

Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease



Abstract

1. Introduction
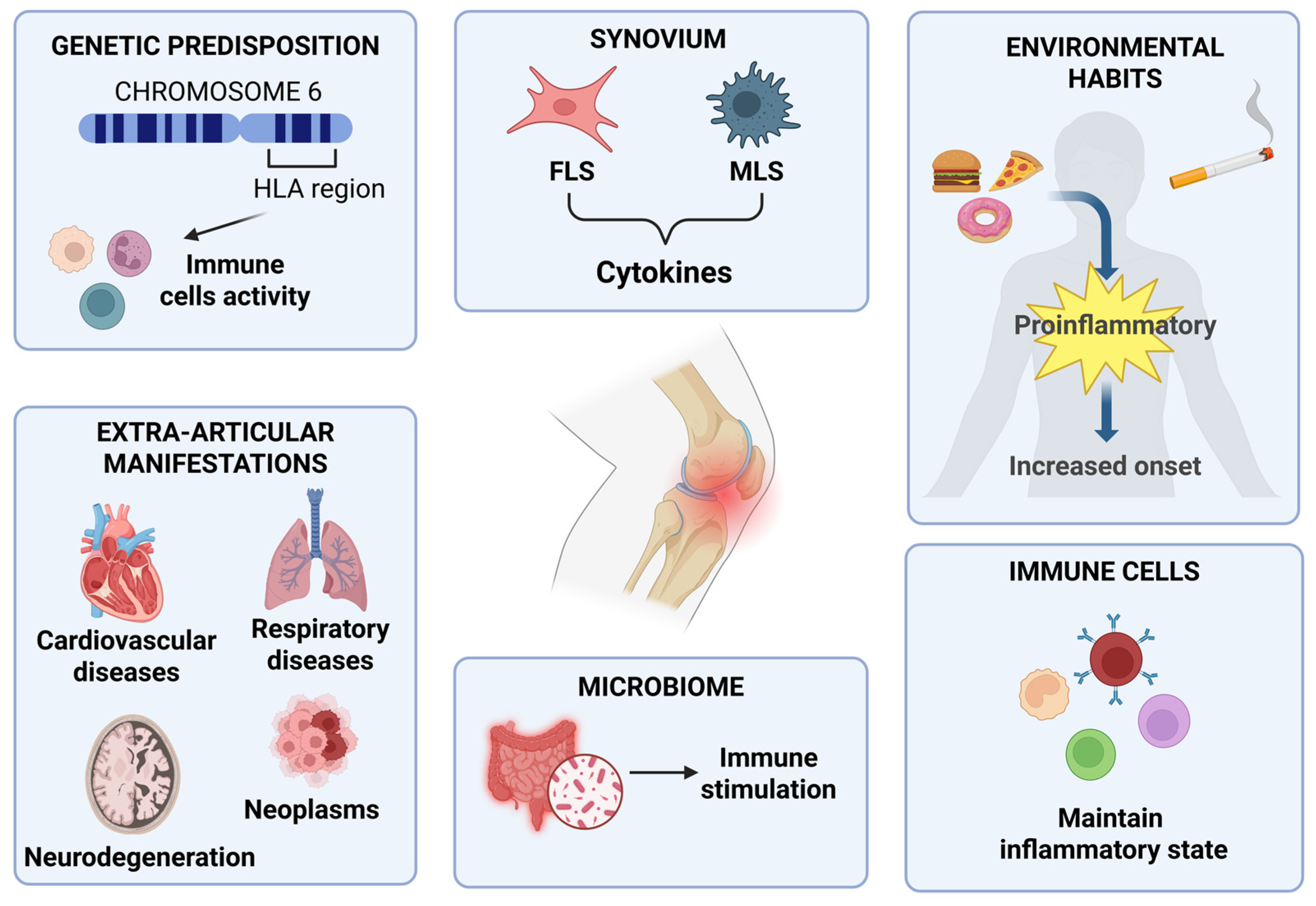
2. Pathophysiology of RA
2.1. Genetic Predisposition
2.2. Environmental Factors
2.3. Immune Trigger and Synovial Cells
2.4. Influence of the Microbiome
2.5. Extra-Articular Manifestations of RA
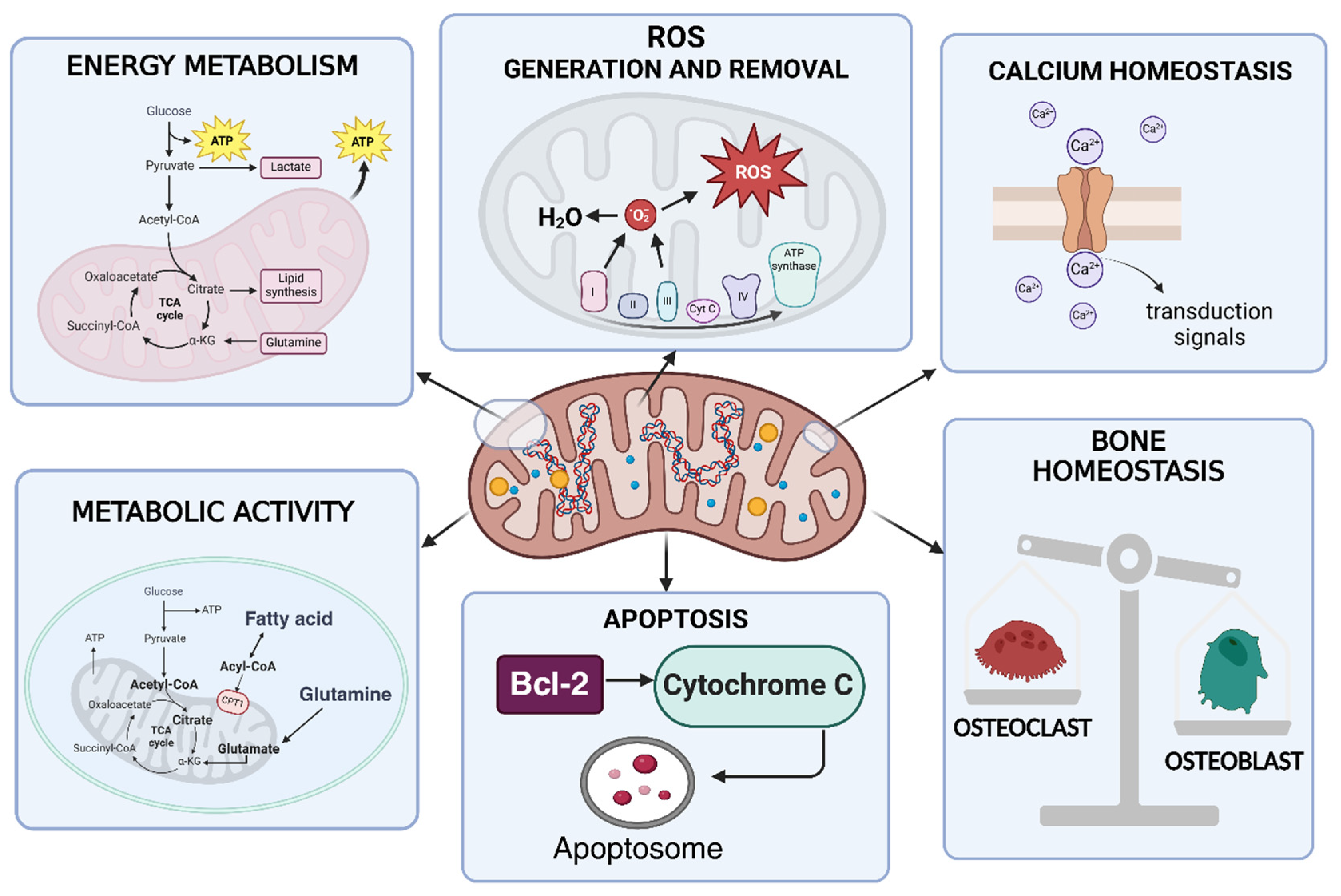
3. Overview of Mitochondrial Functions
4. Mitochondrion Dynamics in RA
4.1. Fusion and Fission Dynamics
4.2. Biogenesis and Mitophagy Processes
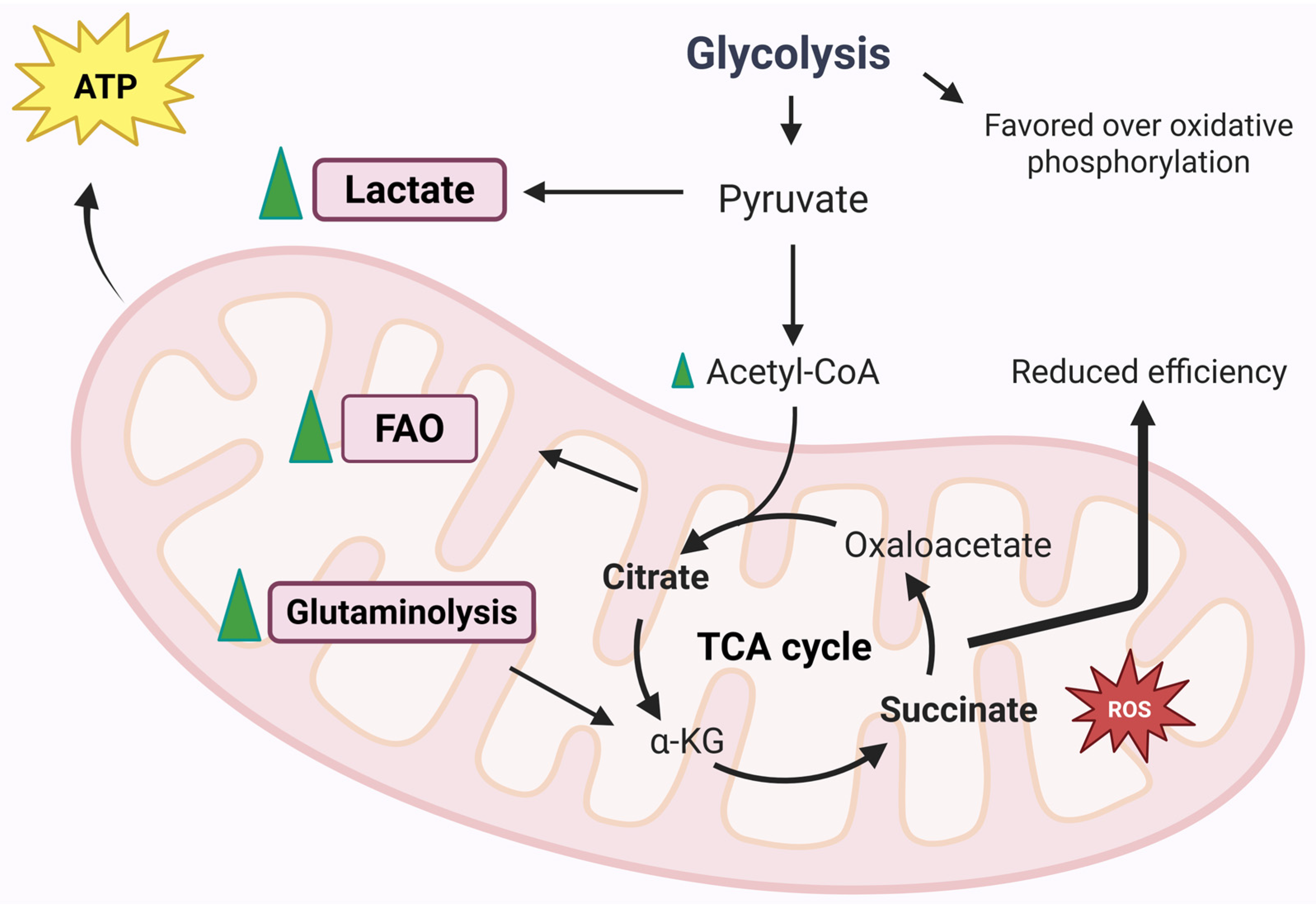
5. Mitochondrial Metabolic Activity and ATP Production in RA
5.1. Glycolysis
5.2. Fatty Acid Oxidation
5.3. Glutaminolysis
5.4. TCA Cycle Metabolites
5.4.1. Succinate
5.4.2. Citrate and Aconitate
6. Mitochondrial Role in RA-Related Immune Inflammation
6.1. NOD-, LRR- and Pyrin Domain-Containing Protein 3 Inflammasome
6.2. Toll-like Receptor 9/Nuclear Factor Kappa B
6.3. Cyclic GMP-AMP Synthase
7. Mitochondrial DAMPS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAMPs | Origin | Role in RA |
|---|---|---|
| ATP | Released from damaged or necrotic cells, or actively from stressed cells. | Extracellular ATP activates purinergic P2X receptors (particularly P2X7) on immune cells, leading to activation of the NLRP3 inflammasome and the release of IL-1β and IL-18. It contributes to synoviocyte proliferation and perpetuation of inflammation [144]. |
| mtDNA | Circular DNA released from damaged or apoptotic mitochondria. | When released into the cytosol or extracellular space, it acts as a DAMP by activating PRRs. It promotes caspase-1 activation, IL-1β and IL-18 release, and contributes to the inflammatory positive feedback loop with mtDNA mutations [145]. |
| Cytochrome c | In the cytosol, it is a key signal for apoptosis, but if released into extracellular space, it becomes a danger signal for the immune system. | When released into extracellular space, it is recognized by activating PRRs receptors. The interaction between cytochrome c and PRRs can trigger a cascade of pro-inflammatory signalling events, leading to the activation of pathways such as NF-κB and the release of pro-inflammatory cytokines (such as IL-1β, TNF-α, IL-6) [146]. |
| HSP (heat shock proteins) | Released from damaged or necrotic cells. | Some members of HSP can act as DAMPs when extracellular, inducing inflammatory responses and stimulating cytokine production [147]. |
7.1. Adenosine Triphosphate
7.2. Cytochrome c
7.3. mtDNA and Mutations
8. Mitochondrial Role on Oxygen Availability in RA
8.1. Oxidative Stress
8.2. Hypoxaemia
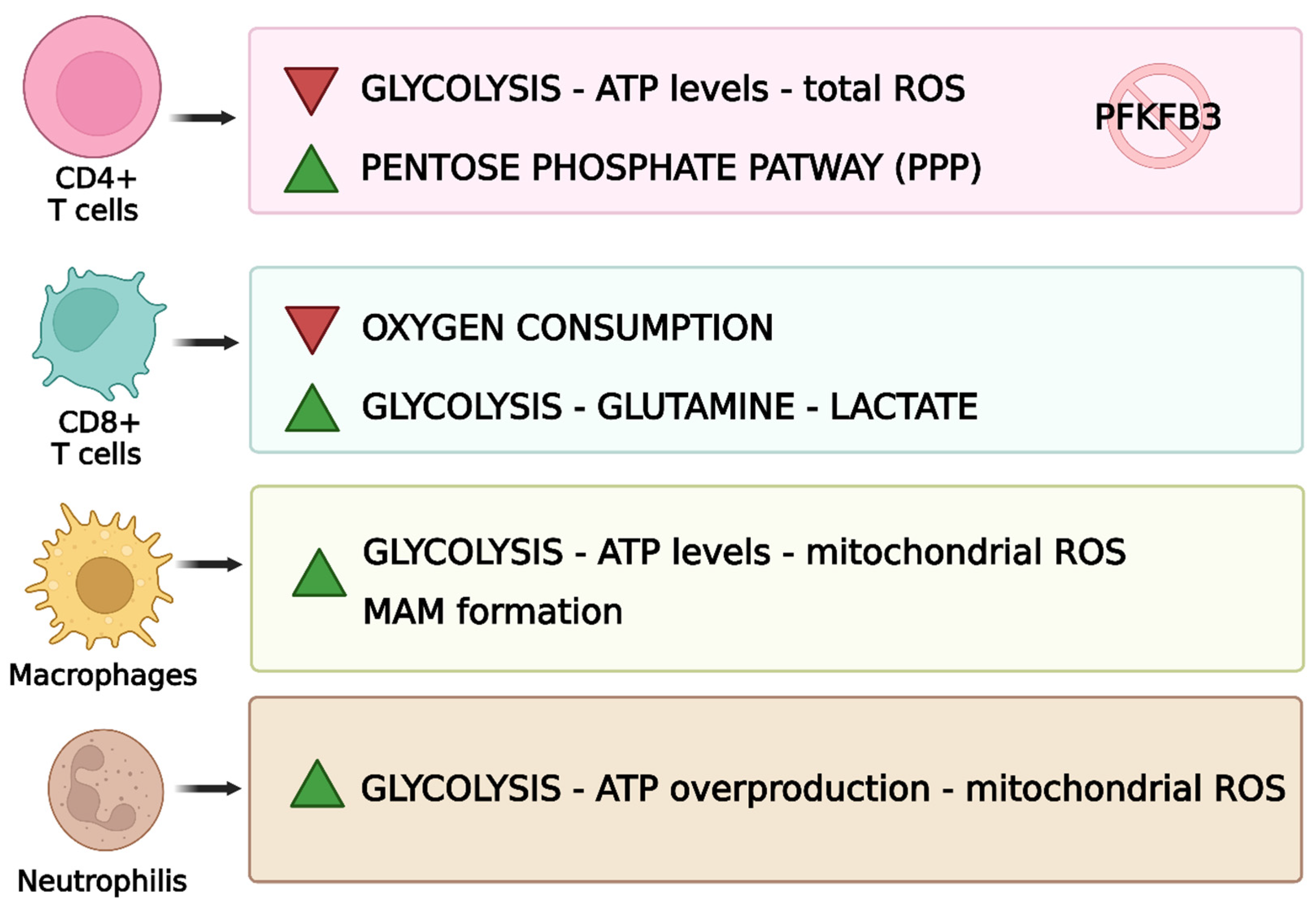
9. Mitochondrial Influence on Immune Cells in RA
9.1. T-Cells
9.1.1. Metabolic Abnormalities in T-Cells
9.1.2. Proinflammatory Cytokines in T-Cells
9.1.3. Regulatory T-Cells
9.2. B-Cells
9.3. Macrophages
9.4. Neutrophils
9.5. Dendritic Cells
10. Mitochondrial Role in Tissues Homeostasis in RA
10.1. Apoptosis
10.2. Bone Homeostasis
10.3. Fibroblast Synoviocites
10.4. Chondrocyte Autophagy
11. Mitochondrial Therapeutic Approaches
11.1. Conventional Synthetic Anti-Rheumatic Drugs (csDMARDs)
11.1.1. Methotrexate
11.1.2. Leflunomide
11.1.3. Sulfasalazine
11.2. Targeted Synthetic DMARDs (tsDMARDs)
JAK/STAT Pathway Inhibitors
11.3. Biological DMARDs (bDMARDs)
12. Materials and Methods
13. Conclusions and Future Prospective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, C.; Jing, W.; Su, C.; Liu, L.; Zhang, X.; Yuan, B.; Du, X.; Wang, H. Rheumatoid Arthritis and Mitochondrial Homeostasis: The Crossroads of Metabolism and Immunity. Front. Med. 2022, 9, 1017650. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Firestein, G.S.; McInnes, I.B. The Pathogenesis of Rheumatoid Arthritis. Immunity 2022, 55, 2255–2270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, Q.-C.; Wang, Q.-C.; Wu, L.; Yu, Q.; He, P.-F. A Compendium of Mitochondrial Molecular Characteristics Provides Novel Perspectives on the Treatment of Rheumatoid Arthritis Patients. J. Transl. Med. 2023, 21, 561. [Google Scholar] [CrossRef]
- Möller, B.; Kollert, F.; Sculean, A.; Villiger, P.M. Infectious Triggers in Periodontitis and the Gut in Rheumatoid Arthritis (RA): A Complex Story About Association and Causality. Front. Immunol. 2020, 11, 1108. [Google Scholar] [CrossRef]
- van der Woude, D.; van der Helm-van Mil, A.H.M. Update on the Epidemiology, Risk Factors, and Disease Outcomes of Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 174–187. [Google Scholar] [CrossRef]
- Ma, C.; Wang, J.; Hong, F.; Yang, S. Mitochondrial Dysfunction in Rheumatoid Arthritis. Biomolecules 2022, 12, 1216. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial Fusion and Fission: The Fine-Tune Balance for Cellular Homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Li, S.; Huo, C.; Liu, A.; Zhu, Y. Mitochondria: A Breakthrough in Combating Rheumatoid Arthritis. Front. Med. 2024, 11, 1439182. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. The Immunology of Rheumatoid Arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef]
- Romão, V.C.; Fonseca, J.E. Etiology and Risk Factors for Rheumatoid Arthritis: A State-of-the-Art Review. Front. Med. 2021, 8, 689698. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and Environmental Risk Factors for Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, L.E. The Genetics of Rheumatoid Arthritis. Rheumatology 2020, 59, 2661–2670. [Google Scholar] [CrossRef]
- Montgomery, R.A.; Tatapudi, V.S.; Leffell, M.S.; Zachary, A.A. HLA in Transplantation. Nat. Rev. Nephrol. 2018, 14, 558–570. [Google Scholar] [CrossRef]
- Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice. Cells 2020, 9, 1127. [Google Scholar] [CrossRef]
- Grievink, H.W.; Smit, V.; Huisman, B.W.; Gal, P.; Yavuz, Y.; Klerks, C.; Binder, C.J.; Bot, I.; Kuiper, J.; Foks, A.C.; et al. Cardiovascular Risk Factors: The Effects of Ageing and Smoking on the Immune System, an Observational Clinical Study. Front. Immunol. 2022, 13, 968815. [Google Scholar] [CrossRef]
- Hasbani, G.E.; Nassar, J.E.; Ali, A.M.E.; Uthman, I.; Jawad, A. The Impact of Nicotine Smoking on Spondyloarthritis and Rheumatoid Arthritis. Reumatismo 2024, 76, 2. [Google Scholar] [CrossRef]
- Arleevskaya, M.; Takha, E.; Petrov, S.; Kazarian, G.; Renaudineau, Y.; Brooks, W.; Larionova, R.; Korovina, M.; Valeeva, A.; Shuralev, E.; et al. Interplay of Environmental, Individual and Genetic Factors in Rheumatoid Arthritis Provocation. Int. J. Mol. Sci. 2022, 23, 8140. [Google Scholar] [CrossRef]
- Chang, K.; Yang, S.M.; Kim, S.H.; Han, K.H.; Park, S.J.; Shin, J.I. Smoking and Rheumatoid Arthritis. Int. J. Mol. Sci. 2014, 15, 22279–22295. [Google Scholar] [CrossRef]
- Ren, J.; Ding, Y.; Zhao, J.; Sun, Y. Impact of Cigarette Smoking on Rheumatoid Arthritis-Associated Lung Diseases: A Retrospective Case Control Study on Clinical and Radiological Features and Prognosis. Rheumatol. Int. 2023, 43, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Nikiphorou, E. Nutrition and Diet in Rheumatoid Arthritis. Nutrients 2022, 14, 888. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Bartoloni, E.; Grassi, D.; Ferri, C.; Gerli, R. Diet in Rheumatoid Arthritis versus Systemic Lupus Erythematosus: Any Differences? Nutrients 2021, 13, 772. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar] [CrossRef]
- Choi, E.; Machado, C.R.L.; Okano, T.; Boyle, D.; Wang, W.; Firestein, G.S. Joint-Specific Rheumatoid Arthritis Fibroblast-like Synoviocyte Regulation Identified by Integration of Chromatin Access and Transcriptional Activity. JCI Insight 2024, 9, e179392. [Google Scholar] [CrossRef]
- Jang, S.; Kwon, E.-J.; Lee, J.J. Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. Int. J. Mol. Sci. 2022, 23, 905. [Google Scholar] [CrossRef]
- Reyes-Castillo, Z.; Valdés-Miramontes, E.; Llamas-Covarrubias, M.; Muñoz-Valle, J.F. Troublesome Friends within Us: The Role of Gut Microbiota on Rheumatoid Arthritis Etiopathogenesis and Its Clinical and Therapeutic Relevance. Clin. Exp. Med. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282. [Google Scholar] [CrossRef]
- Maeda, Y.; Takeda, K. Host-Microbiota Interactions in Rheumatoid Arthritis. Exp. Mol. Med. 2019, 51, 1–6. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, B.; Zhao, L.; Li, H. The Gut Microbiota: Emerging Evidence in Autoimmune Diseases. Trends Mol. Med. 2020, 26, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wei, Y.; Zhu, Y.; Xie, Z.; Hai, Q.; Li, Z.; Qin, D. Gut Microbiota and Rheumatoid Arthritis: From Pathogenesis to Novel Therapeutic Opportunities. Front. Immunol. 2022, 13, 1007165. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, Y.; Chen, X.; Zhao, Y.; Wu, Y.; Li, Y.; Wang, X.; Chen, H.; Xiang, C. Induction of Intestinal Th17 Cells by Flagellins From Segmented Filamentous Bacteria. Front. Immunol. 2019, 10, 2750. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.-X.; Yin, X.-F.; Zhang, M.-X.; Qiao, J.; Xin, X.-H.; Chang, M.-J.; Gao, C.; Li, Y.-F.; Li, X.-F. The Gut Microbiota and Its Relevance to Peripheral Lymphocyte Subpopulations and Cytokines in Patients with Rheumatoid Arthritis. J. Immunol. Res. 2021, 2021, 6665563. [Google Scholar] [CrossRef]
- Figus, F.A.; Piga, M.; Azzolin, I.; McConnell, R.; Iagnocco, A. Rheumatoid Arthritis: Extra-Articular Manifestations and Comorbidities. Autoimmun. Rev. 2021, 20, 102776. [Google Scholar] [CrossRef]
- Bedeković, D.; Bošnjak, I.; Bilić-Ćurčić, I.; Kirner, D.; Šarić, S.; Novak, S. Risk for Cardiovascular Disease Development in Rheumatoid Arthritis. BMC Cardiovasc. Disord. 2024, 24, 291. [Google Scholar] [CrossRef]
- Johri, N.; Varshney, S.; Gandha, S.; Maurya, A.; Mittal, P.; Jangra, S.; Garg, R.; Saraf, A. Association of Cardiovascular Risks in Rheumatoid Arthritis Patients: Management, Treatment and Future Perspectives. Health Sci. Rev. 2023, 8, 100108. [Google Scholar] [CrossRef]
- Inferrera, F.; Marino, Y.; Genovese, T.; Cuzzocrea, S.; Fusco, R.; Di Paola, R. Mitochondrial Quality Control: Biochemical Mechanism of Cardiovascular Disease. Biochim. Biophys. Acta Mol. Cell Res. 2025, 1872, 119906. [Google Scholar] [CrossRef]
- Schäfer, V.S.; Winter, L.; Skowasch, D.; Bauer, C.-J.; Pizarro, C.; Weber, M.; Kütting, D.; Behning, C.; Brossart, P.; Petzinna, S.M. Exploring Pulmonary Involvement in Newly Diagnosed Rheumatoid Arthritis, and Psoriatic Arthritis: A Single Center Study. Rheumatol. Int. 2024, 44, 1975–1986. [Google Scholar] [CrossRef]
- Janssen, I.; Nouri, A.; Tessitore, E.; Meyer, B. Cervical Myelopathy in Patients Suffering from Rheumatoid Arthritis—A Case Series of 9 Patients and A Review of the Literature. J. Clin. Med. 2020, 9, 811. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Ağbuga, Ö.D.; Ersözlü, E.D. Disease and Treatment-Related Comorbidities in Rheumatoid Arthritis. J. Turk. Soc. Rheumatol. 2024, 16, 64–72. [Google Scholar] [CrossRef]
- Beydon, M.; Pinto, S.; De Rycke, Y.; Fautrel, B.; Mariette, X.; Seror, R.; Tubach, F. Risk of Cancer for Patients with Rheumatoid Arthritis versus General Population: A National Claims Database Cohort Study. Lancet Reg. Health Eur. 2023, 35, 100768. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Tan, B.G.; Gustafsson, C.M.; Falkenberg, M. Mechanisms and Regulation of Human Mitochondrial Transcription. Nat. Rev. Mol. Cell Biol. 2024, 25, 119–132. [Google Scholar] [CrossRef]
- Kan, S.; Duan, M.; Liu, Y.; Wang, C.; Xie, J. Role of Mitochondria in Physiology of Chondrocytes and Diseases of Osteoarthritis and Rheumatoid Arthritis. Cartilage 2021, 13 (Suppl. 2), 1102S–1121S. [Google Scholar] [CrossRef]
- Gray, M.W. Lynn Margulis and the Endosymbiont Hypothesis: 50 Years Later. Mol. Biol. Cell 2017, 28, 1285–1287. [Google Scholar] [CrossRef]
- Weyand, C.M.; Wu, B.; Huang, T.; Hu, Z.; Goronzy, J.J. Mitochondria as Disease-Relevant Organelles in Rheumatoid Arthritis. Clin. Exp. Immunol. 2023, 211, 208–223. [Google Scholar] [CrossRef]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and Mechanism of Mitochondrial Electron Transport Chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-Specific Regulation of Cytochrome c by Post-Translational Modifications: Respiration, the Mitochondrial Membrane Potential, ROS, and Apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Deshwal, S.; Onishi, M.; Tatsuta, T.; Bartsch, T.; Cors, E.; Ried, K.; Lemke, K.; Nolte, H.; Giavalisco, P.; Langer, T. Mitochondria Regulate Intracellular Coenzyme Q Transport and Ferroptotic Resistance via STARD7. Nat. Cell Biol. 2023, 25, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The Assembly, Regulation and Function of the Mitochondrial Respiratory Chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted Mitochondria: Moving Mitochondrial Science beyond Function and Dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Guo, H.; Rubinstein, J.L. Structure of ATP Synthase under Strain during Catalysis. Nat. Commun. 2022, 13, 2232. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Ali, A.T.; Boehme, L.; Carbajosa, G.; Seitan, V.C.; Small, K.S.; Hodgkinson, A. Nuclear Genetic Regulation of the Human Mitochondrial Transcriptome. Elife 2019, 8, e41927. [Google Scholar] [CrossRef]
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, Mitochondria and Cell Metabolism: A Functional Triangle in Bioenergetics. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Li, X.; Jiang, O.; Chen, M.; Wang, S. Mitochondrial Homeostasis: Shaping Health and Disease. Curr. Med. 2024, 3, 5. [Google Scholar] [CrossRef]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, R.; Afroz, S.F.; Ramnath, D.; Lawrence, G.M.; Okada, T.; Curson, J.E.; de Bruin, J.; Fairlie, D.P.; Schroder, K.; St John, J.C.; et al. Lipopolysaccharide Promotes Drp1-Dependent Mitochondrial Fission and Associated Inflammatory Responses in Macrophages. Immunol. Cell Biol. 2020, 98, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.-A.; Cho, S.-N.; Son, S.-H.; Song, C.-H. Mitofusin 2-Deficiency Suppresses Mycobacterium Tuberculosis Survival in Macrophages. Cells 2019, 8, 1355. [Google Scholar] [CrossRef]
- Lachmandas, E.; Boutens, L.; Ratter, J.M.; Hijmans, A.; Hooiveld, G.J.; Joosten, L.A.B.; Rodenburg, R.J.; Fransen, J.A.M.; Houtkooper, R.H.; van Crevel, R.; et al. Microbial Stimulation of Different Toll-like Receptor Signalling Pathways Induces Diverse Metabolic Programmes in Human Monocytes. Nat. Microbiol. 2016, 2, 16246. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Dakin, S.G.; Coles, M.; Sherlock, J.P.; Powrie, F.; Carr, A.J.; Buckley, C.D. Pathogenic Stromal Cells as Therapeutic Targets in Joint Inflammation. Nat. Rev. Rheumatol. 2018, 14, 714–726. [Google Scholar] [CrossRef]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial Dynamics Involves Molecular and Mechanical Events in Motility, Fusion and Fission. Front. Cell Dev. Biol. 2022, 10, 1010232. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial Biogenesis and Clearance: A Balancing Act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Kim, E.K.; Kwon, J.-E.; Lee, S.-Y.; Lee, E.-J.; Kim, D.S.; Moon, S.-J.; Lee, J.; Kwok, S.-K.; Park, S.-H.; Cho, M.-L. IL-17-Mediated Mitochondrial Dysfunction Impairs Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts through Activation of Autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef]
- Chadha, S.; Behl, T.; Bungau, S.; Kumar, A.; Kaur, R.; Venkatachalam, T.; Gupta, A.; Kandhwal, M.; Chandel, D. Focus on the Multimodal Role of Autophagy in Rheumatoid Arthritis. Inflammation 2021, 44, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.F.; Abdullah, G.A.; Nolan, A.; Linford, C.; Phelan, M.M.; Wright, H.L. PFKFB3 Is a Critical Regulator of Neutrophil Metabolism and Function in Rheumatoid Arthritis. Prepr. Serv. Health Sci. 2023. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Parkin Clearance of Dysfunctional Mitochondria Regulates ROS Levels and Increases Survival of Human Chondrocytes. Osteoarthr. Cartil. 2018, 26, 1087–1097. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Jin, K.; Wen, Z.; Cao, W.; Wu, B.; Wen, R.; Tian, L.; Berry, G.J.; Goronzy, J.J.; et al. The DNA Repair Nuclease MRE11A Functions as a Mitochondrial Protector and Prevents T Cell Pyroptosis and Tissue Inflammation. Cell Metab. 2019, 30, 477–492.e6. [Google Scholar] [CrossRef]
- Wen, Z.; Jin, K.; Shen, Y.; Yang, Z.; Li, Y.; Wu, B.; Tian, L.; Shoor, S.; Roche, N.E.; Goronzy, J.J.; et al. N-Myristoyltransferase Deficiency Impairs Activation of Kinase AMPK and Promotes Synovial Tissue Inflammation. Nat. Immunol. 2019, 20, 313–325. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Wu, K.; Shieh, J.; Qin, L.; Guo, J.J. Mitochondrial Mechanisms in the Pathogenesis of Chronic Inflammatory Musculoskeletal Disorders. Cell Biosci. 2024, 14, 76. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It Is All about Energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Jutley, G.S.; Sahota, K.; Sahbudin, I.; Filer, A.; Arayssi, T.; Young, S.P.; Raza, K. Relationship Between Inflammation and Metabolism in Patients With Newly Presenting Rheumatoid Arthritis. Front. Immunol. 2021, 12, 676105. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Y.; Liu, H.; Tian, Y.; Yin, G.; Xie, Q. Targeting Glycolytic Pathway in Fibroblast-like Synoviocytes for Rheumatoid Arthritis Therapy: Challenges and Opportunities. Inflamm. Res. 2023, 72, 2155–2167. [Google Scholar] [CrossRef]
- Ziyadullaev, S.K.; Khudaiberdiev, S.S.; Aripova, T.U.; Chirumbolo, S.; Kamalov, Z.S.; Bjørklund, G.; Rizaev, J.A.; Tashkenbaeva, E.N.; Khamidov, O.A.; Gaffarov, U.B. Synovial Fluid as a Crucial Component of the Joint Microenvironment in Rheumatoid Arthritis. Immune Netw. 2025, 25, e2. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, L.C.; Cole, J.; Rattigan, K.M.; Barrett, M.P.; Kurian, N.; McInnes, I.B.; Goodyear, C.S. The Rheumatoid Synovial Environment Alters Fatty Acid Metabolism in Human Monocytes and Enhances CCL20 Secretion. Rheumatology 2020, 59, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Zheng, K.D.; Lin, K.; Zheng, G.; Zou, H.; Wang, J.M.; Lin, Y.Y.; Chuka, C.M.; Ge, R.S.; Zhai, W.; et al. Energy Metabolism Disorder as a Contributing Factor of Rheumatoid Arthritis: A Comparative Proteomic and Metabolomic Study. PLoS ONE 2015, 10, e0132695. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Zengin, G.; Brisc, C.; Brisc, M.C.; Munteanu, M.A.; Nistor-Cseppento, D.C.; Bungau, S. The Lipid Paradox as a Metabolic Checkpoint and Its Therapeutic Significance in Ameliorating the Associated Cardiovascular Risks in Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2020, 21, 9505. [Google Scholar] [CrossRef]
- Shen, Y.; Wen, Z.; Li, Y.; Matteson, E.L.; Hong, J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of the Scaffold Protein TKS5 in Tissue-Invasive, Proinflammatory T Cells. Nat. Immunol. 2017, 18, 1025–1034. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Hu, C.; Xie, Z.; Li, H.; Wei, S.; Wang, D.; Wen, C.; Xu, G. Exploration of the Serum Metabolite Signature in Patients with Rheumatoid Arthritis Using Gas Chromatography-Mass Spectrometry. J. Pharm. Biomed. Anal. 2016, 127, 60–67. [Google Scholar] [CrossRef]
- Kim, S.; Hwang, J.; Xuan, J.; Jung, Y.H.; Cha, H.-S.; Kim, K.H. Global Metabolite Profiling of Synovial Fluid for the Specific Diagnosis of Rheumatoid Arthritis from Other Inflammatory Arthritis. PLoS ONE 2014, 9, e97501. [Google Scholar] [CrossRef]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 Plays a Key Role in the Cell Growth of Fibroblast-like Synoviocytes in Rheumatoid Arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef]
- Li, X.; Peng, X.; Li, Y.; Wei, S.; He, G.; Liu, J.; Li, X.; Yang, S.; Li, D.; Lin, W.; et al. Glutamine Addiction in Tumor Cell: Oncogene Regulation and Clinical Treatment. Cell Commun. Signal. 2024, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic Control of TH17 and Induced Treg Cell Balance by an Epigenetic Mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-Dependent α-Ketoglutarate Production Regulates the Balance between T Helper 1 Cell and Regulatory T Cell Generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [PubMed]
- Souto-Carneiro, M.M.; Klika, K.D.; Abreu, M.T.; Meyer, A.P.; Saffrich, R.; Sandhoff, R.; Jennemann, R.; Kraus, F.V.; Tykocinski, L.; Eckstein, V.; et al. Effect of Increased Lactate Dehydrogenase A Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 2050–2064. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Mun, S.; Lee, Y.-R.; Lee, S.; Kwon, S.; Kim, D.; Lim, M.-K.; Kang, H.-G.; Lee, J. A Discovery of Screening Markers for Rheumatoid Arthritis by Liquid Chromatography Mass Spectrometry: A Metabolomic Approach. Int. J. Rheum. Dis. 2020, 23, 1353–1362. [Google Scholar] [CrossRef]
- Coras, R.; Murillo-Saich, J.D.; Guma, M. Circulating Pro- and Anti-Inflammatory Metabolites and Its Potential Role in Rheumatoid Arthritis Pathogenesis. Cells 2020, 9, 827. [Google Scholar] [CrossRef]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 Senses Extracellular Succinate Released from Inflammatory Macrophages and Exacerbates Rheumatoid Arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Wang, C.; Xia, W.-R.; Zheng, J.-Y.; Yang, J.; Liu, B.; Liu, J.-Q.; Liu, L.-F. Succinate Induces Synovial Angiogenesis in Rheumatoid Arthritis through Metabolic Remodeling and HIF-1α/VEGF Axis. Free Radic. Biol. Med. 2018, 126, 1–14. [Google Scholar] [CrossRef]
- Wu, B.; Qiu, J.; Zhao, T.V.; Wang, Y.; Maeda, T.; Goronzy, I.N.; Akiyama, M.; Ohtsuki, S.; Jin, K.; Tian, L.; et al. Succinyl-CoA Ligase Deficiency in Pro-Inflammatory and Tissue-Invasive T Cells. Cell Metab. 2020, 32, 967–980.e5. [Google Scholar] [CrossRef]
- Saraiva, A.L.; Veras, F.P.; Peres, R.S.; Talbot, J.; de Lima, K.A.; Luiz, J.P.; Carballido, J.M.; Cunha, T.M.; Cunha, F.Q.; Ryffel, B.; et al. Succinate Receptor Deficiency Attenuates Arthritis by Reducing Dendritic Cell Traffic and Expansion of Th17 Cells in the Lymph Nodes. FASEB J. 2018, 32, fj201800285. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wu, B.; Goodman, S.B.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 652771. [Google Scholar] [CrossRef] [PubMed]
- Leblond, A.; Allanore, Y.; Avouac, J. Targeting Synovial Neoangiogenesis in Rheumatoid Arthritis. Autoimmun. Rev. 2017, 16, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef]
- Zhang, A.; Gupte, A.A.; Chatterjee, S.; Li, S.; Ayala, A.G.; Miles, B.J.; Hamilton, D.J. Enhanced Succinate Oxidation with Mitochondrial Complex II Reactive Oxygen Species Generation in Human Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 12168. [Google Scholar] [CrossRef]
- Reddy, A.; Bozi, L.H.M.; Yaghi, O.K.; Mills, E.L.; Xiao, H.; Nicholson, H.E.; Paschini, M.; Paulo, J.A.; Garrity, R.; Laznik-Bogoslavski, D.; et al. pH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 2020, 183, 62–75.e17. [Google Scholar] [CrossRef]
- Zhunussova, A.; Sen, B.; Friedman, L.; Tuleukhanov, S.; Brooks, A.D.; Sensenig, R.; Orynbayeva, Z. Tumor Microenvironment Promotes Dicarboxylic Acid Carrier-Mediated Transport of Succinate to Fuel Prostate Cancer Mitochondria. Am. J. Cancer Res. 2015, 5, 1665–1679. [Google Scholar]
- Certo, M.; Marone, G.; de Paulis, A.; Mauro, C.; Pucino, V. Lactate: Fueling the Fire Starter. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1474. [Google Scholar] [CrossRef]
- Neveu, M.-A.; Beziere, N.; Daniels, R.; Bouzin, C.; Comment, A.; Schwenck, J.; Fuchs, K.; Kneilling, M.; Pichler, B.J.; Schmid, A.M. Lactate Production Precedes Inflammatory Cell Recruitment in Arthritic Ankles: An Imaging Study. Mol. Imaging Biol. 2020, 22, 1324–1332. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Pileri, F.; Gualdrini, F.; Ghisletti, S. Integration of Transcriptional and Metabolic Control in Macrophage Activation. EMBO Rep. 2021, 22, e53251. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4+ T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The Poster Child of Metabolic Reprogramming in Macrophage Function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate Is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Li, C.; Chen, B.; Fang, Z.; Leng, Y.; Wang, D.; Chen, F.; Xu, X.; Sun, Z. Metabolomics in the Development and Progression of Rheumatoid Arthritis: A Systematic Review. Jt. Bone Spine 2020, 87, 425–430. [Google Scholar] [CrossRef]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A Metabolite Regulates Inflammation Response and Oxidative Stress. Oxid. Med. Cell Longev. 2020, 2020, 5404780. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and Perpetuation of NLRP3 Inflammasome Activation and Assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Response by Inhibiting NALP3 Inflammasome-Mediated Mitochondrial DNA Release. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, J.; Lu, G.; Han, C.; Zhang, Y. NLRP3 inflammasome in sepsis. Mol. Med. Rep. 2020, 24, 514. [Google Scholar] [CrossRef]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef]
- Saber, M.M.; Monir, N.; Awad, A.S.; Elsherbiny, M.E.; Zaki, H.F. TLR9: A Friend or a Foe. Life Sci. 2022, 307, 120874. [Google Scholar] [CrossRef]
- Luo, X.; Cui, J.; Long, X.; Chen, Z. TLRs Play Crucial Roles in Regulating RA Synoviocyte. Endocr. Metab. Immune Disord.-Drug Targets 2020, 20, 1156–1165. [Google Scholar] [CrossRef]
- Barrera, M.-J.; Aguilera, S.; Castro, I.; Carvajal, P.; Jara, D.; Molina, C.; González, S.; González, M.-J. Dysfunctional Mitochondria as Critical Players in the Inflammation of Autoimmune Diseases: Potential Role in Sjögren’s Syndrome. Autoimmun. Rev. 2021, 20, 102867. [Google Scholar] [CrossRef]
- Ramos-González, E.J.; Bastian, Y.; Castañeda-Delgado, J.E.; Zapata-Zúñiga, M.; Gómez-Moreno, M.; Castillo-Ortiz, J.D.; Ramos-Remus, C.; Enciso-Moreno, J.A. Overexpression of TLR7 and TLR9 Occurs Before Onset Symptoms In First-Degree Relatives of Rheumatoid Arthritis Patients. Arch. Med. Res. 2022, 53, 86–92. [Google Scholar] [CrossRef]
- Zhu, H.; Kong, B.; Che, J.; Zhao, Y.; Sun, L. Bioinspired Nanogels as Cell-Free DNA Trapping and Scavenging Organelles for Rheumatoid Arthritis Treatment. Proc. Natl. Acad. Sci. USA 2023, 120, e2303385120. [Google Scholar] [CrossRef]
- Liu, X.; Chen, S.; Yan, Y.; Liu, L.; Chen, Y. Nanoparticulate DNA Scavenger Loading Methotrexate Targets Articular Inflammation to Enhance Rheumatoid Arthritis Treatment. Biomaterials 2022, 286, 121594. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Carmody, R.J. Chapter Two-NF-κB and the Transcriptional Control of Inflammation. In International Review of Cell and Molecular Biology; Loos, F., Ed.; Transcriptional Gene Regulation in Health and Disease; Academic Press: Cambridge, MA, USA, 2018; Volume 335, pp. 41–84. [Google Scholar]
- Li, Y.; Liang, Q.; Zhou, L.; Cao, Y.; Yang, J.; Li, J.; Liu, J.; Bi, J.; Liu, Y. An ROS-Responsive Artesunate Prodrug Nanosystem Co-Delivers Dexamethasone for Rheumatoid Arthritis Treatment through the HIF-1α/NF-κB Cascade Regulation of ROS Scavenging and Macrophage Repolarization. Acta Biomater. 2022, 152, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, Q.; Zhang, S.; Sun, L.; Liu, W.; Wang, J. Inhibition of NEMO Alleviates Arthritis by Blocking the M1 Macrophage Polarization. Int. Immunopharmacol. 2023, 117, 109983. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Luo, R.; Lu, H.; Zhang, R.; Deng, G.; Luo, H.; Yu, X.; Wang, C.; Zhang, H.; Zhang, Y.; et al. Activation of cGAS-STING Signaling Pathway Promotes Liver Fibrosis and Hepatic Sinusoidal Microthrombosis. Int. Immunopharmacol. 2023, 125, 111132. [Google Scholar] [CrossRef]
- Zhou, S.; Cheng, F.; Zhang, Y.; Su, T.; Zhu, G. Engineering and Delivery of cGAS-STING Immunomodulators for the Immunotherapy of Cancer and Autoimmune Diseases. Acc. Chem. Res. 2023, 56, 2933–2943. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; Lin, H.; Qiu, Q.; Lao, M.; Zeng, S.; Wang, C.; Xu, S.; Zou, Y.; Shi, M.; et al. Accumulation of Cytosolic dsDNA Contributes to Fibroblast-like Synoviocytes-Mediated Rheumatoid Arthritis Synovial Inflammation. Int. Immunopharmacol. 2019, 76, 105791. [Google Scholar] [CrossRef]
- An, C.; Sun, F.; Liu, C.; Huang, S.; Xu, T.; Zhang, C.; Ge, S. IQGAP1 Promotes Mitochondrial Damage and Activation of the mtDNA Sensor cGAS-STING Pathway to Induce Endothelial Cell Pyroptosis Leading to Atherosclerosis. Int. Immunopharmacol. 2023, 123, 110795. [Google Scholar] [CrossRef]
- Ouyang, W.; Wang, S.; Yan, D.; Wu, J.; Zhang, Y.; Li, W.; Hu, J.; Liu, Z. The cGAS-STING Pathway-Dependent Sensing of Mitochondrial DNA Mediates Ocular Surface Inflammation. Signal Transduct. Target. Ther. 2023, 8, 371. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Peugnet-González, I.; Parada-Venegas, D.; Dijkstra, G.; Faber, K.N. cGAS-STING Signaling Pathway in Intestinal Homeostasis and Diseases. Front. Immunol. 2023, 14, 1239142. [Google Scholar] [CrossRef]
- Hong, Z.; Mei, J.; Guo, H.; Zhu, J.; Wang, C. Intervention of cGAS–STING Signaling in Sterile Inflammatory Diseases. J. Mol. Cell Biol. 2022, 14, mjac005. [Google Scholar] [CrossRef]
- Willemsen, J.; Neuhoff, M.-T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF Leads to mtDNA Release and cGAS/STING-Dependent Interferon Responses That Support Inflammatory Arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging Role of Damage-Associated Molecular Patterns Derived from Mitochondria in Inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Huang, L.; Kuang, Y.; Wu, X.; Ma, X.; Zhao, B.; Lan, J. Unlocking the Therapeutic Potential of P2X7 Receptor: A Comprehensive Review of Its Role in Neurodegenerative Disorders. Front. Pharmacol. 2024, 15, 1450704. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.E.; Shadel, G.S. Mitochondrial DNA Release in Innate Immune Signaling. Annu. Rev. Biochem. 2023, 92, 299–332. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bajwa, E.; Klegeris, A. Cytochrome c Can Be Released into Extracellular Space and Modulate Functions of Human Astrocytes in a Toll-like Receptor 4-Dependent Manner. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129400. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef]
- da Silva, J.L.G.; Passos, D.F.; Bernardes, V.M.; Leal, D.B.R. ATP and Adenosine: Role in the Immunopathogenesis of Rheumatoid Arthritis. Immunol. Lett. 2019, 214, 55–64. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Sandai, D.; Zhang, Z.-W.; Song, Z.-J.; Babu, D.; Tabana, Y.; Dahham, S.S.; Adam Ahmed Adam, M.; Wang, Y.; Wang, W.; et al. Adenosine Triphosphate Induced Cell Death: Mechanisms and Implications in Cancer Biology and Therapy. World J. Clin. Oncol. 2023, 14, 549–569. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Cao, F.; Hu, L.-Q.; Yao, S.-R.; Hu, Y.; Wang, D.-G.; Fan, Y.-G.; Pan, G.-X.; Tao, S.-S.; Zhang, Q.; Pan, H.-F.; et al. P2X7 Receptor: A Potential Therapeutic Target for Autoimmune Diseases. Autoimmun. Rev. 2019, 18, 767–777. [Google Scholar] [CrossRef]
- Fan, Z.-D.; Zhang, Y.-Y.; Guo, Y.-H.; Huang, N.; Ma, H.-H.; Huang, H.; Yu, H.-G. Involvement of P2X7 Receptor Signaling on Regulating the Differentiation of Th17 Cells and Type II Collagen-Induced Arthritis in Mice. Sci. Rep. 2016, 6, 35804. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Arroum, T.; Luo, X.; Kang, R.; Lee, Y.J.; Tang, D.; Hüttemann, M.; Song, X. Diverse Functions of Cytochrome c in Cell Death and Disease. Cell Death Differ. 2024, 31, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef]
- Pullerits, R.; Bokarewa, M.; Jonsson, I.-M.; Verdrengh, M.; Tarkowski, A. Extracellular Cytochrome c, a Mitochondrial Apoptosis-Related Protein, Induces Arthritis. Rheumatology 2005, 44, 32–39. [Google Scholar] [CrossRef]
- Contis, A.; Mitrovic, S.; Lavie, J.; Douchet, I.; Lazaro, E.; Truchetet, M.-E.; Goizet, C.; Contin-Bordes, C.; Schaeverbeke, T.; Blanco, P.; et al. Neutrophil-Derived Mitochondrial DNA Promotes Receptor Activator of Nuclear Factor κB and Its Ligand Signalling in Rheumatoid Arthritis. Rheumatology 2017, 56, 1200–1205. [Google Scholar] [CrossRef]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia Induces Mitochondrial Mutagenesis and Dysfunction in Inflammatory Arthritis. Arthritis Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, Mitochondrial Dysfunction and Synovial Invasiveness in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.J.; Fearon, U. Mitochondrial Mutagenesis Correlates with the Local Inflammatory Environment in Arthritis. Ann. Rheum. Dis. 2012, 71, 582–588. [Google Scholar] [CrossRef]
- Da Sylva, T.R.; Connor, A.; Mburu, Y.; Keystone, E.; Wu, G.E. Somatic Mutations in the Mitochondria of Rheumatoid Arthritis Synoviocytes. Arthritis Res. Ther. 2005, 7, R844. [Google Scholar] [CrossRef]
- Lin, X.; Zhou, Y.; Xue, L. Mitochondrial Complex I Subunit MT-ND1 Mutations Affect Disease Progression. Heliyon 2024, 10, e28808. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Larsson, N.-G.; Gustafsson, C.M. Replication and Transcription of Human Mitochondrial DNA. Annu. Rev. Biochem. 2024, 93, 47–77. [Google Scholar] [CrossRef] [PubMed]
- Antolínez-Fernández, Á.; Esteban-Ramos, P.; Fernández-Moreno, M.Á.; Clemente, P. Molecular Pathways in Mitochondrial Disorders Due to a Defective Mitochondrial Protein Synthesis. Front. Cell Dev. Biol. 2024, 12, 1410245. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Tsuruta, D. What Are Reactive Oxygen Species, Free Radicals, and Oxidative Stress in Skin Diseases? Int. J. Mol. Sci. 2021, 22, 10799. [Google Scholar] [CrossRef]
- Phull, A.-R.; Nasir, B.; ul Haq, I.; Kim, S.J. Oxidative Stress, Consequences and ROS Mediated Cellular Signaling in Rheumatoid Arthritis. Chem.-Biol. Interact. 2018, 281, 121–136. [Google Scholar] [CrossRef]
- Cai, W.; Yu, Y.; Zong, S.; Wei, F. Metabolic Reprogramming as a Key Regulator in the Pathogenesis of Rheumatoid Arthritis. Inflamm. Res. 2020, 69, 1087–1101. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial Dynamics in Health and Disease: Mechanisms and Potential Targets. Signal Transduct. Target. Ther. 2023, 8, 1–25. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Dec, K.; Kałduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive Oxygen Species-Sources, Functions, Oxidative Damage. Pol. Merkur. Lek. 2020, 48, 124–127. [Google Scholar]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating Oxidative Stress in Heart Failure: Past, Present and Future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Y.; Gao, Y.; Qi, D.; Zhao, L.; Zhao, L.; Liu, C.; Tao, T.; Zhou, C.; Sun, X.; et al. NR1D1 Modulates Synovial Inflammation and Bone Destruction in Rheumatoid Arthritis. Cell Death Dis. 2020, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Wu, L.; Wang, J.; Wang, X.; He, Z.; Di, W.; Yang, Q.; Wei, X. Exosomes Derived from Diabetic Serum Accelerate the Progression of Osteoarthritis. Arch. Biochem. Biophys. 2024, 755, 109960. [Google Scholar] [CrossRef] [PubMed]
- Rahane, D.; Dhingra, T.; Chalavady, G.; Datta, A.; Ghosh, B.; Rana, N.; Borah, A.; Saraf, S.; Bhattacharya, P. Hypoxia and Its Effect on the Cellular System. Cell Biochem. Funct. 2024, 42, e3940. [Google Scholar] [CrossRef]
- Li, M.; Luo, X.; Long, X.; Jiang, P.; Jiang, Q.; Guo, H.; Chen, Z. Potential Role of Mitochondria in Synoviocytes. Clin. Rheumatol. 2021, 40, 447–457. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Fernández-Rodríguez, J.A.; Blanco, F.J. Mitochondrial Dysfunction and Oxidative Stress in Rheumatoid Arthritis. Antioxidants 2022, 11, 1151. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in the Development of Rheumatoid Arthritis. Immunol. Rev. 2020, 294, 177–187. [Google Scholar] [CrossRef]
- Telang, S.; Clem, B.F.; Klarer, A.C.; Clem, A.L.; Trent, J.O.; Bucala, R.; Chesney, J. Small Molecule Inhibition of 6-Phosphofructo-2-Kinase Suppresses t Cell Activation. J. Transl. Med. 2012, 10, 95. [Google Scholar] [CrossRef]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase Deficiency Impairs ATP Generation, Autophagy, and Redox Balance in Rheumatoid Arthritis T Cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring Oxidant Signaling Suppresses Proarthritogenic T Cell Effector Functions in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef] [PubMed]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation—Lessons From Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tian, J.; Wang, S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 1112. [Google Scholar] [CrossRef]
- Gremese, E.; Tolusso, B.; Bruno, D.; Perniola, S.; Ferraccioli, G.; Alivernini, S. The Forgotten Key Players in Rheumatoid Arthritis: IL-8 and IL-17-Unmet Needs and Therapeutic Perspectives. Front. Med. 2023, 10, 956127. [Google Scholar] [CrossRef]
- Sharma, A.; Rudra, D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef]
- Yan, S.; Kotschenreuther, K.; Deng, S.; Kofler, D.M. Regulatory T Cells in Rheumatoid Arthritis: Functions, Development, Regulation, and Therapeutic Potential. Cell Mol. Life Sci. 2022, 79, 533. [Google Scholar] [CrossRef]
- Han, A.; Peng, T.; Xie, Y.; Zhang, W.; Sun, W.; Xie, Y.; Ma, Y.; Wang, C.; Xie, N. Mitochondrial-Regulated Tregs: Potential Therapeutic Targets for Autoimmune Diseases of the Central Nervous System. Front. Immunol. 2023, 14, 1301074. [Google Scholar] [CrossRef]
- Li, P.; Zhou, M.; Wang, J.; Tian, J.; Zhang, L.; Wei, Y.; Yang, F.; Xu, Y.; Wang, G. Important Role of Mitochondrial Dysfunction in Immune Triggering and Inflammatory Response in Rheumatoid Arthritis. J. Inflamm. Res. 2024, 17, 11631–11657. [Google Scholar] [CrossRef]
- Alissafi, T.; Kalafati, L.; Lazari, M.; Filia, A.; Kloukina, I.; Manifava, M.; Lim, J.-H.; Alexaki, V.I.; Ktistakis, N.T.; Doskas, T.; et al. Mitochondrial Oxidative Damage Underlies Regulatory T Cell Defects in Autoimmunity. Cell Metab. 2020, 32, 591–604.e7. [Google Scholar] [CrossRef]
- Volkov, M.; van Schie, K.A.; van der Woude, D. Autoantibodies and B Cells: The ABC of Rheumatoid Arthritis Pathophysiology. Immunol. Rev. 2020, 294, 148–163. [Google Scholar] [CrossRef]
- Rönnelid, J.; Turesson, C.; Kastbom, A. Autoantibodies in Rheumatoid Arthritis—Laboratory and Clinical Perspectives. Front. Immunol. 2021, 12, 685312. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Perdriger, A.; Amé, P. Definition of B Cell Helper T Cells in Rheumatoid Arthritis and Their Behavior during Treatment. Semin. Arthritis Rheum. 2020, 50, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Bankó, Z.; Pozsgay, J.; Szili, D.; Tóth, M.; Gáti, T.; Nagy, G.; Rojkovich, B.; Sármay, G. Induction and Differentiation of IL-10–Producing Regulatory B Cells from Healthy Blood Donors and Rheumatoid Arthritis Patients. J. Immunol. 2017, 198, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wei, K.; Jiang, P.; Zhao, J.; Shan, Y.; Shi, Y.; Zhao, F.; Chang, C.; Li, Y.; Zhou, M.; et al. Macrophage Polarization in Rheumatoid Arthritis: Signaling Pathways, Metabolic Reprogramming, and Crosstalk with Synovial Fibroblasts. Front. Immunol. 2024, 15, 1394108. [Google Scholar] [CrossRef]
- Schett, G.; Gravallese, E. Bone Erosion in Rheumatoid Arthritis: Mechanisms, Diagnosis and Treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Zeisbrich, M.; Yanes, R.E.; Zhang, H.; Watanabe, R.; Li, Y.; Brosig, L.; Hong, J.; Wallis, B.B.; Giacomini, J.C.; Assimes, T.L.; et al. Hypermetabolic Macrophages in Rheumatoid Arthritis and Coronary Artery Disease Due to Glycogen Synthase Kinase 3b Inactivation. Ann. Rheum. Dis. 2018, 77, 1053–1062. [Google Scholar] [CrossRef]
- Li, H.; Feng, Y.; Zheng, X.; Jia, M.; Mei, Z.; Wang, Y.; Zhang, Z.; Zhou, M.; Li, C. M2-Type Exosomes Nanoparticles for Rheumatoid Arthritis Therapy via Macrophage Re-Polarization. J. Control. Release 2022, 341, 16–30. [Google Scholar] [CrossRef]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic Reticulum-Mitochondria Signaling in Neurons and Neurodegenerative Diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation. Int. J. Mol. Sci. 2020, 21, 287. [Google Scholar] [CrossRef]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 Takes Charge during Neutrophil Activation: Impact of PAD4 Mediated NET Formation on Immune-mediated Disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochem. Mosc. 2020, 85, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Oliveira, C.B.; Wang, X.; Navarrete, M.; Barrera-Vargas, A.; Merayo-Chalico, J.; Aljahdali, R.; Aguirre-Aguilar, E.; Carlucci, P.; Kaplan, M.J.; et al. Neutrophil Extracellular Trap-Associated Carbamylation and Histones Trigger Osteoclast Formation in Rheumatoid Arthritis. Ann. Rheum. Dis. 2023, 82, 630–638. [Google Scholar] [CrossRef]
- Schneider, A.H.; Machado, C.C.; Veras, F.P.; Maganin, A.G.d.M.; de Souza, F.F.L.; Barroso, L.C.; de Oliveira, R.D.R.; Alves-Filho, J.C.; Cunha, T.M.; Fukada, S.Y.; et al. Neutrophil Extracellular Traps Mediate Joint Hyperalgesia Induced by Immune Inflammation. Rheumatology 2021, 60, 3461–3473. [Google Scholar] [CrossRef]
- MacDonald, L.; Elmesmari, A.; Somma, D.; Frew, J.; Di Mario, C.; Madhu, R.; Paoletti, A.; Simakou, T.; Hardy, O.M.; Tolusso, B.; et al. Synovial Tissue Myeloid Dendritic Cell Subsets Exhibit Distinct Tissue-Niche Localization and Function in Health and Rheumatoid Arthritis. Immunity 2024, 57, 2843–2862.e12. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W.; Li, W.; Zhao, Y. NLRP3 Inflammasome: Checkpoint Connecting Innate and Adaptive Immunity in Autoimmune Diseases. Front. Immunol. 2021, 12, 732933. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 809806. [Google Scholar] [CrossRef]
- Wu, Q.; Zhong, Z.-M.; Zhu, S.-Y.; Liao, C.-R.; Pan, Y.; Zeng, J.-H.; Zheng, S.; Ding, R.-T.; Lin, Q.-S.; Ye, Q.; et al. Advanced Oxidation Protein Products Induce Chondrocyte Apoptosis via Receptor for Advanced Glycation End Products-Mediated, Redox-Dependent Intrinsic Apoptosis Pathway. Apoptosis 2016, 21, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, Z.; Liu, X. Mitochondrial Dysfunction and Therapeutic Perspectives in Osteoporosis. Front. Endocrinol. 2024, 15, 1325317. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, L.; Duan, X.; Niu, Y.; Li, M.; Yun, L.; Sun, H.; Ma, Y.; Guo, Y. Sirtuins Mediate Mitochondrial Quality Control Mechanisms: A Novel Therapeutic Target for Osteoporosis. Front. Endocrinol. 2023, 14, 1281213. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Jeong, D.; Kang, H.K.; Jung, S.Y.; Kang, S.S.; Min, B.-M. Osteoclast Precursors Display Dynamic Metabolic Shifts toward Accelerated Glucose Metabolism at an Early Stage of RANKL-Stimulated Osteoclast Differentiation. Cell Physiol. Biochem. 2007, 20, 935–946. [Google Scholar] [CrossRef]
- Srinivasan, S.; Koenigstein, A.; Joseph, J.; Sun, L.; Kalyanaraman, B.; Zaidi, M.; Avadhani, N.G. Role of Mitochondrial Reactive Oxygen Species in Osteoclast Differentiation. Ann. N. Y. Acad. Sci. 2010, 1192, 245–252. [Google Scholar] [CrossRef]
- Bertero, E.; Popoiu, T.-A.; Maack, C. Mitochondrial Calcium in Cardiac Ischemia/Reperfusion Injury and Cardioprotection. Basic Res. Cardiol. 2024, 119, 569–585. [Google Scholar] [CrossRef]
- Zhu, Y.-S.; Mo, T.-T.; Jiang, C.; Zhang, J.-N. Osteonectin Bidirectionally Regulates Osteoblast Mineralization. J. Orthop. Surg. Res. 2023, 18, 761. [Google Scholar] [CrossRef]
- Lee, S.-Y.; An, H.-J.; Kim, J.M.; Sung, M.-J.; Kim, D.K.; Kim, H.K.; Oh, J.; Jeong, H.Y.; Lee, Y.H.; Yang, T.; et al. PINK1 Deficiency Impairs Osteoblast Differentiation through Aberrant Mitochondrial Homeostasis. Stem Cell Res. Ther. 2021, 12, 589. [Google Scholar] [CrossRef]
- Al-Azab, M.; Qaed, E.; Ouyang, X.; Elkhider, A.; Walana, W.; Li, H.; Li, W.; Tang, Y.; Adlat, S.; Wei, J.; et al. TL1A/TNFR2-Mediated Mitochondrial Dysfunction of Fibroblast-like Synoviocytes Increases Inflammatory Response in Patients with Rheumatoid Arthritis via Reactive Oxygen Species Generation. FEBS J. 2020, 287, 3088–3104. [Google Scholar] [CrossRef]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of Mitochondrial DNA Escape and Its Relationship with Different Metabolic Diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- Taams, L.S. Interleukin-17 in Rheumatoid Arthritis: Trials and Tribulations. J. Exp. Med. 2020, 217, e20192048. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Hatton, R.D.; Mangan, P.R.; Harrington, L.E. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu. Rev. Immunol. 2007, 25, 821–852. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the Pathogenesis of Rheumatoid Arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Promila, L.; Joshi, A.; Khan, S.; Aggarwal, A.; Lahiri, A. Role of Mitochondrial Dysfunction in the Pathogenesis of Rheumatoid Arthritis: Looking Closely at Fibroblast- like Synoviocytes. Mitochondrion 2023, 73, 62–71. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of Matrix Metalloproteinase Gene Expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix Metalloproteinases: Role in Arthritis. Front. Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Seol, D.-W. The Role of Mitochondria in Apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Hollander, J.M.; Zeng, L. The Emerging Role of Glucose Metabolism in Cartilage Development. Curr. Osteoporos. Rep. 2019, 17, 59–69. [Google Scholar] [CrossRef]
- Wang, S.; Deng, Z.; Ma, Y.; Jin, J.; Qi, F.; Li, S.; Liu, C.; Lyu, F.-J.; Zheng, Q. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 2020, 16, 2675–2691. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Caramés, B.; Martín, M.A.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F.J. Mitochondrial Activity Is Modulated by TNFα and IL-1β in Normal Human Chondrocyte Cells. Osteoarthr. Cartil. 2006, 14, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and Mechanism of the Mitochondrial Ca2+ Uniporter Holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Zhang, J.; Li, Y.; Chen, Y.-X.; Wu, X.-M.; Li, X.; Zhang, X.-F.; Ma, L.-Z.; Yang, Y.-Z.; Zheng, K.-M.; et al. Advanced Oxidation Protein Products Induce G1/G0-Phase Arrest in Ovarian Granulosa Cells via the ROS-JNK/P38 MAPK-P21 Pathway in Premature Ovarian Insufficiency. Oxidative Med. Cell. Longev. 2021, 2021, 6634718. [Google Scholar] [CrossRef]
- Babaahmadi, M.; Tayebi, B.; Gholipour, N.M.; Kamardi, M.T.; Heidari, S.; Baharvand, H.; Eslaminejad, M.B.; Hajizadeh-Saffar, E.; Hassani, S.-N. Rheumatoid Arthritis: The Old Issue, the New Therapeutic Approach. Stem Cell Res. Ther. 2023, 14, 268. [Google Scholar] [CrossRef]
- Arnold, K.; Liao, Y.-E.; Liu, J. Potential Use of Anti-Inflammatory Synthetic Heparan Sulfate to Attenuate Liver Damage. Biomedicines 2020, 8, 503. [Google Scholar] [CrossRef]
- Kitas, G.D.; Nightingale, P.; Armitage, J.; Sattar, N.; Belch, J.J.F.; Symmons, D.P.M.; Consortium, T.R.C.R. A Multicenter, Randomized, Placebo-Controlled Trial of Atorvastatin for the Primary Prevention of Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1437–1449. [Google Scholar] [CrossRef]
- Sjúrðarson, T.; Larsen, S.; Jensen, S.B.K.; Bejder, J.; Rasmussen, J.; Á Borg, S.; Kristiansen, J.; Meinhardsson, J.M.; Olsen, H.W.; Ellingsgaard, H.; et al. Investigating the Combined Effects of Statins and Exercise on Skeletal Muscle Mitochondrial Content and Function, Cardiorespiratory Fitness and Quality of Life in Individuals with Dyslipidaemia: Protocol for a Randomised Placebo-Controlled Trial. BMJ Open 2025, 15, e101425. [Google Scholar] [CrossRef]
- Promila, L.; Sarkar, K.; Guleria, S.; Rakshit, A.; Rathore, M.; Singh, N.C.; Khan, S.; Tomar, M.S.; Ammanathan, V.; Barthwal, M.K.; et al. Mitochondrial Calcium Uniporter Regulates Human Fibroblast-like Synoviocytes Invasion via Altering Mitochondrial Dynamics and Dictates Rheumatoid Arthritis Pathogenesis. Free Radic. Biol. Med. 2025, 234, 55–71. [Google Scholar] [CrossRef]
- Peng, Y.; Lian, X.; Wang, Y.; Li, X. Matrine, a Potential Drug for the Treatment of Rheumatoid Arthritis. Rheumatol. Autoimmun. 2024, 4, 11–19. [Google Scholar] [CrossRef]
- Benjamin, O.; Goyal, A.; Lappin, S.L. Disease-Modifying Antirheumatic Drugs (DMARD). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Heidari, R.; Ahmadi, A.; Mohammadi, H.; Ommati, M.M.; Azarpira, N.; Niknahad, H. Mitochondrial Dysfunction and Oxidative Stress Are Involved in the Mechanism of Methotrexate-Induced Renal Injury and Electrolytes Imbalance. Biomed. Pharmacother. 2018, 107, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Al Maruf, A.; O’Brien, P.J.; Naserzadeh, P.; Fathian, R.; Salimi, A.; Pourahmad, J. Methotrexate Induced Mitochondrial Injury and Cytochrome c Release in Rat Liver Hepatocytes. Drug Chem. Toxicol. 2018, 41, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Hemshekhar, M.; Thushara, R.M.; Sundaram, M.S.; NaveenKumar, S.K.; Naveen, S.; Devaraja, S.; Somyajit, K.; West, R.; Basappa; et al. Methotrexate Promotes Platelet Apoptosis via JNK-Mediated Mitochondrial Damage: Alleviation by N-Acetylcysteine and N-Acetylcysteine Amide. PLoS ONE 2015, 10, e0127558. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Ren, Z.; Qing, T.; Couch, L.; Shi, L.; Tolleson, W.H.; Guo, L. Mitochondrial Dysfunction Induced by Leflunomide and Its Active Metabolite. Toxicology 2018, 396–397, 33–45. [Google Scholar] [CrossRef]
- Niknahad, H.; Heidari, R.; Mohammadzadeh, R.; Ommati, M.M.; Khodaei, F.; Azarpira, N.; Abdoli, N.; Zarei, M.; Asadi, B.; Rasti, M.; et al. Sulfasalazine Induces Mitochondrial Dysfunction and Renal Injury. Ren. Fail. 2017, 39, 745–753. [Google Scholar] [CrossRef]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.J.; Fearon, U. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, S.-H.; Lee, S.W.; Lee, S.H.; Son, M.K.; Choi, Y.H.; Chung, W.T.; Yoo, Y.H. Synoviocyte Apoptosis May Differentiate Responder and Non-Responder Patients to Methotrexate Treatment in Rheumatoid Arthritis. Arch. Pharm. Res. 2014, 37, 1286–1294. [Google Scholar] [CrossRef]
- Gaies, E.; Jebabli, N.; Trabelsi, S.; Salouage, I.; Charfi, R.; Lakhal, M.; Klouz, A. Methotrexate Side Effects: Review Article. J. Drug Metab. Toxicol. 2012, 3, 4. [Google Scholar] [CrossRef]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Eschborn, M.; Lindner, M.; Liebmann, M.; Herold, M.; Janoschka, C.; Torres Garrido, B.; Schulte-Mecklenbeck, A.; Gross, C.C.; Breuer, J.; et al. Teriflunomide Treatment for Multiple Sclerosis Modulates T Cell Mitochondrial Respiration with Affinity-Dependent Effects. Sci. Transl. Med. 2019, 11, eaao5563. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-Orotate Dehydrogenase Is Physically Associated with the Respiratory Complex and Its Loss Leads to Mitochondrial Dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef]
- Liptay, S.; Fulda, S.; Schanbacher, M.; Bourteele, S.; Ferri, K.F.; Kroemer, G.; Adler, G.; Debatin, K.M.; Schmid, R.M. Molecular Mechanisms of Sulfasalazine-Induced T-Cell Apoptosis. Br. J. Pharmacol. 2002, 137, 608–620. [Google Scholar] [CrossRef]
- Jie, L.; Du, H.; Huang, Q.; Wei, S.; Huang, R.; Sun, W. Tanshinone IIA Induces Apoptosis in Fibroblast-Like Synoviocytes in Rheumatoid Arthritis via Blockade of the Cell Cycle in the G2/M Phase and a Mitochondrial Pathway. Biol. Pharm. Bull. 2014, 37, 1366–1372. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Zhang, Y.; Zhang, J.; Cao, W.; Chen, X. Effect of Lentivirus-Mediated Overexpression or Silencing of MnSOD on Apoptosis of Resveratrol-Treated Fibroblast-like Synoviocytes in Rheumatoid Arthritis. Eur. J. Pharmacol. 2019, 844, 65–72. [Google Scholar] [CrossRef]
- Parisi, S.; Bortoluzzi, A.; Sebastiani, G.D.; Conti, F.; Caporali, R.; Ughi, N.; Prevete, I.; Ariani, A.; Manara, M.; Carrara, G.; et al. The Italian Society for Rheumatology Clinical Practice Guidelines for Rheumatoid Arthritis. Reumatismo 2019, 71, 22–49. [Google Scholar] [CrossRef]
- Pang, M.; Sun, Z.; Zhang, H. Biologic DMARDs and Targeted Synthetic DMARDs and the Risk of All-Cause Mortality in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Medicine 2022, 101, e29838. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, Oxidative Stress and Inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Costa, N.T.; Iriyoda, T.M.V.; Alfieri, D.F.; Simão, A.N.C.; Dichi, I. Influence of Disease-Modifying Antirheumatic Drugs on Oxidative and Nitrosative Stress in Patients with Rheumatoid Arthritis. Inflammopharmacology 2018, 26, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Saki, A.; Rajaei, E.; Rahim, F. Safety and Efficacy of Tocilizumab for Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Clinical Trial Studies. Reumatologia 2021, 59, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Parisi, S.; Ditto, M.C.; Ghellere, F.; Panaro, S.; Piccione, F.; Borrelli, R.; Fusaro, E. Update on Tocilizumab in Rheumatoid Arthritis: A Narrative Review. Front. Immunol. 2025, 16, 1470488. [Google Scholar] [CrossRef] [PubMed]
- Biniecka, M.; Kennedy, A.; Ng, C.T.; Chang, T.C.; Balogh, E.; Fox, E.; Veale, D.J.; Fearon, U.; O’Sullivan, J.N. Successful Tumour Necrosis Factor (TNF) Blocking Therapy Suppresses Oxidative Stress and Hypoxia-Induced Mitochondrial Mutagenesis in Inflammatory Arthritis. Arthritis Res. Ther. 2011, 13, R121. [Google Scholar] [CrossRef]
- Meugnier, E.; Coury, F.; Tebib, J.; Ferraro-Peyret, C.; Rome, S.; Bienvenu, J.; Vidal, H.; Sibilia, J.; Fabien, N. Gene Expression Profiling in Peripheral Blood Cells of Patients with Rheumatoid Arthritis in Response to Anti-TNF-α Treatments. Physiol. Genom. 2011, 43, 365–371. [Google Scholar] [CrossRef]
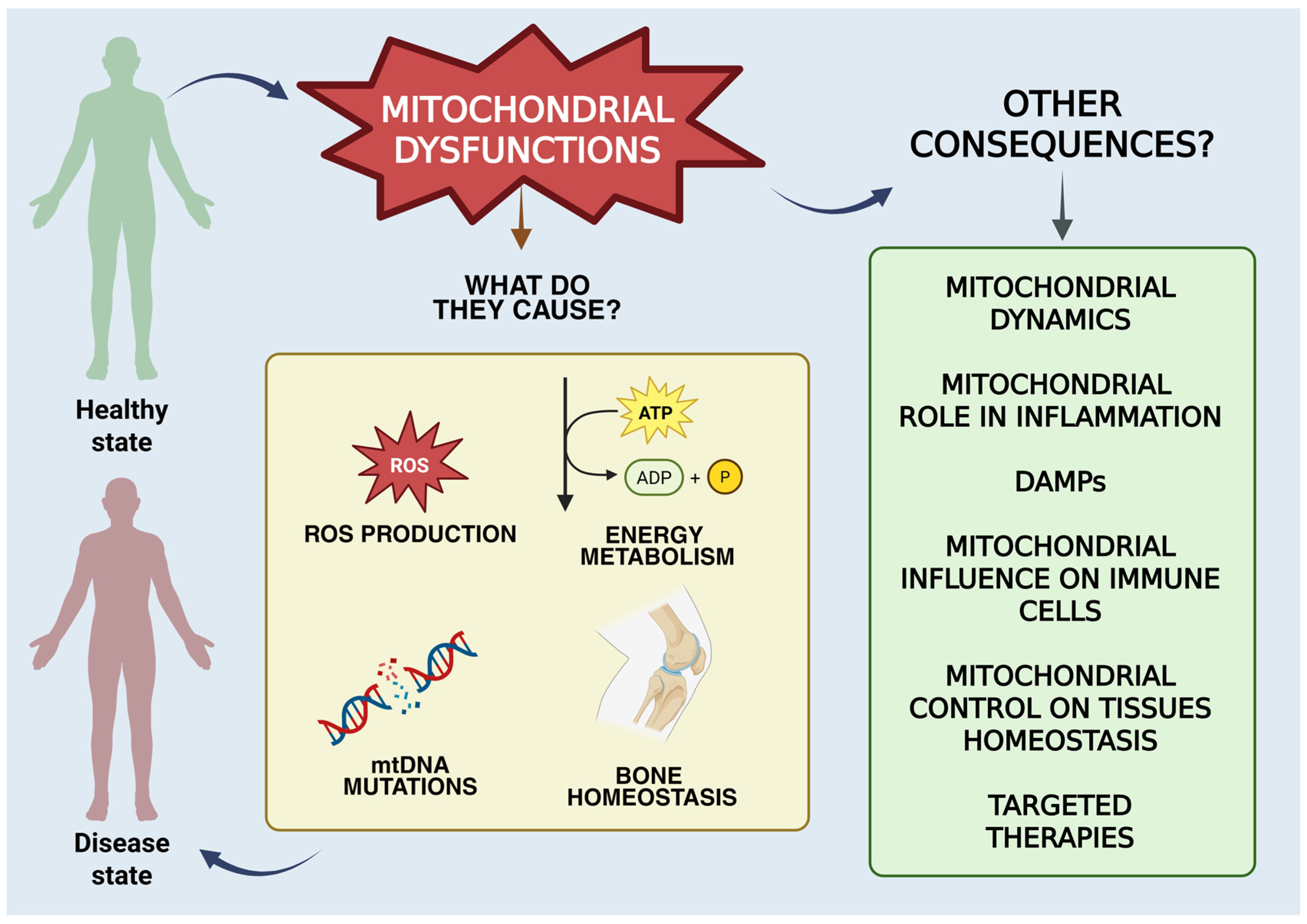
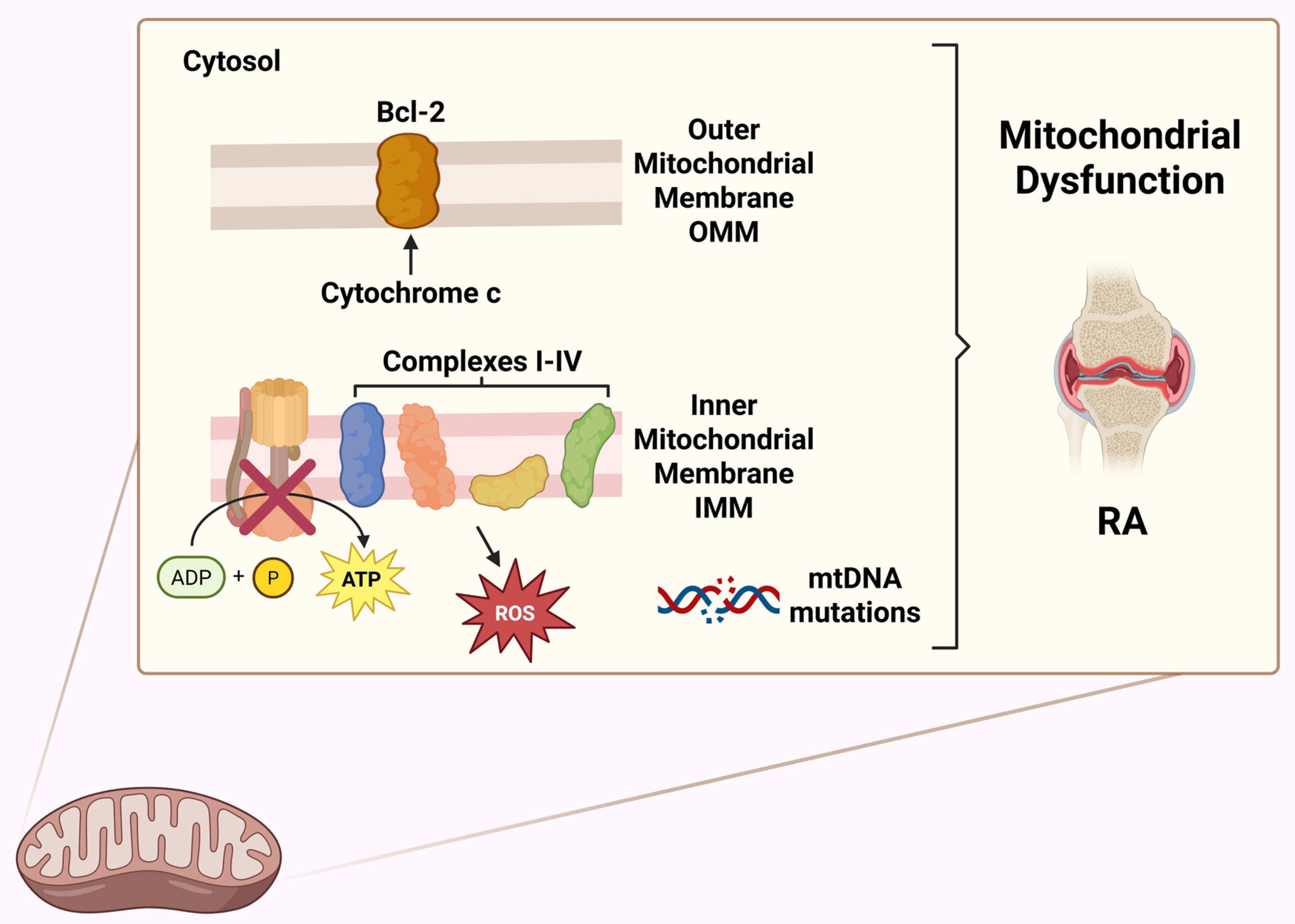

| Mitochondrial Gene/Locus | Mutation Type/Description | Role in RA |
|---|---|---|
| mtDNA | Increased frequency of somatic mutations in mtDNA in synovial tissue. | Mitochondrial dysfunction, increased ROS and release of mtDAMPs, driving chronic synovial inflammation. Mutations also lead to impaired ATP production and altered cellular signalling [57]. |
| MT-ND1 (NADH dehydrogenase 1) | Somatic mutations in this gene, encoding subunit one of Complex I of the electron transport chain. | Mutations in MT-ND1 can lead to dysfunctional Complex I, impairing oxidative phosphorylation and increasing ROS production [162]. |
| Mitochondrial D-loop Region | Various point mutations and deletions. | The D-loop is a non-coding region involved in mtDNA replication and transcription. Mutations here can affect mtDNA copy number and overall mitochondrial function [163]. |
| Genes encoding tRNA or rRNA | Point mutations in genes for mitochondrial transfer RNAs (tRNAs) or ribosomal RNAs (rRNAs). | Mutations in these genes can impair mitochondrial protein synthesis, leading to widespread defects in the electron transport chain complexes and other mitochondrial proteins [164]. |
| Genes of the ETC | Mutations in genes encoding other subunits of Complexes I-IV and ATP synthase. | Dysfunction in any part of the ETC can lead to inefficient ATP production and increased electron leakage, resulting in higher ROS levels, worsening the inflammatory state [7]. |
| Patient (P) | Intervention (I) | Comparison (C) | Outcome (O) | Study Design | Trial Registration Number |
|---|---|---|---|---|---|
| RA patients | Drugs that directly modulate mitochondrial dynamics | Placebo or standard therapy | Reduction in disease severity, inhibition of inflammatory mediators. Restoration of mitochondrial morphology, reduction in migration and invasiveness. Mainly pre-clinical phase [57]. | Preclinical studies (mouse models of CIA) and in vitro studies (RA-FLS) [57]. | Currently unavailable |
| RA patients | JAK inhibitors | Placebo or conventional/biologic DMARDs | Improved patient-reported outcomes. Mitochondrial implications: some studies suggest that JAK inhibitors may modulate the energy metabolism of immune cells, including mitochondria. Status: numerous trials completed with positive results and approved drugs [237]. | Preclinical studies [237]. | NCT01359150 (Tofacitinib) NCT01721057 (Baricitinib) NCT02706852 (Upadacitinib) |
| RA patients | Statins (e.g., Atorvastatin) | Placebo or standard therapy | Statins may modulate mitochondrial function (e.g., reduction in ROS formation and decreased swollen joints). Status: research is focused on modulating mitochondrial dynamics as the primary endpoint [238,239]. | In vitro and preclinical studies [238,239]. | NCT06841536 (Statins) ISRCTN41829447 (Atorvastatin) |
| RA patients | Targeting the mitochondrial calcium uniporter (MCU) | Standard therapy | Inhibition of MCU reduced migration and invasiveness of RA-FLS, restored altered mitochondrial dynamics and reduced mitochondrial ROS. Status: primarily preclinical research not registered [240]. | Preclinical studies (mouse and FLS models of RA [240]. | Currently unavailable |
| RA patients | Matrine (traditional Chinese medicine compound) | Placebo or standard therapy | Evidence of efficacy in animal models and some initial clinical studies in RA. Status: research in progress [241]. | Limited preclinical and clinical studies [241]. | No specific registered trials were found |
| Category | Drug | Main Side Effects |
|---|---|---|
| Conventional Synthetic DMARDs | Methotrexate | Nephrotoxic [243,244,245] Hepatotoxic [243,244,245] |
| Leflunomide | Hepatotoxic [246] | |
| Sulfasalazine | Nephrotoxic [247] | |
| Targeted Synthetic DMARDs | Tofacitinib | Headaches [248] Nausea [248] Risk of infections [248] |
| Biological DMARDs | Tocilizumab | Subcutaneous tissue disorders [232] Neutropenia [233] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iaconis, A.; Molinari, F.; Fusco, R.; Di Paola, R. Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease. Biomedicines 2025, 13, 1708. https://doi.org/10.3390/biomedicines13071708
Iaconis A, Molinari F, Fusco R, Di Paola R. Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease. Biomedicines. 2025; 13(7):1708. https://doi.org/10.3390/biomedicines13071708
Chicago/Turabian StyleIaconis, Antonella, Francesco Molinari, Roberta Fusco, and Rosanna Di Paola. 2025. "Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease" Biomedicines 13, no. 7: 1708. https://doi.org/10.3390/biomedicines13071708
APA StyleIaconis, A., Molinari, F., Fusco, R., & Di Paola, R. (2025). Mitochondria as a Disease-Relevant Organelle in Rheumatoid Arthritis: A Key Breakout in Fight Against the Disease. Biomedicines, 13(7), 1708. https://doi.org/10.3390/biomedicines13071708