on previously activated GlyRs in Div14 neurons indicated that GlyR activation is not essential for their maintenance at the synapse. Importantly, the alteration in cluster size was not dependent on CaMKII activity but was shown to be regulated by protein kinase C (PKC) in a Ca2+-independent manner. Cytosolic PKC activated by diacylglycerol or other lipid- or non-lipid molecules translocates to the plasma membrane [30,31], where it phosphorylates specific serine residues of the GlyR, changing channel properties and receptor trafficking as well as interactions between GlyRs and proteins like gephyrin [32]. In this context, it is of relevance that PKC phosphorylation at the Ser403 site was found to reduce the binding affinity between the GlyR and gephyrin, resulting in acceleration of the receptor’s diffusion in the plasma membrane and reduced GlyRs accumulation at synapses [33].
Further, it is worth mentioning that the in vitro strychnine inhibition experiment with mature state spinal cord neurons also revealed additional differences in comparison to the earlier findings in young spinal cord neurons. First of all, the strychnine treatment did not alter the number of either total or synaptic GlyR clusters in mature cultures. Additionally, site-specific application of glycine successfully induced the formation of new GlyR clusters in the mature-state spinal cord neurons, an experimental result that was reported in one of the first GlyR cluster studies from 1998 to be not effective in immature neurons [20].
In conclusion, a limited number of publications have shown until now that transmitter-dependent GlyR activation affects GlyR clustering both in the early membrane depolarization and later hyperpolarization states by regulating the initial formation and maintenance of GlyR clusters at the postsynaptic membrane site or GlyR receptor cluster size, respectively. Mechanistically, the activation might imply different kinds of phosphorylations through either CaMKII in early or PKC in later developmental stages, resulting in modulation of the lateral mobility of GlyRs, i.e., persistence of the GlyRs at the postsynaptic membrane specialization defined probably through the submembranous scaffold of gephyrin. Whether or not GlyR cluster formation can occur without any glycine binding is not known.
5. Gephyrin Cluster Formation at Glycinergic Synapses
In contrast to the obvious dependence of GlyR cluster formation and stability at postsynaptic membrane sites on glycinergic transmission described above, for the scaffold protein gephyrin a different scenario emerges. At postsynaptic sites, all GlyRs and the majority of GABAARs connect to gephyrin, which is thought to be the major scaffold protein of the postsynaptic density at inhibitory synapses, although meanwhile numerous additional proteins were identified to contribute to the proteome of inhibitory synapses [34,35]. Gephyrin has been demonstrated to regulate diffusion properties of GlyR and GABAAR receptors, thus influencing the size and density of postsynaptic receptor clusters [17,18]. The mechanisms underlying gephyrin-receptor interactions leading to receptor clustering are relatively well defined; in particular, the binding domains of both gephyrin and the intracellular linker region of the GlyRβ subunit could be determined through crystallography and in vitro binding assays at atomic resolution level [36].
However, how these interactions and the subcellular distribution of gephyrin are affected by receptor activation is not well understood. Structurally, gephyrin consists of an N-terminal trimer forming the G-domain, a C-terminal dimer forming the E-domain, and a connecting linker region. A number of studies clearly disclosed that these domains interact to create a hexagonal lattice that can anchor inhibitory receptors at postsynaptic sites and interact with various synaptic and cytoskeletal proteins (reviewed in 18). Later studies evidenced that this lattice can undergo phase separation, leading to the formation of condensed sheet-like structures termed inhibitory postsynaptic density (iPSD), particularly when gephyrin is bound to GlyRs or GABAARs, which can be further fine-tuned by different gephyrin-binding proteins such as neuroligin 2 (NLG2) [37,38]. Since the specific interaction of the dimeric E-domain of gephyrin with GlyRs or GABAAR multimers seems to be required for the iPSD formation, linker region phosphorylation at different residues or binding of other synaptic proteins regulates gephyrin-mediated GlyR and GABAAR clustering [37]. Phosphorylation of specific serine and threonine residues on gephyrin involving various kinases such as Glycogen Synthase Kinase 3 beta (GSK3β), Cyclin-dependent kinase 5 (CDK5), protein kinase A (PKA), and CaMKII can modulate its interaction with both receptors and other proteins involved in synapse formation and plasticity [28,39,40,41,42].
The postsynaptic accumulation of gephyrin, in comparison to GlyR clusters, was observed also upon chronic strychnine treatment in both immature (Div 7) [19] and mature spinal cord neurons (Div 14) [28]. In these studies, neither chronic strychnine application nor the washout of chronic strychnine with subsequent GlyR activation modified the subcellular distribution and the number of gephyrin clusters detected by the mAb7 antibody. In contrast, the study of Kirsch et al., reported that similarly to the GlyR clusters, gephyrin, revealed by the same monoclonal antibody 7a, “was no longer detected at postsynaptic sites of DIV8 spinal cord neurons after chronic strychnine treatment but was seen instead at sites of focal cell attachment”. Consistently, Ca2+-channel blockers affected the gephyrin distribution in the same way [20]. How these different observations might be explained is not totally clear. At a later time point it was shown that the binding of the monoclonal antibody mAb7a to gephyrin is dependent on the phosphorylation of gephyrin at Ser270 [42]. Since the same developmental stage spinal cord neurons were used in both of these studies, some differences in the cell culture system conditions (e.g., cell density, medium composition) might have resulted in different phosphorylation states of gephyrin at this site, causing discrepancies in the detection pattern of gephyrin clusters by the mAb7a antibody. Additionally, it is known that even within the same culture, different neurons can have varying levels of differentiation and maturity, leading to inconsistencies between experiments. Nevertheless, the latter data are in some way supported by the study of Maas, C et al., showing in hippocampal neurons that inhibition of the GlyRs reduced the anterograde transport of gephyrin-GlyR complexes on microtubule tracks into the dendrites, thus resulting in significantly decreased gephyrin signal number and fluorescence intensity in neurites and in a net accumulation of newly synthesized gephyrin in the cytoplasmic compartment [43]. The alteration in microtubule-dependent transport activity seen with the strychnine-mediated GlyR blockade could be mimicked by excitatory (AMPA receptor) activity. This suggests that the observed transport deficits were also in these cases probably indirectly caused by the GlyR activation, namely due to the depolarization of the postsynaptic membrane inducing Ca2+ influx by voltage-dependent Ca2+ channels characteristic of young stages of spinal cord and also hippocampal neurons. Unfortunately, in this study the subcellular distribution of GlyRs was not analyzed; thus, it remains unclear whether the large intracellular GlyR accumulations seen in the studies from the Kirsch and Triller laboratories [19,20] would have been confirmed, which would be of interest, as in the former studies with strychnine inhibition conditions, on the other hand, no large intracellular gephyrin clusters were observed.
In conclusion, a handful of studies analyzing the accumulation of gephyrin and its effect on GlyR clustering at the postsynaptic sites in correlation with glycine-dependent synaptic activity reported contrasting results in regard to gephyrin clustering. Further studies have to clarify whether or not and when and how glycine-dependent neurotransmission might affect gephyrin aggregation supporting GlyR clustering at glycinergic synapses. Anyhow, the findings of these studies indicate that synaptic activity could alter the distribution of GlyR receptors and eventually other postsynaptic signaling molecules without affecting that of gephyrin, suggesting that distinct regulatory mechanisms might be involved in controlling the behavior of the different postsynaptic site components.
6. Postsynaptic Clustering of the GABAA Receptors In Vitro and In Vivo
Two years after the two crucial studies showing that in cultured spinal cord neurons GlyR activation is needed for the formation and stabilization of postsynaptic GlyR clusters, a study published in the year 2000 using cultures of mature rat hippocampal neurons (21–28 Div) reported that GABA input is not necessary for clustering the GABAARs in pyramidal cells [44]. More exactly, the study revealed that isolated hippocampal pyramidal cells in microisland cultures thus deprived of GABA input formed GABAAR clusters, evidenced in this study by immunostaining for the β2/3 subunits of the GABAAR. Even more, the authors indicated that in the absence of input from GABAergic axons, these receptor clusters were localized opposite to glutamatergic presynaptic terminals, strongly indicating that the neurotransmitter itself does not represent the specificity-conferring signal for GABAAR clustering at the postsynaptic site. Shortly after, this statement was supported by two other studies in in vivo experimental conditions. In the first in vivo study, the analysis of Munc18-1 deficient mice revealed that even in the case of a complete loss of neurotransmitter secretion during development, including both evoked and spontaneous vesicular release, functional GABAAR clusters at postsynaptic sites are formed in the brain of these mice [45]. Munc18-1 is a presynaptic protein with multiple activities during soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-regulated exocytosis, including docking the neurotransmitter-transporting presynaptic vesicle, thus essential for membrane fusion and neurotransmitter release. Loss of Munc18-1 causes severe defects in neurotransmitter release [46]. GABA iontophoresis in Munc-18 mutant brain slices caused a normal postsynaptic response. The protein composition of the GABAergic postsynaptic sites was not addressed in this study. However, the severe neurodegeneration observed after the initial successful assembly of synaptic connectivity suggested that since the synapse formation might not require neurotransmitter secretion, it might be important for the maintenance of already established synaptic connections. In the second in vivo study three years later, the authors analyzed the formation of GABAergic neuromuscular junctions in Caenorhabditis elegans with the unc-25 mutation lacking any GABA synthesis [47]. Unc genes were originally identified in the nematode worm Caenorhabditis elegans and are a group of genes that, when mutated, result in uncoordinated (hence “UNC”) movement. Specifically, the unc-25 gene encodes glutamic acid decarboxylase (GAD), the biosynthetic enzyme for GABA [48]. By means of immunohistochemistry and GFP-tagged proteins, it turned out that unc-25 mutants formed GABAAR clusters at postsynaptic sites in the absence of GABA, and these clusters were indistinguishable from those of wild-type animals, including cluster size and synapse densities all along the time course evaluated. Shortly afterwards, the work of Harms and Craig [49] and then a more recent study [50] evidenced, just in line with the in vivo findings, that despite early block of any neurotransmitter release, GABAergic inhibitory synapses are also established in cultured hippocampal neurons (16–18 Div). In these experiments tetanus toxin was used to chronically cleave vesicle-associated membrane protein 2 (VAMP2), a central protein for SNARE complex assembly, vesicle fusion, and transmitter release. Tetanus toxin is a metalloprotease that specifically targets VAMP (also known as synaptobrevin), an essential component of the neurotransmitter release machinery. It inhibits particularly the release of GABA and glycine at inhibitory synapses, leading to increased excitability and development of spontaneous seizures, making the tetanus toxin model a useful tool for studying epilepsy [51,52]. This model also allows us to investigate the mechanisms of synaptic transmission, including the impacts of disrupted neurotransmitter release both in vivo and in vitro. However, it is important to be aware of the model’s limitations, including concentration-dependent effects, retrograde transport, and potential for off-target effects [53,54]. Immunolabeling for the GABAAR α2 and γ2 subunits evidenced receptor clustering opposite to GAD-positive terminals in tetanus toxin-treated as in control neurons. Neither GABAAR distribution nor size of GABAAR clusters was affected by inactivity [49] (Figure 2); however, when three-dimensional, structured illumination microscopy (3D-SIM) was used to analyze the nanoscale architecture of the inhibitory postsynaptic regions, the summed GABAAR (γ2) volume at the subsynaptic domains per synapse was significantly decreased at chronically silenced synapses, probably due to discrete decreases in both the number/synapse and individual volume of GABAAR subsynaptic domains [55]. This could be of importance since in the last years super-resolution microscopy methods like 3D-SIM and dSTORM could evidence the nanoarchitecture of synapses in which neurotransmitter receptors, scaffold proteins, and adhesion molecules cluster into nanoscale subsynaptic domains (SSD) [56,57]. Recent cryo-electron tomography (cryo-ET) studies could confirm the precise nanoscale organization of proteins in SSDs [58] that seems to be crucial for efficient and regulated synaptic transmission. Changes in the nanoscale organization of synapses, including GABAARs, such as those induced by activity, might enable dynamic regulation of synaptic function [57,59].
Although in the experimental models established to inhibit presynaptic neurotransmitter secretion and used in the above-mentioned studies, a release of residual GABA or other factors that might activate GABAA receptors cannot be totally excluded, altogether these studies disclaim the requirement of local GABAA receptor activation for the postsynaptic clustering of GABAA receptors, as was the case for glycine receptors. This assigns a different role for the ligand binding with respect to the assembly of postsynaptic sites at GABAergic than at glycinergic synapses. In this context, of relevance is the information provided by the study of Levi et al., comprising a comparative analysis of receptor movement by single-molecule tracking technique in cultured spinal cord neurons (Div 10–13) combined with functional electrophysiological analysis and showing that GlyR but not GABAAR movement in and out of the postsynaptic membrane specialization is regulated by neuronal activity [60]. The main finding of this study was that GlyRs and GABAARs differ in the regulation of their diffusion properties by activity, the activity-dependent modulation of GlyR diffusion properties leading to changes in GlyR numbers at synapses, but without affecting gephyrin cluster immunoreactivities. Moreover, this study also demonstrated a specific NMDAR-dependent calcium-mediated regulation of GlyRs but not of GABAARs lateral diffusion, synaptic density, and mIPSC amplitude. This might represent a possible rapid homeostatic regulation mechanism at the inhibitory glycinergic or mixed glycine-GABAergic synapses to increased excitation [60]. In addition, specific phosphorylation of GABAAR subunits was indicated as another molecular mechanism that can govern receptors synaptic targeting, including binding to gephyrin [14,61]. Namely, cAMP-dependent PKA-mediated phosphorylation of the α2 subunit at Ser359 was identified to regulate scaffold binding and GABAAR cluster density at inhibitory synapses, thereby regulating the strength of inhibition in rat primary and cortical cell cultures and mouse brain slices [62].
7. Gephyrin Clustering at GABAergic Synapses
Unfortunately, only a few studies analyzed both GABAAR and gephyrin clustering under similar conditions of altered neurotransmitter-dependent synaptic activity. The early study of Rao et al. reported that in the absence of GABA input, not only GABAAR clusters but also gephyrin clusters are formed [44]. In isolated mature hippocampal pyramidal neurons, both GABAAR and gephyrin clusters were found opposed to presynaptic synaptophysin puncta in the absence of GABAergic innervation, although a detailed qualitative and quantitative analysis for gephyrin clusters was not carried out in this study. In tetanus toxin-treated hippocampal neurons, gephyrin clusters were also successfully recruited at postsynaptic sites, where they co-clustered with α2 and γ2 GABAAR subunits. No changes in gephyrin cluster density were noticed in the absence of GABA release [49]. However, in a later study when under the same experimental conditions of chronic tetanus toxin inhibiting hippocampal neurons (Div16), the postsynaptic architecture of inhibitory synapses was analyzed by super-resolution microscope (SIM). By unchanged number, the average size of gephyrin subsynaptic domains was markedly reduced, in contrast to the discrete changes of GABAAR subsynaptic domains, and resulted in significantly smaller postsynaptic gephyrin volume [55]. Altogether, these results suggested that neurotransmission might not be essential neither for GABAAR nor for gephyrin cluster formation at GABAergic synapses, but it might play a role in recruiting further gephyrin molecules to already formed postsynaptic sites and the organization of gephyrin SSDs.
This view is, however, challenged by a study analyzing the formation of GABAergic postsynaptic assemblies in organotypic slice cultures of mouse cortex and in vivo by local release of GABA near dendritic branches by two-photon photolysis under conditions of high-frequency uncaging (HFU) during development [63]. Under both experimental conditions new gephyrin clusters were induced in distal dendritic areas of cortical neurons. The success rate of gephyrin cluster formation was in the range of about 55% in younger slices (6–8 Div) and was reduced in older stages (14–18 Div). Gephyrin clustering was not detected after glutamate HFU, but further pharmacological experiments with this system confirmed the dependence of the gephyrin cluster formation on Ca2+ influx through voltage-dependent L- and T-type Ca2+ channels, leading to the conclusion that the early depolarizing action of GABA can promote local synaptogenesis during cortical development. Unfortunately, GABAAR structural changes were not analyzed by immunofluorescence microscopy in this study, but functional analysis by measuring uncaging-evoked inhibitory postsynaptic currents (uIPSCs) from newly formed gephyrin puncta supported the presence of functional GABAAR clusters at these sites. Tetanus toxin-induced prevention of GABA release resulted in this study in reduced density of gephyrin puncta in distal apical dendrites of young cortical layer 2/3 pyramidal neurons and decreased the frequency of both miniature IPSCs (mIPSCs) and miniature EPSCs (mEPSCs) in slices and in vivo. Thus, these results support the view that early GABAergic inputs from cortical interneurons are essential for inhibitory (and even excitatory) synaptic connections during cortical development. A quite different experimental system was used in a later study, namely stem cell-derived human neurons as well as in vivo mouse neurons of purely glutamatergic identity, which were reprogrammed for ectopic GABA synthesis and release from presynaptic terminals [64]. This led to accumulation and activation of postsynaptic GABAARs, generating fully functional GABAergic synapses in adjacent neurons. In more detail, the formation of both gephyrin and GABAAR (alpha3) clusters was newly induced both at the dendritic segments as well as perisomatic regions of glutamatergic neurons without affecting cluster size, thus supporting that presynaptic GABA release itself can enable the organization of the corresponding postsynaptic apparatus. Noteworthy, at the end of the discussion part of the manuscript, the authors claimed that important levels of both gephyrin and GABAAR clusters were already present even in neurons that were not exposed to presynaptic GABA release. In this context, although neither the number nor the size of these preexisting clusters was apparently evaluated within this work, the findings of another study published more than 20 years before the latter manuscript are worth considering [65]. The authors of this manuscript communicated that in low-density hippocampal cultures (Div19) in the absence of GABA release at the surface of dendrites and soma of pyramidal cells, small (0.36 +/− 0.01 micrometer diameter) GABAAR clusters with heterogenous subunit composition were monitored. Upon GABAergic innervation the same cells formed 3–4-fold larger GABAAR clusters with higher receptor density and more defined subunit composition. Large clusters of GABAARs are opposed to GABAergic boutons and colocalized with large gephyrin clusters, whereas small clusters of GABAARs are colocalized with small clusters of gephyrin. Since the lateral diffusion of such large aggregates in the membrane is unlikely, the authors proposed that the local signals generated at GABAergic synapses induced the assembly of large synaptic GABAAR clusters and the parallel disappearance of the small GABAAR clusters around the synaptic area.
Thus, in the absence of GABAergic innervation, other signals, e.g., those generated at glutamatergic synapses, might induce the formation of small gephyrin and GABAAR clusters that can be associated also with other than GABAergic presynaptic contacts or might even have no presynaptic partners. In this line a basic level of preexisting GABAA receptor clusters at the neuron’s surface is very likely to be present even in the absence of GABA; however, an instructive role of GABA to induce real newly formed GABAergic synaptic clusters is strongly supported by these studies. Interestingly enough, two years after informing about the signaling role of the neurotransmitter itself for postsynaptic accumulation of gephyrin and GABAARs, the same lab brought out a publication reporting that gephyrin promotes autonomous assembly and synaptic localization of GABAergic postsynaptic components without presynaptic GABA release [66]. In this study, the authors analyzed the cellular distribution of several GABAergic postsynaptic proteins in purely glutamatergic human neuronal cultures derived from induced pluripotent stem cells (iPS), showing that various GABAAR subunits, gephyrin, and even cell-adhesion molecules can coaggregate also at GABA-deficient subsynaptic domains. However, delayed vesicular GABA supply led to a significant increase in gephyrin cluster density without affecting cluster size, supporting that gephyrin–GABAAR clusters, developed in the absence of GABA transmission, can be further modulated by post-hoc GABA release.
Taken together, this latest study somehow confirmed the first set of publications that intrinsic molecular mechanisms promote gephyrin and GABAAR assembly to form postsynaptic clusters well suited to establish functional inhibitory synapses. In addition, similar to the instructive function of glycine to induce the formation of GlyR clusters, GABA also has the capability to promote the formation of GABAAR clusters and contribute to their maintenance at the postsynapse, probably by modulating gephyrin interactions with its multiple binding partners [64,66]. Several studies have shown that phosphorylation of gephyrin depending on the kinase and the phosphorylation site can either increase or reduce GABAAR clustering [42,67], but how these molecular mechanisms relate to neurotransmitter-induced synaptic activity was in general not studied. Activity-dependent GABAAR synapse remodeling was demonstrated by gephyrin phosphorylation involving CAMKII [27]. How ligand binding affects the binding of gephyrin to its other partner proteins, such as adhesion molecules, collybistin, a brain-specific guanine nucleotide exchange factor, and cytoskeletal proteins [17,18,68], or how activity might directly modulate these gephyrin binding partners to control postsynaptic plasticity is even less studied. Gephyrin’s direct or indirect interactions with adhesion molecules that mediate initial contacts between pre- and post-synaptic membranes (like latrophilins, neuroligins, IgSF9b, and N-cadherin) are thought to be important for receptor localization stabilizing the nascent synaptic structure [68] (Figure 2). Specifically, NLG2 has been shown to be localized at GABAergic inhibitory post-synapses and interacts with presynaptic neurexins to form a trans-synaptic complex [69]. Moreover, NLG2 stabilizes the open/active conformation of collybistin, allowing increased gephyrin clustering [70]. Importantly, NLG2 overexpression caused an increase in synapse number (synapsin stain), and elevation of iPSC amplitudes in hippocampal neurons that were reversed after chronic inhibition of neuronal network activities, indicating that NLG2 might mediate activity-dependent specification and validation of inhibitory synapses [71]. However, phosphorylation of NLG2 by PKA disrupted its interaction with the scaffold protein and decreased GABAAR abundance [72]. Even more, super-resolution microscopy studies have shown that NLGN2 forms SSDs that significantly overlap with gephyrin SSDs at the nanoscale level [73], and that acute cleavage of the extracellular domain of NLGN2 resulted in dispersion of GABAARs, leading to impaired synaptic transmission [59]. Interestingly, recent studies provided evidence for several transmembrane proteins such as LHFPL4/GARLH, Clptm1, and Shisa-7 that interact with GABAARs, modulating their trafficking and function [74], altogether further underscoring the complexity of inhibitory synapse biology.
8. Limitations and Perspectives
It is important to realize that in addition to the very different ways to approach the topic of neurotransmitter-dependent clustering of GlyRs or GABAARs at the postsynaptic sites, all these experiments were conducted in somewhat restricted conditions, mostly missing the entire complex environment where synapse formation normally happens, and therefore not all whole-context-dependent mechanisms might be sufficiently recognized. Especially, the in vitro studies might lack the effects of the microvasculature as well as complex cellular interactions and neuronal networks found in the brain and spinal cord in vivo. The modulations through glial cells often remain unnoticed since glial cells are mostly reduced or altered in these cell culture systems, even more so in primary hippocampal cell cultures containing in general fewer glial cells compared to spinal cord cultures. The sometimes contradictory findings obtained in apparently similar conditions raise the need for more standardized methods in association with multimodal approaches in basic experimental research and data interpretation leading to useful and valid conclusions and enhancing data reproducibility. Furthermore, the main majority of these experiments used rodent-derived cells or rodent models for their studies. Species-dependent structural and functional differences have to be taken into account when these results are translated to humans [75,76].
Herein it is also notable that several other studies point to further proteins present at the postsynaptic regions, such as scaffolding proteins and adhesion molecules, having the major impact on the induction of pre- and postsynaptic structures that were presented in great detail in a recent review, even proposing the molecular interactions of different classes of cell adhesion molecules as an independent mechanism of receptor and gephyrin cluster formation without any GABA or glycine release [8]. Moreover, super-resolution imaging techniques have shown that in addition to receptor clustering at the iPSD, the subsynaptic nanoscale organization of inhibitory receptors and gephyrin in SSDs opposite presynaptic transmitter release sites is also an important factor shaping synaptic strength and plasticity. Activity-dependent growth or shrinkage of inhibitory synapses might involve addition or removal, respectively, of these postsynaptic SSDs [10,58,59]. Whether these SSDs at inhibitory synapses are plastic also in vivo remains to be determined. Only the inclusion of all these components might allow in the future to develop the complete picture of GABAergic and glycinergic synapse formation.
9. Clinical Relevance
The activation of GlyRs and, to a certain degree, GABAARs by ligand binding generates molecular and cellular signals that contribute also to synaptogenesis and circuit computation. Dysfunctions within these steps due to various factors such as genetics, drug use, aging, and viral infections might have devastating effects on cellular communication, leading to network disruption within the central neural systems and muscle devastation in the periphery, underlying different pathologies. GlyR mutations resulting in dysfunction of the GlyRs are responsible for hyperekplexia, or startle disease, a neurological locomotion disorder, and are implicated in autism spectrum disorders. Some studies have also reported the role of glycine receptors in certain autoimmune diseases such as stiff person syndrome and high anxiety associated with Alzheimer’s disease [12]. It is important to note the value of studies carried out on mutated GlyRs to better understand GlyR dynamics at the synaptic level. Specifically, the work of Piro et al. shows how GlyR mutations cause structural changes in the interactions of GlyRβ with gephyrin, concluding that alterations in synaptic clustering of GlyR may potentially underlie the pathology of startle disease in patients that have such mutations in GlyRβ [77]. Further studies should clarify whether an altered ligand binding activation of mutated receptors is also part of the pathology. Recent advances in structural biology techniques have greatly enhanced understanding of the structural-functional properties of GyRs and elucidated novel allosteric binding sites as potential drug targets. Recently, several positive allosteric modulators of GlyRs were shown to have promising analgesic activity; however, their therapeutic application is impeded by difficulties in obtaining potent, selective modulators with favorable pharmacokinetic profiles and without side effects [78].
Ionotropic GABAARs are thought to be implicated in an even larger range of neurological disorders, including epilepsy, anxiety, depression, and sleep disorders, and to contribute to cognitive dysfunctions in Alzheimer’s disease, Parkinson’s disease, autism spectrum disorder, schizophrenia, etc. [79]. GABAARs can be positively or negatively modulated by different classes of endogenous and exogenous molecules, such as neurosteroids and benzodiazepines, showing therapeutic effects in some of these pathologic conditions. For example, the widely used benzodiazepines that target the α-γ subunit interface in the extracellular domain of the receptor [80] are attenuating seizures [81] and act as effective anxiolytics [82]. The potentiation of GABA-mediated currents by benzodiazepines and neurosteroids is caused by increased frequency of single-channel openings and duration of channel opening [80]. However, whether or not the potentiated ion currents of GABAARs might cause an increase in cluster size/number is not known; instead, a recent report has shown a time- and dose-dependent decrease in surface levels of GABAARs resulting in a loss of GABAergic synapses upon prolonged exposure to diazepam, one major benzodiazepine [83]. Since the pharmacological properties of GABAARs are determined by their subunit composition, due to the reduced subunit selectivity of presently available GABAAR-targeting drugs, they have several side effects, including dependence and withdrawal symptoms, and limited efficacy. Future studies have to decode the molecular mechanisms of subtype-specific inhibitory synapse formation and provide novel GABAAR pharmaceuticals to treat neurological disorders.
Dysfunctions related to gephyrin-mediated neurotransmission have been implicated in several neurological disorders, such as neurodevelopmental disorders, epilepsy, and even neurodegenerative diseases like Alzheimer’s [84,85,86,87,88]. Since extensive studies have been performed to recognize the pharmacological potential of inhibitory post-synaptic receptors, small molecules that target gephyrin were not identified until recently. Lately, the anti-malarial drug artemisinin and its semi-synthetic derivatives, collectively referred to as artemisinins, were discovered to directly interact with gephyrin and to modulate GABAAR signaling in pancreatic cells [89,90]. In our experiments artemisinins, when used in low doses, restored the number of postsynaptic gephyrin as well as GABAAR and GyR clusters in mouse models of cerebral amyloidosis [91,92]. Thus, these molecules could serve as potent gephyrin-specific modulators with therapeutic benefits in several neurological disorders [93].
In summary, deciphering molecular mechanisms that mediate the organization of pre- and postsynaptic specializations during inhibitory synapse formation, as well as understanding the relationships between GlyR and GABAAR deficits and neural system disorders, are essential for getting deeper insight into disease pathogenesis and for discovering new potential therapeutic targets, but also for improving existing therapeutics. In this context, a possible molecular mechanism linking the potentiation of GABAAR currents to alteration of receptor cluster sizes or even cluster number should be studied in more detail.
10. Conclusions
Reviewing the key publications from more than 25 years of research on the role of neurotransmitter-induced activity for the initiation and maintenance of glycine- and GABAA-receptor clustering at inhibitory synapses (Table 1), the following answers can be formulated to the initial questions that prompted this work:

Table 1.
Major publications presented in chronological order reporting the functional impact of the neurotransmitters glycine and GABA on clustering of the strychnine-sensitive GlyRs and GABAARs, respectively, as well as gephyrin in vitro and in vivo systems. Column 5 describes the most relevant results since in column 6 are cited original conclusions of the authors from the respective publications. Munc18-1: mammalian uncoordinated-18 protein; unc-25: Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase; hpf: hours post fertilization; dpf: days post-fertilization; vGAT: vesicular GABA transporter; GAD65: Glutamate decarboxylase 65 kDa isoform.
- The neurotransmitter-dependent activation of GlyRs seems to be essential for GlyR cluster formation, whereas several publications support that in the case of GABAAR cluster formation, the ligand-dependent activation is not essential for the initial postsynaptic clustering of these receptors (Figure 1 and Figure 2). Whether or not the capacity of GABAARs to form clusters independently from GABA binding is relevant under certain physiological conditions remains to be further elucidated.
- It is very likely that under physiological conditions, the release of the neurotransmitter on both synapse types could regulate the postsynaptic receptor cluster formation: both transmitters are able to induce the formation of new GlyR or GABAAR clusters, and both transmitters seem to be sufficient to modulate the size of existing GlyRs or GABAAR clusters, respectively.
- Glycine seems to be essential for the maintenance of existing GlyRs, whereas GABA seems to be dispensable for the maintenance of GABAAR clusters. Again, the interesting question of whether the independence of GABAAR clusters from continuous GABA activation is an important distinct feature of GABAergic synapses in comparison to GlyRs with relevancy in certain physiological conditions cannot be answered based on present knowledge.
Future experiments providing the missing elements are acutely needed to completely understand the cell biology of inhibitory synapse assembly, especially considering the pharmacological importance of both GlyRs and GABAARs [12,13].
Author Contributions
E.K. and J.K. (Jochen Kuhse) conceptualized, designed and wrote the primary draft of the manuscript. S.K. and J.K. (Joachim Kirsch) edited and revised the final version manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant from the Romanian Ministry of Research and Innovation CNCS- UEFISCDI (PN-III-P4-PCE-2021-1089) to E.K.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| cAMP | cyclic adenosine monophosphate |
| CaMKII | Ca2+/calmodulin-dependent protein kinase type II |
| CDK5 | cyclin-dependent kinase 5 |
| Clptm1 | cleft lip and palate transmembrane protein 1 |
| 3D-SIM | 3D structured illumination microscope |
| dSTORM | direct Stochastic Optical Reconstruction Microscopy |
| EPSC | excitatory postsynaptic currents |
| GAD | glutamic acid decarboxylase |
| GABA | γ-aminobutyric acid |
| GABAAR | γ-aminobutyric acid A-receptors |
| GARLH | GABAAR regulatory LHPFL family protein |
| GyR | glycine receptor |
| GSK3β | glycogen Synthase Kinase 3 beta |
| HFU | High-frequency uncaging |
| iPSD | inhibitory postsynaptic density |
| IPSC | inhibitory postsynaptic currents |
| LHFPL4 | lipoma HMGIC fusion partner-like 4 |
| Munc18-1 | mammal unc-18-1 (Munc18-1 or n-Sec1; herein M18) |
| NLG2 | neuroligin 2 |
| NMDA | N-methyl D-aspartate |
| PKA | protein kinase A |
| PKC | protein kinase C |
| RER | rough endoplasmic reticulum |
| SIM | Structured Illumination Microscopy |
| Shisa-7 | Shisa family member 7 |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| SSD | subsynaptic domain |
| uIPSCs | uncaging-evoked inhibitory postsynaptic currents |
| VIAAT | Vesicular inhibitory amino acid transporter |
| VAMP2 | Vesicle-associated membrane protein 2 |
References
- Nusser, Z.; Hájos, N.; Somogyi, P.; Mody, I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature 1998, 395, 172–177. [Google Scholar] [CrossRef]
- Kilman, V.; van Rossum, M.C.; Turrigiano, G.G. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J. Neurosci. 2002, 22, 1328–1337. [Google Scholar] [CrossRef]
- Barberis, A. Postsynaptic plasticity of GABAergic synapses. Neuropharmacology 2020, 169, 107643. [Google Scholar] [CrossRef]
- Chiu, C.Q.; Martenson, J.S.; Yamazaki, M.; Natsume, R.; Sakimura, K.; Tomita, S.; Tavalin, S.J.; Higley, M.J. Input-Specific NMDAR-Dependent Potentiation of Dendritic GABAergic Inhibition. Neuron 2018, 97, 368–377.e3. [Google Scholar] [CrossRef] [PubMed]
- Welle, T.M.; Rajgor, D.; Garcia, J.D.; Kareemo, D.; Zych, S.M.; Gookin, S.E.; Martinez, T.P.; Dell’Acqua, M.L.; Ford, C.P.; Kennedy, M.J.; et al. miRNA-mediated control of gephyrin synthesis drives sustained inhibitory synaptic plasticity. EMBO Rep. 2024, 25, 5141–5168. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, N.; Roemer, V.; Janzen, D.; Villmann, C. Impaired Glycine Receptor Trafficking in Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef]
- Südhof, T.C. The cell biology of synapse formation. J. Cell Biol. 2021, 220, e202103052. [Google Scholar] [CrossRef]
- Qi, C.; Luo, L.D.; Feng, I.; Ma, S. Molecular mechanisms of synaptogenesis. Front. Synaptic Neurosci. 2022, 14, 939793. [Google Scholar] [CrossRef]
- Welle, T.M.; Smith, K.R. Release your inhibitions: The cell biology of GABAergic postsynaptic plasticity. Curr. Opin. Neurobiol. 2025, 90, 102952. [Google Scholar] [CrossRef]
- Betz, H. Ligand-gated ion channels in the brain: The amino acid receptor superfamily. Neuron 1990, 5, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, N.; Blundell, R. Glycine receptors: Structure, function, and therapeutic implications. Mol. Aspects Med. 2025, 103, 101360. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef]
- Gravielle, M.C. Regulation of GABAA Receptors Induced by the Activation of L-Type Voltage-Gated Calcium Channels. Membranes 2021, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.P.; Skelding, K.A. Calcium/Calmodulin-Stimulated Protein Kinase II (CaMKII): Different Functional Outcomes from Activation, Depending on the Cellular Microenvironment. Cells 2023, 12, 401. [Google Scholar] [CrossRef]
- Maynard, S.A.; Triller, A. Inhibitory Receptor Diffusion Dynamics. Front. Mol. Neurosci. 2019, 12, 313. [Google Scholar] [CrossRef]
- Tyagarajan, S.K.; Fritschy, J.M. Gephyrin: A master regulator of neuronal function? Nat. Rev. Neurosci. 2014, 15, 141–156. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Trattnig, C.; Kuhse, J.; Nawrotzki, R.A.; Kirsch, J. Gephyrin: A key regulatory protein of inhibitory synapses and beyond. Histochem. Cell Biol. 2018, 150, 489–508. [Google Scholar] [CrossRef]
- Lévi, S.; Vannier, C.; Triller, A. Strychnine-sensitive stabilization of postsynaptic glycine receptor clusters. J. Cell Sci. 1998, 111 Pt 3, 335–345. [Google Scholar] [CrossRef]
- Kirsch, J.; Betz, H. Glycine-receptor activation is required for receptor clustering in spinal neurons. Nature 1998, 392, 717–720. [Google Scholar] [CrossRef]
- Du, J.; Lü, W.; Wu, S.; Cheng, Y.; Gouaux, E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 2015, 526, 224–229. [Google Scholar] [CrossRef]
- Zhu, H. Structure and Mechanism of Glycine Receptor Elucidated by Cryo-Electron Microscopy. Front. Pharmacol. 2022, 13, 925116. [Google Scholar] [CrossRef] [PubMed]
- Betz, H.; Kuhse, J.; Fischer, M.; Schmieden, V.; Laube, B.; Kuryatov, A.; Langosch, D.; Meyer, G.; Bormann, J.; Rundström, N.; et al. Structure, diversity and synaptic localization of inhibitory glycine receptors. J. Physiol. 1994, 88, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Barron, S.E.; Guth, P.S. Uses and limitations of strychnine as a probe in neurotransmission. Trends Pharmacol. Sci. 1987, 8, 204–206. [Google Scholar] [CrossRef]
- Rasmussen, H.; Rasmussen, T.; Triller, A.; Vannier, C. Strychnine-blocked glycine receptor is removed from synapses by a shift in insertion/degradation equilibrium. Mol. Cell. Neurosci. 2002, 19, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, I.; Miki, M.; Asakawa, K.; Kawakami, K.; Oda, Y.; Hirata, H. Glycinergic transmission and postsynaptic activation of CaMKII are required for glycine receptor clustering in vivo. Genes Cells Devoted Mol. Cell. Mech. 2013, 18, 211–224. [Google Scholar] [CrossRef]
- Flores, C.E.; Nikonenko, I.; Mendez, P.; Fritschy, J.M.; Tyagarajan, S.K.; Muller, D. Activity-dependent inhibitory synapse remodeling through gephyrin phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E65–E72. [Google Scholar] [CrossRef]
- Ogino, K.; Yamada, K.; Nishioka, T.; Oda, Y.; Kaibuchi, K.; Hirata, H. Phosphorylation of Gephyrin in Zebrafish Mauthner Cells Governs Glycine Receptor Clustering and Behavioral Desensitization to Sound. J. Neurosci. 2019, 39, 8988–8997. [Google Scholar] [CrossRef]
- Nakahata, Y.; Eto, K.; Murakoshi, H.; Watanabe, M.; Kuriu, T.; Hirata, H.; Moorhouse, A.J.; Ishibashi, H.; Nabekura, J. Activation-Dependent Rapid Postsynaptic Clustering of Glycine Receptors in Mature Spinal Cord Neurons. eNeuro 2017, 4, e0194-16.2017. [Google Scholar] [CrossRef]
- Lipp, P.; Reither, G. Protein kinase C: The “masters” of calcium and lipid. Cold Spring Harb. Perspect. Biol. 2011, 3, a004556. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Breitinger, U.; Bahnassawy, L.M.; Janzen, D.; Roemer, V.; Becker, C.M.; Villmann, C.; Breitinger, H.G. PKA and PKC Modulators Affect Ion Channel Function and Internalization of Recombinant Alpha1 and Alpha1-Beta Glycine Receptors. Front. Mol. Neurosci. 2018, 11, 154. [Google Scholar] [CrossRef]
- Specht, C.G.; Grünewald, N.; Pascual, O.; Rostgaard, N.; Schwarz, G.; Triller, A. Regulation of glycine receptor diffusion properties gephyrin interactions by protein kinase C. EMBO J. 2011, 30, 3842–3853. [Google Scholar] [CrossRef] [PubMed]
- Uezu, A.; Kanak, D.J.; Bradshaw, T.W.; Soderblom, E.J.; Catavero, C.M.; Burette, A.C.; Weinberg, R.J.; Soderling, S.H. Identification of an elaborate complex mediating postsynaptic inhibition. Science 2016, 353, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Krueger-Burg, D. Understanding GABAergic synapse diversity and its implications for GABAergic pharmacotherapy. Trends Neurosci. 2025, 48, 47–61. [Google Scholar] [CrossRef]
- Kasaragod, V.B.; Schindelin, H. Structure-Function Relationships of Glycine and GABAA Receptors and Their Interplay With the Scaffolding Protein Gephyrin. Front. Mol. Neurosci. 2018, 11, 317. [Google Scholar] [CrossRef]
- Bai, G.; Wang, Y.; Zhang, M. Gephyrin-mediated formation of inhibitory postsynaptic density sheet via phase separation. Cell Res. 2021, 31, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Kim, S.; Hwang, D.E.; Eom, Y.G.; Jang, G.; Park, H.Y.; Choi, J.M.; Ko, J.; Shin, Y. Thermodynamic modulation of gephyrin condensation by inhibitory synapse components. Proc. Natl. Acad. Sci. USA 2024, 121, e2313236121. [Google Scholar] [CrossRef]
- Zacchi, P.; Antonelli, R.; Cherubini, E. Gephyrin phosphorylation in the functional organization and plasticity of GABAergic synapses. Front. Cell. Neurosci. 2014, 8, 103. [Google Scholar] [CrossRef]
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2020, 439, 125–136. [Google Scholar] [CrossRef]
- Zhou, L.; Kiss, E.; Demmig, R.; Kirsch, J.; Nawrotzki, R.A.; Kuhse, J. Binding of gephyrin to microtubules is regulated by its phosphorylation at Ser270. Histochem. Cell Biol. 2021, 156, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Kuhse, J.; Kalbouneh, H.; Schlicksupp, A.; Mükusch, S.; Nawrotzki, R.; Kirsch, J. Phosphorylation of gephyrin in hippocampal neurons by cyclin-dependent kinase CDK5 at Ser-270 is dependent on collybistin. J. Biol. Chem. 2012, 287, 30952–30966. [Google Scholar] [CrossRef]
- Maas, C.; Belgardt, D.; Lee, H.K.; Heisler, F.F.; Lappe-Siefke, C.; Magiera, M.M.; van Dijk, J.; Hausrat, T.J.; Janke, C.; Kneussel, M. Synaptic activation modifies microtubules underlying transport of postsynaptic cargo. Proc. Natl. Acad. Sci. USA 2009, 106, 8731–8736. [Google Scholar] [CrossRef]
- Rao, A.; Cha, E.M.; Craig, A.M. Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J. Neurosci. 2000, 20, 8344–8353. [Google Scholar] [CrossRef] [PubMed]
- Verhage, M.; Maia, A.S.; Plomp, J.J.; Brussaard, A.B.; Heeroma, J.H.; Vermeer, H.; Toonen, R.F.; Hammer, R.E.; van den Berg, T.K.; Missler, M.; et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000, 287, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, C. Neuronal SNARE complex assembly guided by Munc18-1 and Munc13-1. FEBS Open Bio 2022, 12, 1939–1957. [Google Scholar] [CrossRef]
- Gally, C.; Bessereau, J.L. GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J. Neurosci. 2003, 23, 2591–2599. [Google Scholar] [CrossRef]
- Jin, Y.; Jorgensen, E.; Hartwieg, E.; Horvitz, H.R. The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J. Neurosci. 1999, 19, 539–548. [Google Scholar] [CrossRef]
- Harms, K.J.; Craig, A.M. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J. Comp. Neurol. 2005, 490, 72–84. [Google Scholar] [CrossRef]
- Ramsay, H.J.; Gookin, S.E.; Ramsey, A.M.; Kareemo, D.J.; Crosby, K.C.; Stich, D.G.; Olah, S.S.; Actor-Engel, H.S.; Smith, K.R.; Kennedy, M.J. AMPA and GABAA receptor nanodomains assemble in the absence of synaptic neurotransmitter release. Front. Mol. Neurosci. 2023, 16, 1232795. [Google Scholar] [CrossRef]
- Humeau, Y.; Doussau, F.; Grant, N.J.; Poulain, B. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie 2000, 82, 427–446. [Google Scholar] [CrossRef]
- Benke, T.A.; Swann, J. The tetanus toxin model of chronic epilepsy. Adv. Exp. Med. Biol. 2004, 548, 226–238. [Google Scholar] [CrossRef]
- Louis, E.D.; Williamson, P.D.; Darcey, T.M. Experimental models of chronic focal epilepsy: A critical review of four models. Yale J. Biol. Med. 1987, 60, 255–272. [Google Scholar]
- Megighian, A.; Pirazzini, M.; Fabris, F.; Rossetto, O.; Montecucco, C. Tetanus and tetanus neurotoxin: From peripheral uptake to central nervous tissue targets. J. Neurochem. 2021, 158, 1244–1253. [Google Scholar] [CrossRef]
- Chen, H.; Tang, A.H.; Blanpied, T.A. Subsynaptic spatial organization as a regulator of synaptic strength and plasticity. Curr. Opin. Neurobiol. 2018, 51, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Crosby, K.C.; Gookin, S.E.; Garcia, J.D.; Hahm, K.M.; Dell’Acqua, M.L.; Smith, K.R. Nanoscale Subsynaptic Domains Underlie the Organization of the Inhibitory Synapse. Cell Rep. 2019, 26, 3284–3297.e3. [Google Scholar] [CrossRef]
- Held, R.G.; Liang, J.; Brunger, A.T. Nanoscale architecture of synaptic vesicles and scaffolding complexes revealed by cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2024, 121, e2403136121. [Google Scholar] [CrossRef]
- Olah, S.S.; Kareemo, D.J.; Buchta, W.C.; Sinnen, B.L.; Miller, C.N.; Actor-Engel, H.S.; Gookin, S.E.; Winborn, C.S.; Kleinjan, M.S.; Crosby, K.C.; et al. Acute reorganization of postsynaptic GABAA receptors reveals the functional impact of molecular nanoarchitecture at inhibitory synapses. Cell Rep. 2023, 42, 113331. [Google Scholar] [CrossRef]
- Xu, N.; Chen, S.Y.; Tang, A.H. Tuning synapse strength by nanocolumn plasticity. Trends Neurosci. 2025, 48, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Lévi, S.; Schweizer, C.; Bannai, H.; Pascual, O.; Charrier, C.; Triller, A. Homeostatic regulation of synaptic GlyR numbers driven by lateral diffusion. Neuron 2008, 59, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Comenencia-Ortiz, E.; Moss, S.J.; Davies, P.A. Phosphorylation of GABAA receptors influences receptor trafficking and neurosteroid actions. Psychopharmacology 2014, 231, 3453–3465. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Morrow, D.H.; Nathanson, A.J.; Henley, J.M.; Wilkinson, K.A.; Moss, S.J. Phosphorylation on Ser-359 of the α2 subunit in GABA type A receptors down-regulates their density at inhibitory synapses. J. Biol. Chem. 2020, 295, 12330–12342. [Google Scholar] [CrossRef]
- Oh, W.C.; Lutzu, S.; Castillo, P.E.; Kwon, H.B. De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 2016, 353, 1037–1040. [Google Scholar] [CrossRef]
- Burlingham, S.R.; Wong, N.F.; Peterkin, L.; Lubow, L.; Dos Santos Passos, C.; Benner, O.; Ghebrial, M.; Cast, T.P.; Xu-Friedman, M.A.; Südhof, T.C.; et al. Induction of synapse formation by de novo neurotransmitter synthesis. Nat. Commun. 2022, 13, 3060. [Google Scholar] [CrossRef]
- Christie, S.B.; Miralles, C.P.; De Blas, A.L. GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J. Neurosci. 2002, 22, 684–697. [Google Scholar] [CrossRef]
- Carricaburu, E.; Benner, O.; Burlingham, S.R.; Dos Santos Passos, C.; Hobaugh, N.; Karr, C.H.; Chanda, S. Gephyrin promotes autonomous assembly and synaptic localization of GABAergic postsynaptic components without presynaptic GABA release. Proc. Natl. Acad. Sci. USA 2024, 121, e2315100121. [Google Scholar] [CrossRef]
- Tyagarajan, S.K.; Ghosh, H.; Yévenes, G.E.; Nikonenko, I.; Ebeling, C.; Schwerdel, C.; Sidler, C.; Zeilhofer, H.U.; Gerrits, B.; Muller, D.; et al. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. USA 2011, 108, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Choii, G.; Ko, J. Gephyrin: A central GABAergic synapse organizer. Exp. Mol. Med. 2015, 47, e158. [Google Scholar] [CrossRef]
- Dong, N.; Qi, J.; Chen, G. Molecular reconstitution of functional GABAergic synapses with expression of neuroligin-2 and GABAA receptors. Mol. Cell. Neurosci. 2007, 35, 14–23. [Google Scholar] [CrossRef]
- Soykan, T.; Schneeberger, D.; Tria, G.; Buechner, C.; Bader, N.; Svergun, D.; Tessmer, I.; Poulopoulos, A.; Papadopoulos, T.; Varoqueaux, F.; et al. A conformational switch in collybistin determines the differentiation of inhibitory postsynapses. EMBO J. 2014, 33, 2113–2133. [Google Scholar] [CrossRef] [PubMed]
- Chubykin, A.A.; Atasoy, D.; Etherton, M.R.; Brose, N.; Kavalali, E.T.; Gibson, J.R.; Südhof, T.C. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 2007, 54, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Halff, E.F.; Hannan, S.; Kwanthongdee, J.; Lesept, F.; Smart, T.G.; Kittler, J.T. Phosphorylation of neuroligin-2 by PKA regulates its cell surface abundance and synaptic stabilization. Sci. Signal. 2022, 15, eabg2505. [Google Scholar] [CrossRef]
- Gookin, S.E.; Taylor, M.R.; Schwartz, S.L.; Kennedy, M.J.; Dell’Acqua, M.L.; Crosby, K.C.; Smith, K.R. Complementary Use of Super-Resolution Imaging Modalities to Study the Nanoscale Architecture of Inhibitory Synapses. Front. Synaptic Neurosci. 2022, 14, 852227. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shepard, R.D.; Lu, W. Regulation of GABAARs by Transmembrane Accessory Proteins. Trends Neurosci. 2021, 44, 152–165. [Google Scholar] [CrossRef]
- Sun, C.; Zhu, H.; Clark, S.; Gouaux, E. Cryo-EM structures reveal native GABAA receptor assemblies and pharmacology. Nature 2023, 622, 195–201. [Google Scholar] [CrossRef]
- Zhou, J.; Noviello, C.M.; Teng, J.; Moore, H.; Lega, B.; Hibbs, R.E. Resolving native GABAA receptor structures from the human brain. Nature 2025, 638, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Piro, I.; Eckes, A.L.; Kasaragod, V.B.; Sommer, C.; Harvey, R.J.; Schaefer, N.; Villmann, C. Novel Functional Properties of Missense Mutations in the Glycine Receptor β Subunit in Startle Disease. Front. Mol. Neurosci. 2021, 14, 745275. [Google Scholar] [CrossRef]
- Gallagher, C.I.; Ha, D.A.; Harvey, R.J.; Vandenberg, R.J. Positive Allosteric Modulators of Glycine Receptors and Their Potential Use in Pain Therapies. Pharmacol. Rev. 2022, 74, 933–961. [Google Scholar] [CrossRef]
- Zhang, W.; Xiong, B.R.; Zhang, L.Q.; Huang, X.; Yuan, X.; Tian, Y.K.; Tian, X.B. The role of the GABAergic system in diseases of the central nervous system. Neuroscience 2021, 470, 88–99. [Google Scholar] [CrossRef]
- Kim, J.J.; Hibbs, R.E. Direct Structural Insights into GABAA Receptor Pharmacology. Trends Biochem. Sci. 2021, 46, 502–517. [Google Scholar] [CrossRef]
- Greenfield, L.J., Jr. Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 2013, 22, 589–600. [Google Scholar] [CrossRef]
- Balon, R.; Starcevic, V. Role of Benzodiazepines in Anxiety Disorders. Adv. Exp. Med. Biol. 2020, 1191, 367–388. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Sweeney, A.; Pekle, E.; Alam, S.; Ali, A.B.; Duchen, M.; Jovanovic, J.N. Diazepam-induced loss of inhibitory synapses mediated by PLCδ/Ca2+/calcineurin signalling downstream of GABAA receptors. Mol. Psychiatry 2018, 23, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Tannenberg, R.K.; Dodd, P.R. Reduced expression of the inhibitory synapse scaffolding protein gephyrin in Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 14, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Rees, H.; Seyfried, N.T.; Dammer, E.B.; Duong, D.M.; Gearing, M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Levey, A.I.; et al. Abnormal gephyrin immunoreactivity associated with Alzheimer disease pathologic changes. J. Neuropathol. Exp. Neurol. 2013, 72, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Shen, L.; Yin, H.; Pan, Y.M.; Wang, L.; Chen, D.; Xi, Z.Q.; Xiao, Z.; Wang, X.F.; Zhou, S.N. Downregulation of gephyrin in temporal lobe epilepsy neurons in humans and a rat model. Synapse 2011, 65, 1006–1014. [Google Scholar] [CrossRef]
- Kiss, E.; Gorgas, K.; Schlicksupp, A.; Groß, D.; Kins, S.; Kirsch, J.; Kuhse, J. Biphasic Alteration of the Inhibitory Synapse Scaffold Protein Gephyrin in Early and Late Stages of an Alzheimer Disease Model. Am. J. Pathol. 2016, 186, 2279–2291. [Google Scholar] [CrossRef]
- Kim, S.; Kang, M.; Park, D.; Lee, A.R.; Betz, H.; Ko, J.; Chang, I.; Um, J.W. Impaired formation of high-order gephyrin oligomers underlies gephyrin dysfunction-associated pathologies. iScience 2021, 24, 102037. [Google Scholar] [CrossRef]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair α Cell Identity. Cell 2017, 168, 86–100.e15. [Google Scholar] [CrossRef]
- Kasaragod, V.B.; Hausrat, T.J.; Schaefer, N.; Kuhn, M.; Christensen, N.R.; Tessmer, I.; Maric, H.M.; Madsen, K.L.; Sotriffer, C.; Villmann, C.; et al. Elucidating the Molecular Basis for Inhibitory Neurotransmission Regulation by Artemisinins. Neuron 2019, 101, 673–689.e11. [Google Scholar] [CrossRef]
- Kiss, E.; Kins, S.; Zöller, Y.; Schilling, S.; Gorgas, K.; Groß, D.; Schlicksupp, A.; Rosner, R.; Kirsch, J.; Kuhse, J. Artesunate restores the levels of inhibitory synapse proteins and reduces amyloid-β and C-terminal fragments (CTFs) of the amyloid precursor protein in an AD-mouse model. Mol. Cell. Neurosci. 2021, 113, 103624. [Google Scholar] [CrossRef] [PubMed]
- Kuhse, J.; Groeneweg, F.; Kins, S.; Gorgas, K.; Nawrotzki, R.; Kirsch, J.; Kiss, E. Loss of Extrasynaptic Inhibitory Glycine Receptors in the Hippocampus of an AD Mouse Model Is Restored by Treatment with Artesunate. Int. J. Mol. Sci. 2023, 24, 4623. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Kins, S.; Gorgas, K.; Venczel Szakács, K.H.; Kirsch, J.; Kuhse, J. Another Use for a Proven Drug: Experimental Evidence for the Potential of Artemisinin and Its Derivatives to Treat Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 4165. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).