

Unidirectional Crosstalk Between NTRK1 and IGF2 Drives ER Stress in Chronic Pain

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design Overview
2.2. Animals and Ethical Compliance
2.3. Skin/Muscle Incision and Retraction (SMIR) Model
2.4. Intrathecal Catheterization
2.5. Drug Administration
2.6. Mechanical Allodynia Assessment
2.7. Quantitative Real-Time PCR (qRT-PCR)
2.8. Western Blot Analysis
2.9. Immunofluorescence Staining
2.10. Experimental Designs and Animal Groups
- Experiment 1: Assessment of NTRK1, ER stress, and IGF2 alterations in the spinal cord of rats with SMIR.
- Experiment 2: Evaluation of NTRK1 inhibitor effects on mechanical allodynia and ER stress expression in rats with SMIR.
- Experiment 3: Determination of the effective dose of IGF2 siRNA on mechanical allodynia in rats with SMIR
- Experiment 4: Comparison of the effects of NTRK1 inhibitor and IGF2 siRNA on the expression of IGF2 and NTRK1, and their relationship in rats with SMIR
2.11. Statistical Analyses
3. Results
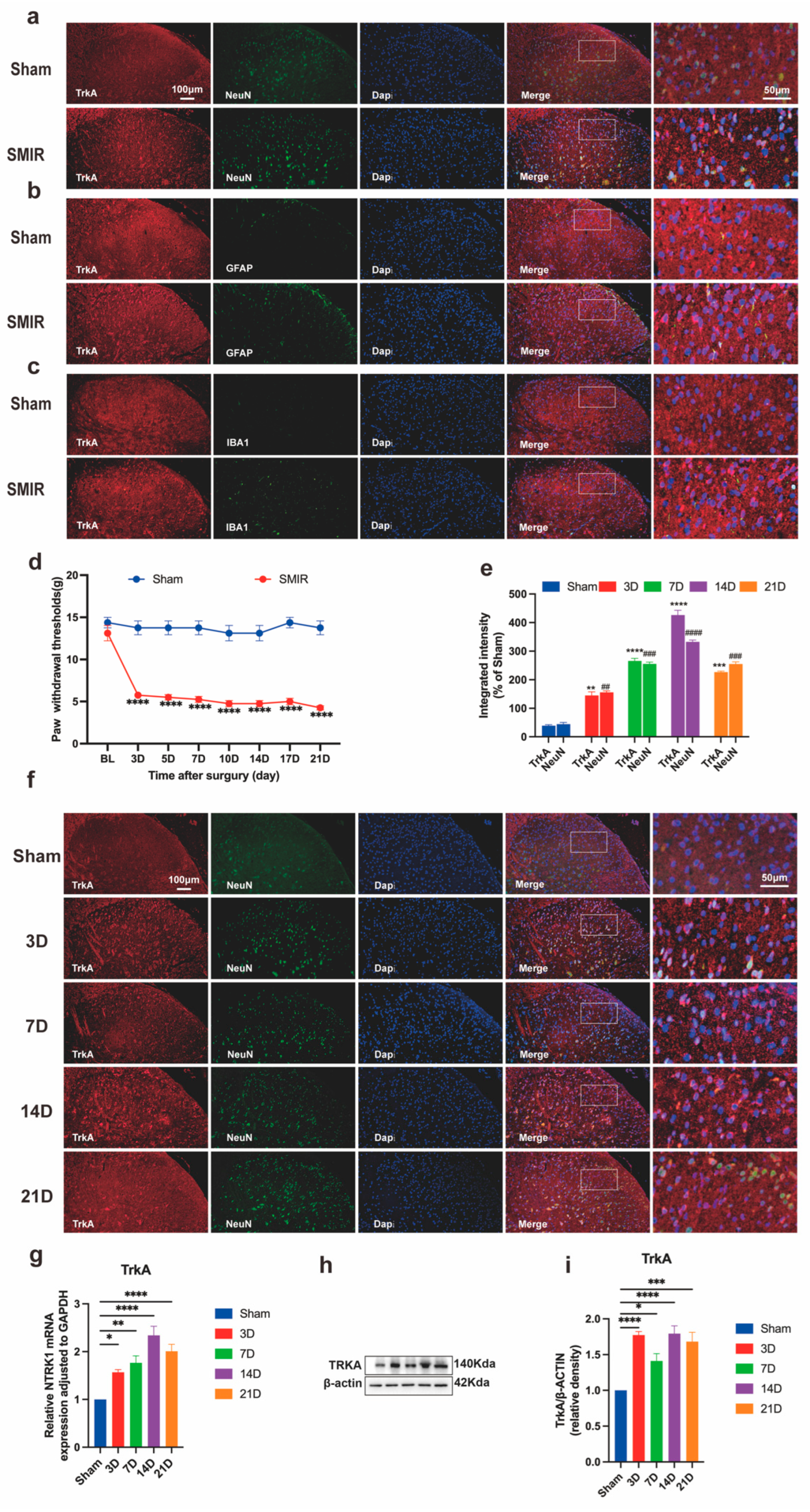
3.1. Temporal Upregulation of Neuronal NTRK1 Drives CPSP Pathogenesis
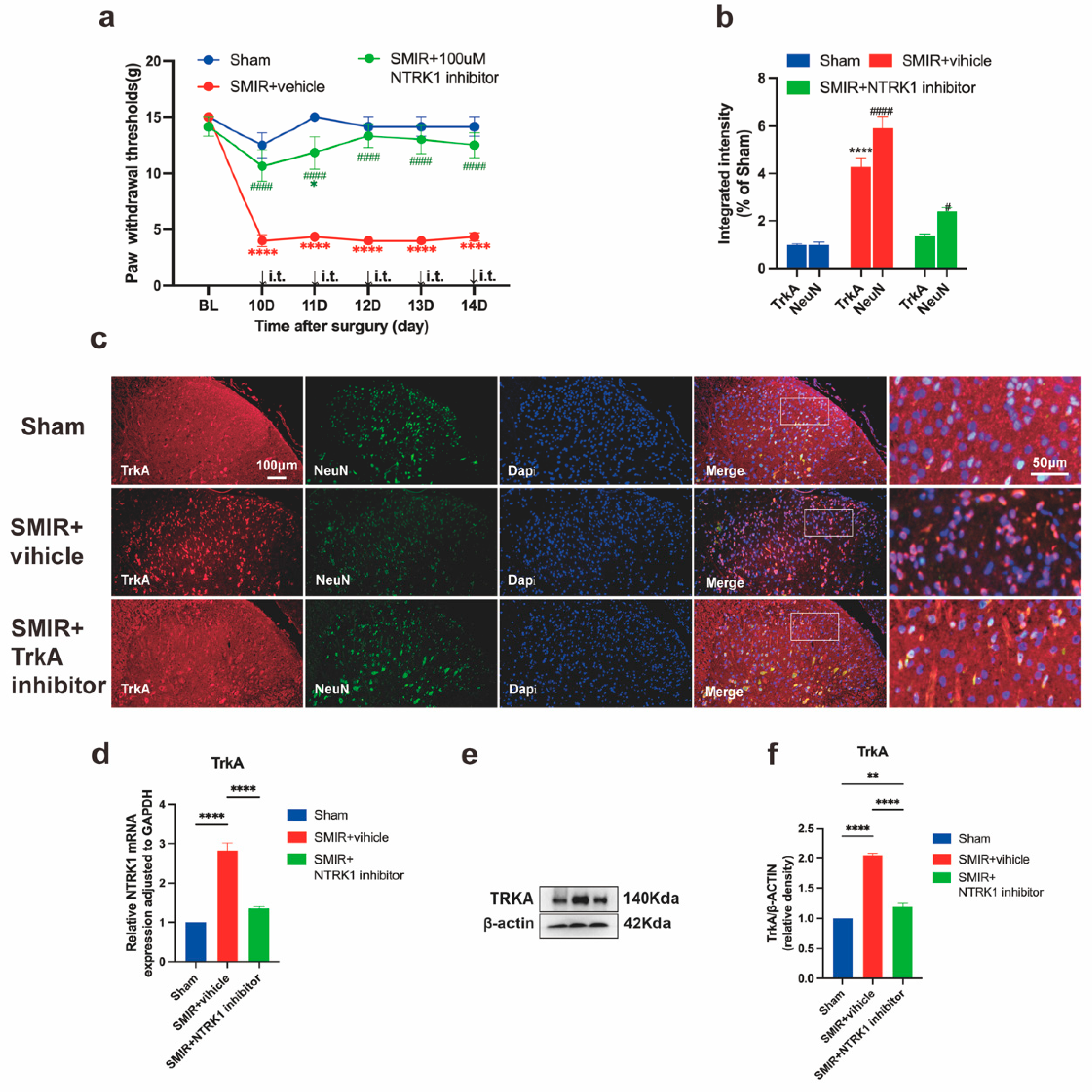
3.2. NTRK1 Inhibition Reverses Mechanical Hypersensitivity
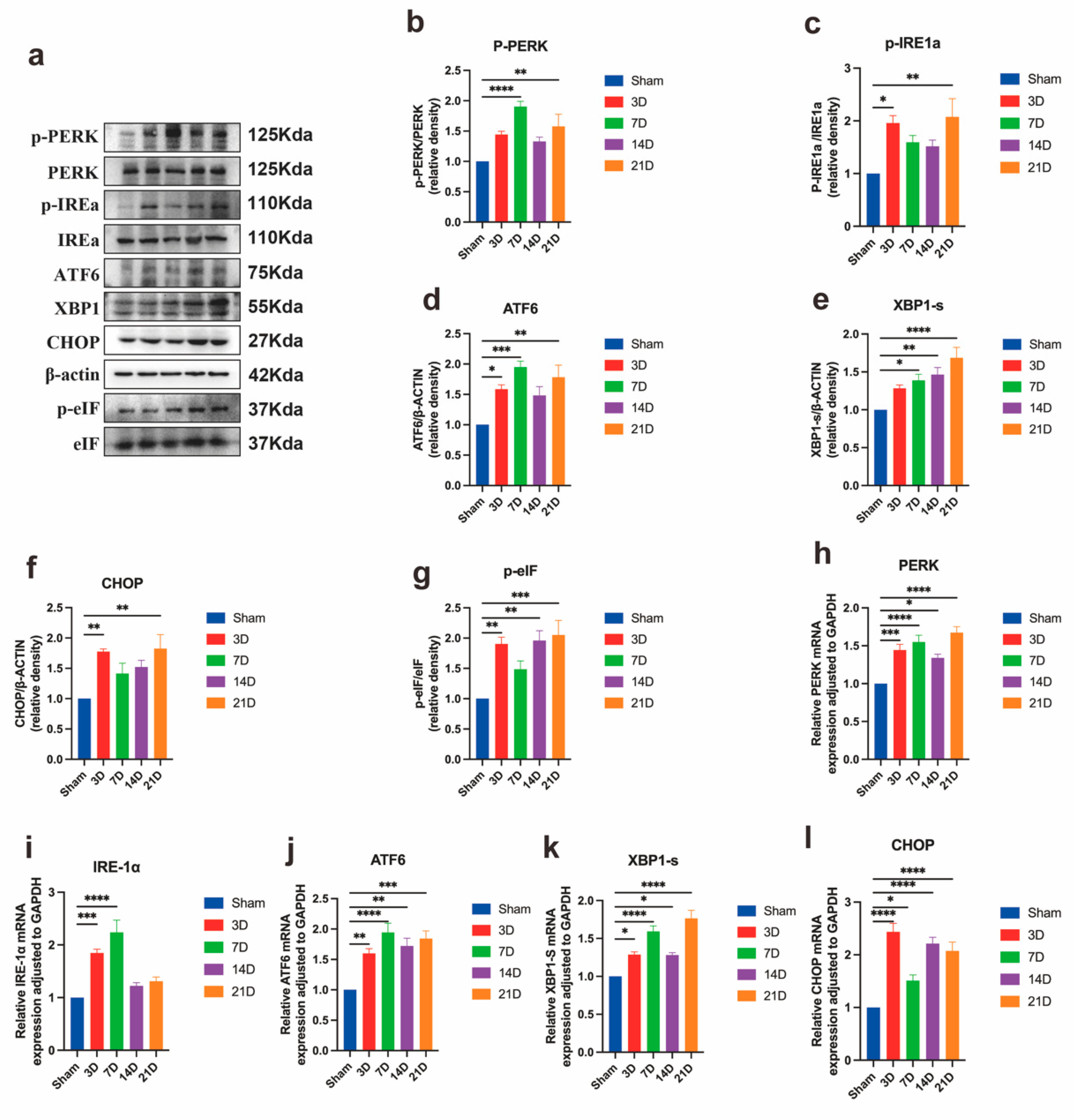
3.3. Neuronal ER Stress Activation Underlies CPSP Pathophysiology
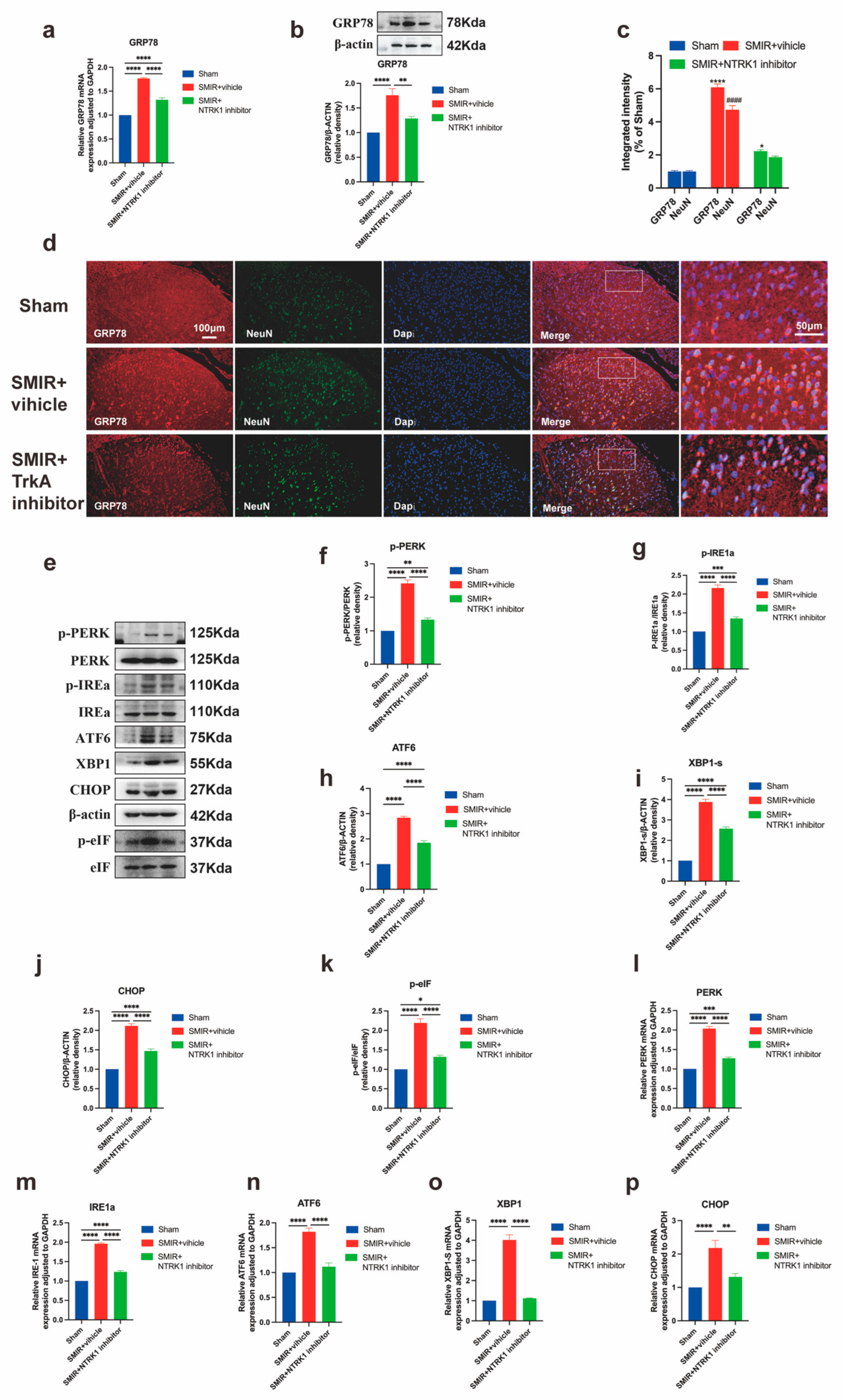
3.4. NTRK1 Inhibition Attenuates ER Stress in Spinal Dorsal Horn
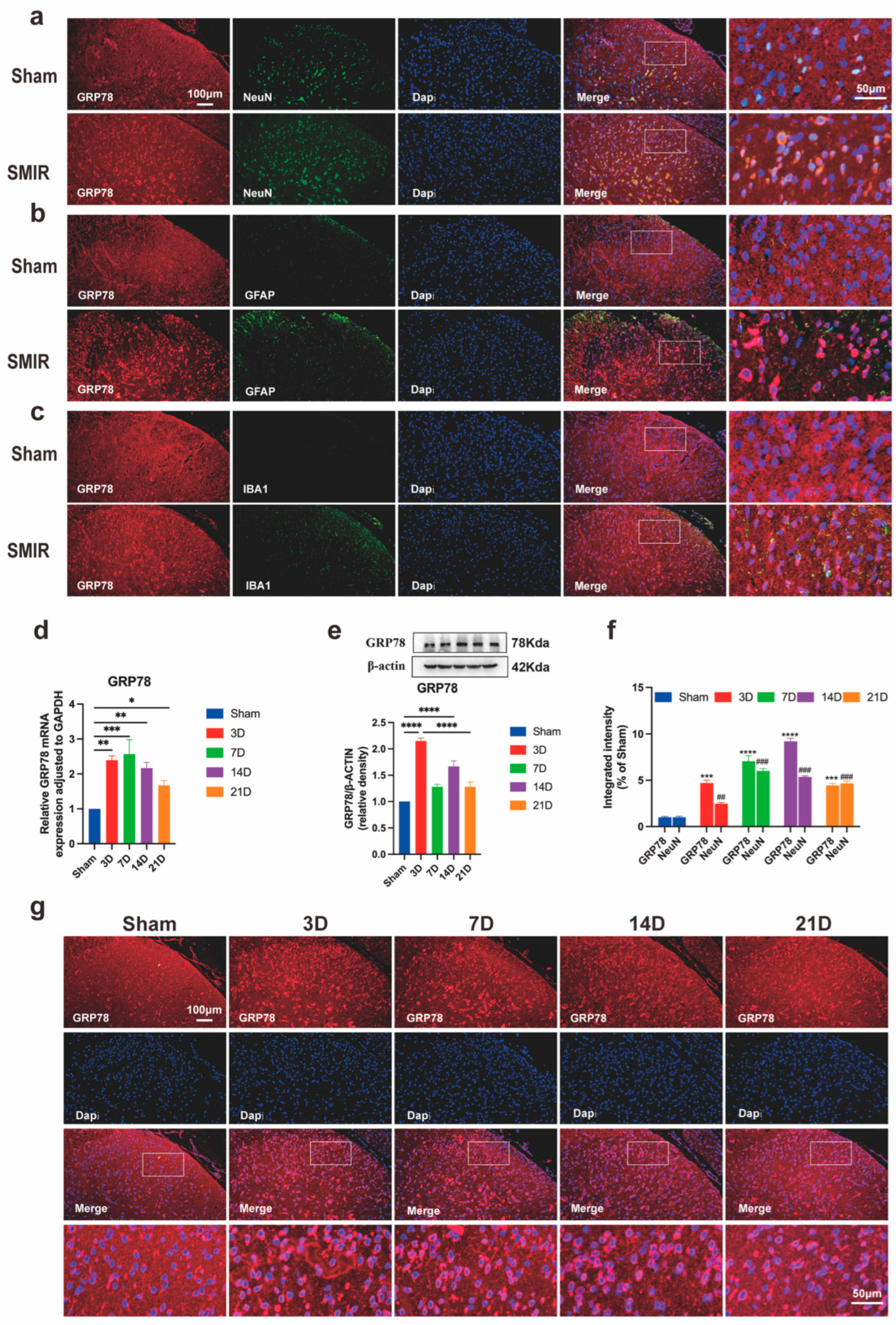
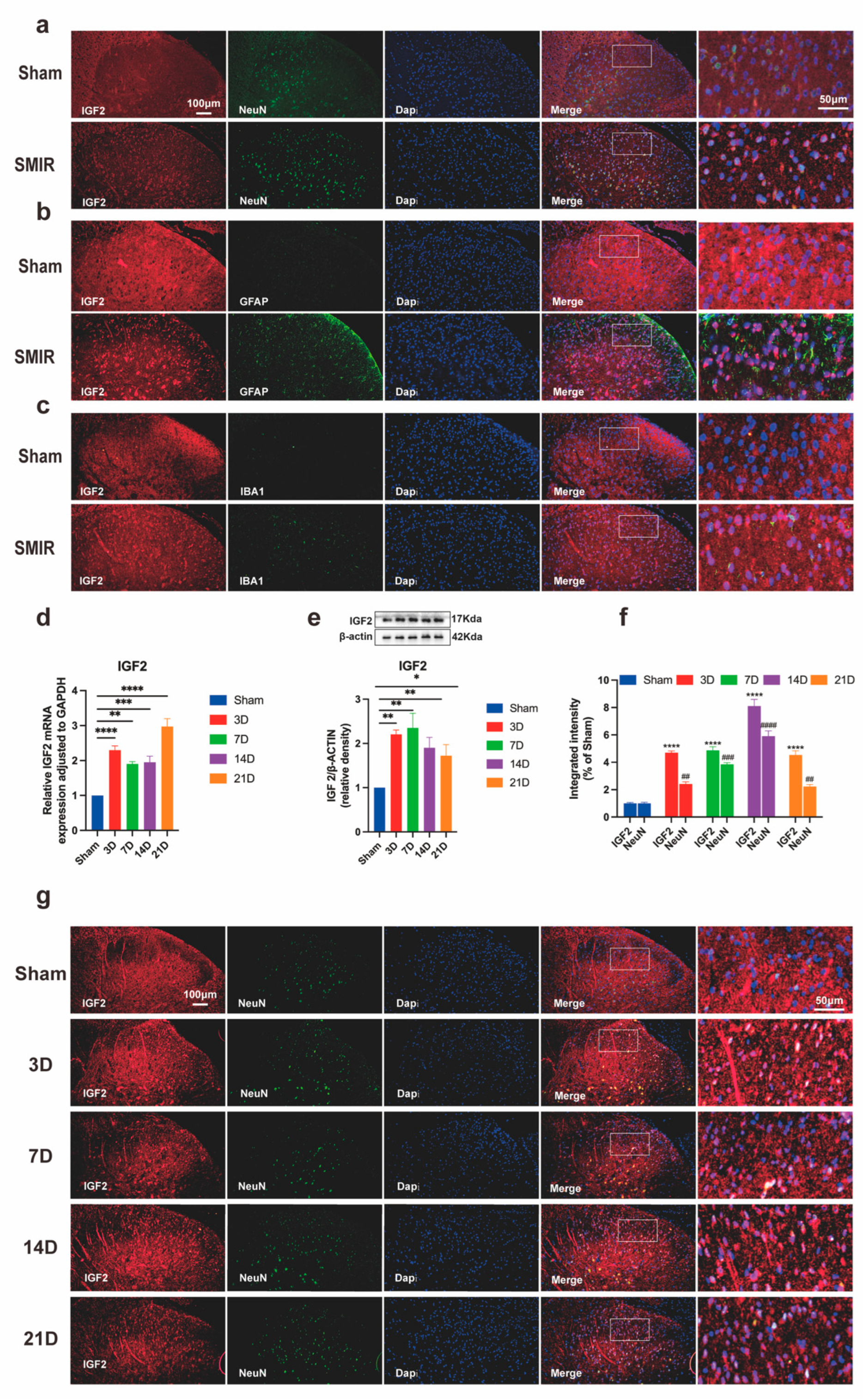
3.5. SMIR Surgery Upregulates Neuronal IGF2 in Spinal Dorsal Horn
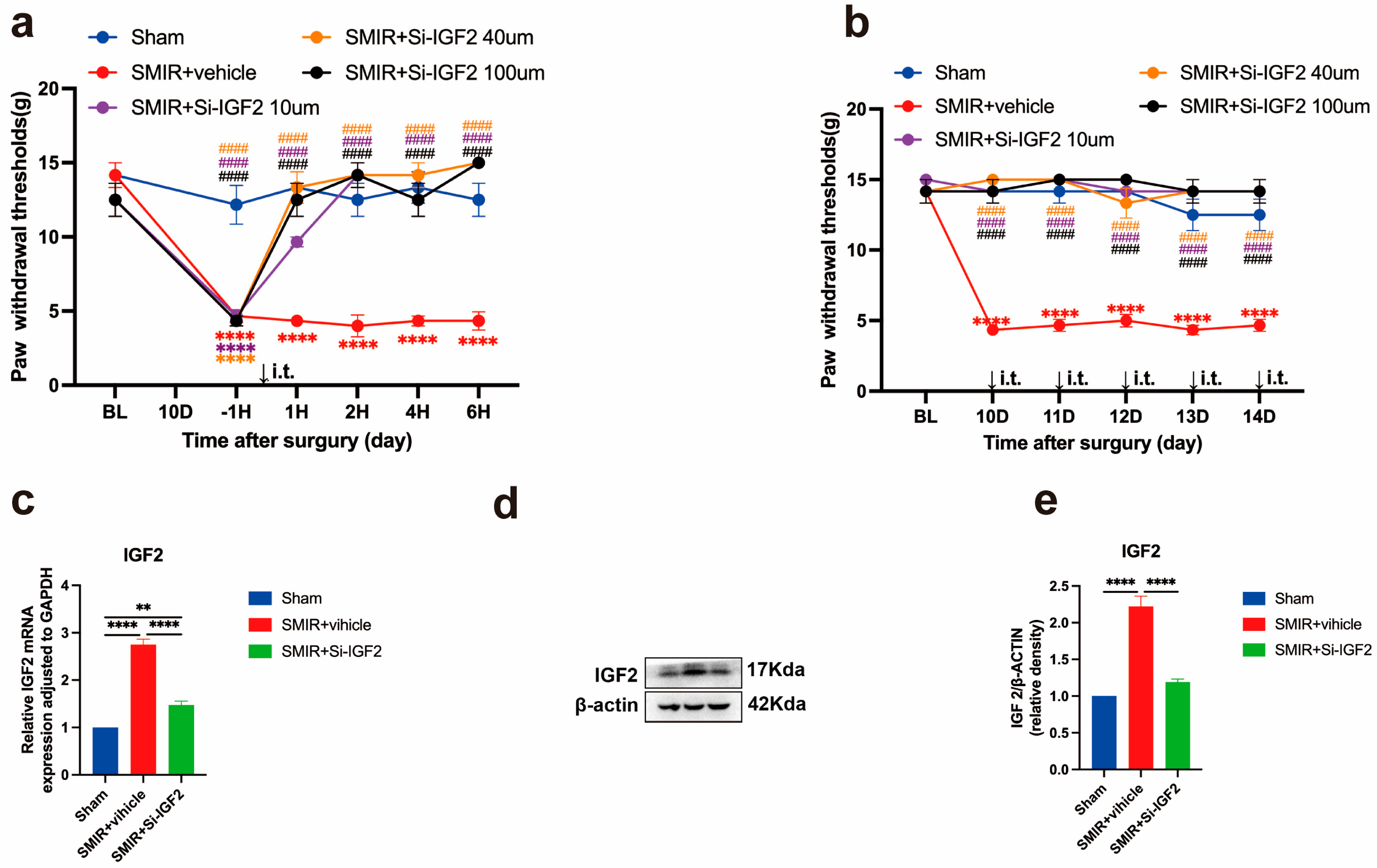
3.6. IGF2 Silencing Reverses Mechanical Hypersensitivity
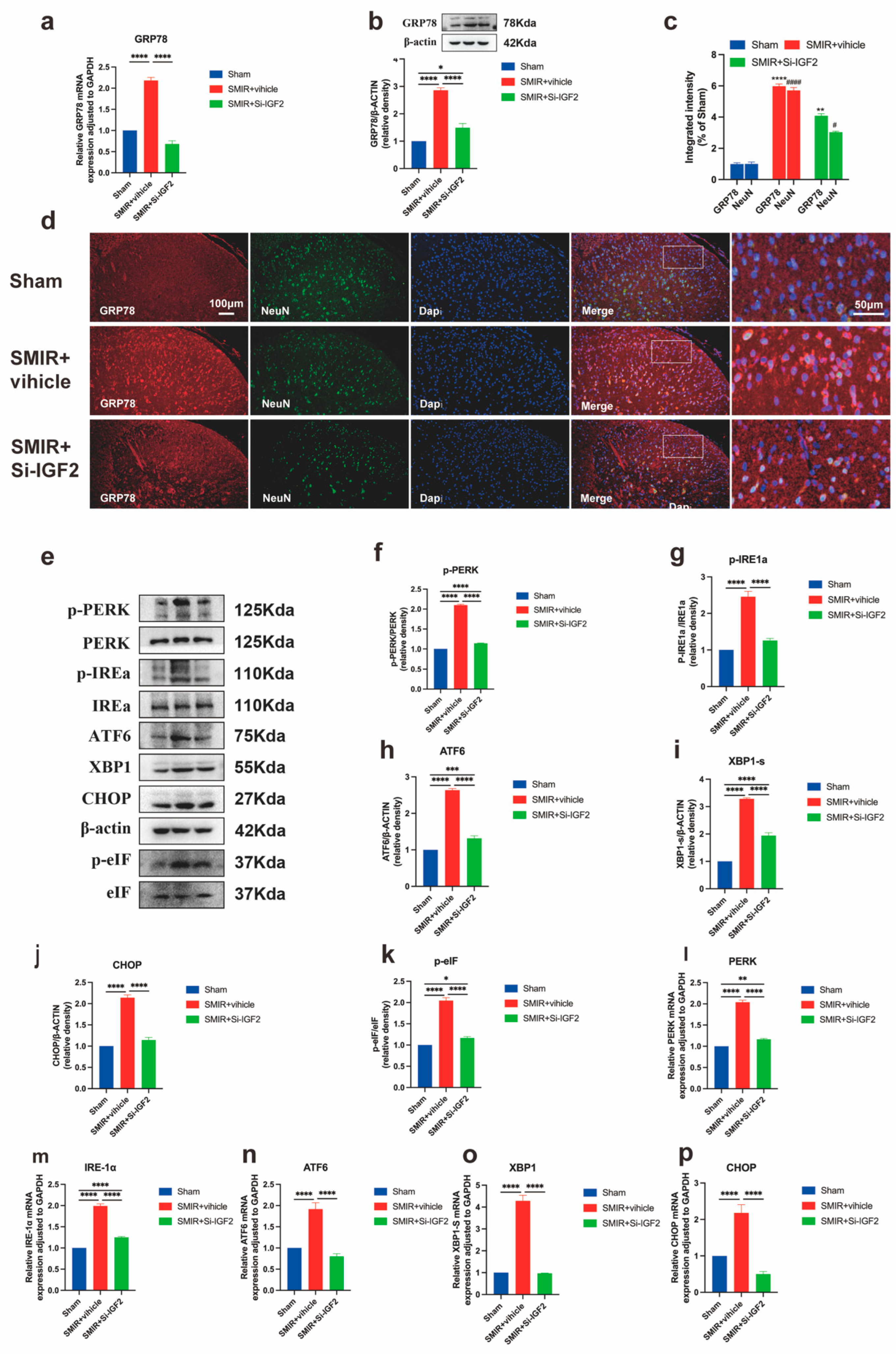
3.7. IGF2 Knockdown Attenuates ER Stress via GRP78 and UPR Pathways
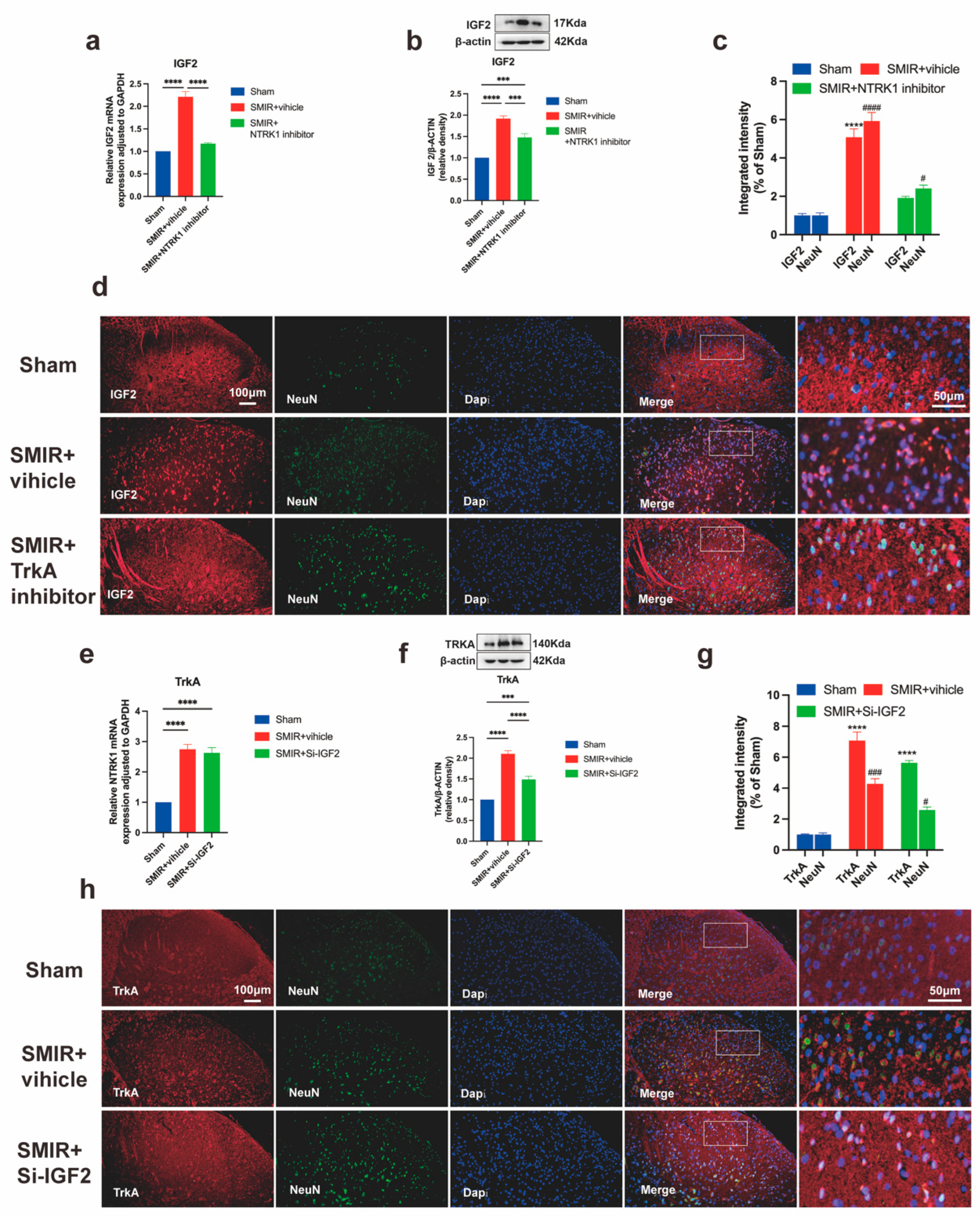
3.8. Unidirectional NTRK1→IGF2 Regulation in CPSP Pathogenesis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF6 | Activating Transcription Factor 6 |
| CHOP | C/EBP Homologous Protein C/EBP |
| CPSP | Chronic Post-surgical Pain |
| e-IF2α | Eukaryotic Initiation Factor 2α |
| ER stress | Endoplasmic Reticulum stress |
| GRP78 | Glucose-Regulated Protein 78 |
| IF | Immunofluorescence |
| IGF2 | Insulin-Like Growth Factor II |
| IRE1α | Inositol-Requiring Enzyme 1α |
| PERK | PKR-like ER Kinase |
| RT-qPCR | Real Time Quantitative polymerase chain reaction |
| MPWT | Mechanical paw withdraw threshold |
| NTRK1 | Neurotrophic Tyrosine Kinase Receptor Type 1 |
| SMIR | Skin/Muscle Incision and Retraction |
| TrkA | Tyrosine kinase receptor A |
| UPR | Unfolded Protein Response |
| WB | Western Blot |
| XBP1 | X-box Binding Protein 1 X |
References
- Kehlet, H.; Jensen, T.S.; Woolf, C.J. Persistent postsurgical pain: Risk factors and prevention. Lancet 2006, 367, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Steyaert, A.; De Kock, M. Chronic postsurgical pain. Curr. Opin. Anesthesiol. 2012, 25, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Treede, R.D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.F.; Plumb, A.N.; Berardi, G.; Sluka, K.A. Sex differences in the transition to chronic pain. J. Clin. Investig. 2025, 135, e191931. [Google Scholar] [CrossRef]
- Varallo, G.; Giusti, E.M.; Manna, C.; Castelnuovo, G.; Pizza, F.; Franceschini, C.; Plazzi, G. Sleep disturbances and sleep disorders as risk factors for chronic postsurgical pain: A systematic review and meta-analysis. Sleep. Med. Rev. 2022, 63, 101630. [Google Scholar] [CrossRef]
- Su, H.P.; Rickert, K.; Burlein, C.; Narayan, K.; Bukhtiyarova, M.; Hurzy, D.M.; Stump, C.A.; Zhang, X.; Reid, J.; Krasowska-Zoladek, A.; et al. Structural characterization of nonactive site, TrkA-selective kinase inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, E297–E306. [Google Scholar] [CrossRef]
- Solomon, J.P.; Benayed, R.; Hechtman, J.F.; Ladanyi, M. Identifying patients with NTRK fusion cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef]
- Apostolou, A.; Shen, Y.; Liang, Y.; Luo, J.; Fang, S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp. Cell Res. 2008, 314, 2454–2467. [Google Scholar] [CrossRef]
- He, Q.; Wang, T.; Ni, H.; Liu, Q.; An, K.; Tao, J.; Chen, Y.; Xu, L.; Zhu, C.; Yao, M. Endoplasmic reticulum stress promoting caspase signaling pathway-dependent apoptosis contributes to bone cancer pain in the spinal dorsal horn. Mol. Pain 2019, 15, 1744806919876150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef]
- Lin, T.T.; Qu, J.; Wang, C.Y.; Yang, X.; Hu, F.; Hu, L.; Wu, X.F.; Jiang, C.Y.; Liu, W.T.; Han, Y. Rescue of HSP70 in Spinal Neurons Alleviates Opioids-Induced Hyperalgesia via the Suppression of Endoplasmic Reticulum Stress in Rodents. Front. Cell Dev. Biol. 2020, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zhang, F.; Feng, N.; Kuang, B.; Fan, M.; Chen, C.; Pan, Y.; Zhou, P.; Geng, N.; Li, X.; et al. IRE1α protects against osteoarthritis by regulating progranulin-dependent XBP1 splicing and collagen homeostasis. Exp. Mol. Med. 2023, 55, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Zuo, J.; Fang, Q.; Yuan, M.; Xia, Y.; Jin, Q.; Liu, Y.; Wang, Y.; Zhang, Z.; Liu, W.; et al. Erbin protects against sepsis-associated encephalopathy by attenuating microglia pyroptosis via IRE1α/Xbp1s-Ca(2+) axis. J. Neuroinflamm. 2022, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Zhang, C.; Jiao, B.; Cao, X.; Zhang, W.; Yu, S.; Zhang, K.; Zhang, M.; Zhang, X. NTRK1-mediated protection against manganese-induced neurotoxicity and cell apoptosis via IGF2 in SH-SY5Y cells. Biomed. Pharmacother. 2023, 169, 115889. [Google Scholar] [CrossRef]
- Rosenberger, D.C.; Pogatzki-Zahn, E.M. Chronic Post-surgical Pain—Update on incidence, risk factors and preventive treatment options. BJA Educ. 2022, 22, 190–196. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; LeQuang, J.A.; Magnusson, P.; Varrassi, G. Identifying risk factors for chronic postsurgical pain and preventive measures: A comprehensive update. Expert Rev. Neurother. 2023, 23, 1297–1310. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflamm. 2009, 6, 41. [Google Scholar] [CrossRef]
- An, J.; Zhang, X.; Jia, K.; Zhang, C.; Zhu, L.; Cheng, M.; Li, F.; Zhao, S.; Hao, J. Trichostatin A increases BDNF protein expression by improving XBP-1s/ATF6/GRP78 axis in Schwann cells of diabetic peripheral neuropathy. Biomed. Pharmacother. 2021, 133, 111062. [Google Scholar] [CrossRef]
- Zhang, S.; Tian, W.; Duan, X.; Zhang, Q.; Cao, L.; Liu, C.; Li, G.; Wang, Z.; Zhang, J.; Li, J.; et al. Melatonin attenuates diabetic cardiomyopathy by increasing autophagy of cardiomyocytes via regulation of VEGF-B/GRP78/PERK signaling pathway. Cardiovasc. Diabetol. 2024, 23, 19. [Google Scholar] [CrossRef]
- Zhou, F.; Zhang, W.; Zhou, J.; Li, M.; Zhong, F.; Zhang, Y.; Liu, Y.; Wang, Y. Involvement of endoplasmic reticulum stress in formalin-induced pain is attenuated by 4-phenylbutyric acid. J. Pain Res. 2017, 10, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, M.S.; Maguire, A.D.; Simmen, T.; Kerr, B.J. Endoplasmic reticulum-mitochondria interplay in chronic pain: The calcium connection. Molecular Pain 2020, 16, 1744806920946889. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, H.; Sun, X.; Li, J.; Shan, W.; Yang, J.; Zuo, Z. Endoplasmic Reticulum Stress-Activated Neuronal and Microglial Autophagy Contributes to Postoperative Cognitive Dysfunction in Neonatal rats. Neurochem. Res. 2023, 48, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Zhang, M.; Zhang, C.; Cao, X.; Liu, B.; Li, N.; Sun, J.; Zhang, X. Transcriptomics reveals the effects of NTRK1 on endoplasmic reticulum stress response-associated genes in human neuronal cell lines. PeerJ 2023, 11, e15219. [Google Scholar] [CrossRef]
- Cunningham, M.E.; Greene, L.A. A function-structure model for NGF-activated TRK. EMBO J. 1998, 17, 7282–7293. [Google Scholar] [CrossRef]
- Cherief, M.; Negri, S.; Qin, Q.; Pagani, C.A.; Lee, S.; Yang, Y.P.; Clemens, T.L.; Levi, B.; James, A.W. TrkA+ Neurons Induce Pathologic Regeneration After Soft Tissue Trauma. Stem Cells Transl. Med. 2022, 11, 1165–1176. [Google Scholar] [CrossRef]
- Indo, Y. Nerve growth factor, pain, itch and inflammation: Lessons from congenital insensitivity to pain with anhidrosis. Expert Rev. Neurother. 2010, 10, 1707–1724. [Google Scholar] [CrossRef]
- Liu, B.W.; Zhang, J.; Hong, Y.S.; Li, N.B.; Liu, Y.; Zhang, M.; Wu, W.Y.; Zheng, H.; Lampert, A.; Zhang, X.W. NGF-Induced Nav1.7 Upregulation Contributes to Chronic Post-surgical Pain by Activating SGK1-Dependent Nedd4-2 Phosphorylation. Mol. Neurobiol. 2021, 58, 964–982. [Google Scholar] [CrossRef]
- Franco, M.L.; Melero, C.; Sarasola, E.; Acebo, P.; Luque, A.; Calatayud-Baselga, I.; García-Barcina, M.; Vilar, M. Mutations in TrkA Causing Congenital Insensitivity to Pain with Anhidrosis (CIPA) Induce Misfolding, Aggregation, and Mutation-dependent Neurodegeneration by Dysfunction of the Autophagic Flux. J. Biol. Chem. 2016, 291, 21363–21374. [Google Scholar] [CrossRef]
- Klein, R.; Jing, S.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Fagan, A.M.; Zhang, H.; Landis, S.; Smeyne, R.J.; Silos-Santiago, I.; Barbacid, M. TrkA, but not TrkC, receptors are essential for survival of sympathetic neurons in vivo. J. Neurosci. 1996, 16, 6208–6218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.; Huang, X.; Sun, H.; Xu, J.; Qu, H.; Yan, X.; Shi, W.; Teng, W.; Jin, X.; et al. Engineered Sensory Nerve Guides Self-Adaptive Bone Healing via NGF-TrkA Signaling Pathway. Adv. Sci. 2023, 10, e2206155. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, M.A.; Greco, A. Oncogenic rearrangements of the NTRK1/NGF receptor. Cancer Lett. 2006, 232, 90–98. [Google Scholar] [CrossRef]
- Thiels, C.A.; Habermann, E.B.; Hooten, W.M.; Jeffery, M.M. Chronic use of tramadol after acute pain episode: Cohort study. BMJ 2019, 365, l1849. [Google Scholar] [CrossRef]
- Choi, S.; Hachisuka, J.; Brett, M.A.; Magee, A.R.; Omori, Y.; Iqbal, N.U.; Zhang, D.; DeLisle, M.M.; Wolfson, R.L.; Bai, L.; et al. Parallel ascending spinal pathways for affective touch and pain. Nature 2020, 587, 258–263. [Google Scholar] [CrossRef]
- Pan, Z.; Shao, M.; Zhao, C.; Yang, X.; Li, H.; Cui, G.; Liang, X.; Yu, C.W.; Ye, Q.; Gao, C.; et al. J24335 exerts neuroprotective effects against 6-hydroxydopamine-induced lesions in PC12 cells and mice. Eur. J. Pharm. Sci. 2024, 194, 106696. [Google Scholar] [CrossRef]
- Suo, D.; Park, J.; Harrington, A.W.; Zweifel, L.S.; Mihalas, S.; Deppmann, C.D. Coronin-1 is a neurotrophin endosomal effector that is required for developmental competition for survival. Nat. Neurosci. 2014, 17, 36–45. [Google Scholar] [CrossRef]
- Beletskiy, A.; Chesnokova, E.; Bal, N. Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer-Its Comparative Expression, Processing and Signaling in Mammalian CNS. Int. J. Mol. Sci. 2021, 22, 1849. [Google Scholar] [CrossRef]
- Pardo, M.; Cheng, Y.; Sitbon, Y.H.; Lowell, J.A.; Grieco, S.F.; Worthen, R.J.; Desse, S.; Barreda-Diaz, A. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci. Res. 2019, 149, 1–13. [Google Scholar] [CrossRef]
- Cline, B.H.; Steinbusch, H.W.; Malin, D.; Revishchin, A.V.; Pavlova, G.V.; Cespuglio, R.; Strekalova, T. The neuronal insulin sensitizer dicholine succinate reduces stress-induced depressive traits and memory deficit: Possible role of insulin-like growth factor 2. BMC Neurosci. 2012, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Huerta, P.; Troncoso-Escudero, P.; Wu, D.; Thiruvalluvan, A.; Cisternas-Olmedo, M.; Henriquez, D.R.; Plate, L.; Chana-Cuevas, P.; Saquel, C.; Thielen, P.; et al. Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 2020, 140, 737–764. [Google Scholar] [CrossRef] [PubMed]
- Dikkes, P.; Jaffe, D.B.; Guo, W.H.; Chao, C.; Hemond, P.; Yoon, K.; Zurakowski, D.; Lopez, M.F. IGF2 knockout mice are resistant to kainic acid-induced seizures and neurodegeneration. Brain Res. 2007, 1175, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Bella, P.; Farini, A.; Banfi, S.; Parolini, D.; Tonna, N.; Meregalli, M.; Belicchi, M.; Erratico, S.; D’Ursi, P.; Bianco, F.; et al. Blockade of IGF2R improves muscle regeneration and ameliorates Duchenne muscular dystrophy. EMBO Mol. Med. 2020, 12, e11019. [Google Scholar] [CrossRef]
- Terauchi, A.; Johnson-Venkatesh, E.M.; Bullock, B.; Lehtinen, M.K.; Umemori, H. Retrograde fibroblast growth factor 22 (FGF22) signaling regulates insulin-like growth factor 2 (IGF2) expression for activity-dependent synapse stabilization in the mammalian brain. Elife Sci. 2016, 5, e12151. [Google Scholar] [CrossRef]
- Longato, L.; Ripp, K.; Setshedi, M.; Dostalek, M.; Akhlaghi, F.; Branda, M.; Wands, J.R.; de la Monte, S.M. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxidative Med. Cell. Longev. 2012, 2012, 479348. [Google Scholar] [CrossRef]
- Guo, D.; Xu, Y.; Liu, Z.; Wang, Y.; Xu, X.; Li, C.; Li, S.; Zhang, J.; Xiong, T.; Cao, W.; et al. IGF2 inhibits hippocampal over-activated microglia and alleviates depression-like behavior in LPS- treated male mice. Brain Res. Bull. 2023, 194, 1–12. [Google Scholar] [CrossRef]
- Jin, Y.; Kotler, J.L.M.; Wang, S.; Huang, B.; Halpin, J.C.; Street, T.O. The ER Chaperones BiP and Grp94 Regulate the Formation of Insulin-Like Growth Factor 2 (IGF2) Oligomers. J. Mol. Biol. 2021, 433, 166963. [Google Scholar] [CrossRef]
- Sélénou, C.; Brioude, F.; Giabicani, E.; Sobrier, M.L.; Netchine, I. IGF2: Development, Genetic and Epigenetic Abnormalities. Cells 2022, 11, 1886. [Google Scholar] [CrossRef]
- Yeh, C.C.; Sun, H.L.; Huang, C.J.; Wong, C.S.; Cherng, C.H.; Huh, B.K.; Wang, J.S.; Chien, C.C. Long-Term Anti-Allodynic Effect of Immediate Pulsed Radiofrequency Modulation through Down-Regulation of Insulin-Like Growth Factor 2 in a Neuropathic Pain Model. Int. J. Mol. Sci. 2015, 16, 27156–27170. [Google Scholar] [CrossRef]
- Chan, W.H.; Huang, N.C.; Lin, Y.W.; Lin, F.Y.; Tsai, C.S.; Yeh, C.C. Intrathecal IGF2 siRNA injection provides long-lasting anti-allodynic effect in a spared nerve injury rat model of neuropathic pain. PLoS ONE 2021, 16, e0260887. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J. Characterization of a model of persistent postoperative pain evoked by skin/muscle incision and retraction (SMIR). Pain 2008, 135, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Walla, B.C.; Diaz, M.F.; Fuller, G.N.; Gutstein, H.B. Intermittent lumbar puncture in rats: A novel method for the experimental study of opioid tolerance. Anesth. Analg. 2006, 103, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Fujii, K.; Mita, Y.; Watahiki, H.; Fukagawa, T.; Kitayama, T.; Mizuno, N.; Nakahara, H.; Sekiguchi, K. Development and validation of a SYBR green-based mitochondrial DNA quantification method by following the MIQE and other guidelines. Leg. Med. 2022, 58, 102096. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.; Li, N.; Zhang, J.; Zhang, X. Oxycodone regulates incision-induced activation of neurotrophic factors and receptors in an acute post-surgery pain rat model. J. Pain Res. 2018, 11, 2663–2674. [Google Scholar] [CrossRef]
- O-Sullivan, I.; Kc, R.; Singh, G.; Das, V.; Ma, K.; Li, X.; Mwale, F.; Votta-Velis, G.; Bruce, B.; Natarajan Anbazhagan, A.; et al. Sensory Neuron-Specific Deletion of Tropomyosin Receptor Kinase A (TrkA) in Mice Abolishes Osteoarthritis (OA) Pain via NGF/TrkA Intervention of Peripheral Sensitization. Int. J. Mol. Sci. 2022, 23, 12076. [Google Scholar] [CrossRef]
- Hsieh, Y.-L.; Kan, H.-W.; Chiang, H.; Lee, Y.-C.; Hsieh, S.-T. Distinct TrkA and Ret modulated negative and positive neuropathic behaviors in a mouse model of resiniferatoxin-induced small fiber neuropathy. Exp. Neurol. 2018, 300, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Tabata, M.; Murata, E.; Ueda, K.; Kato-Kogoe, N.; Kuroda, Y.; Hirose, M. Effects of TrkA inhibitory peptide on cancer-induced pain in a mouse melanoma model. J. Anesth. 2012, 26, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998, 143, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Zhang, W.; Zhang, C.; Zhang, K.; Cao, X.; Yu, S.; Zhang, X. Protein tyrosine phosphatase 1B contributes to neuropathic pain by aggravating NF-κB and glial cells activation-mediated neuroinflammation via promoting endoplasmic reticulum stress. CNS Neurosci. Ther. 2024, 30, e14609. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Oh-Hashi, K.; Matsuoka, Y.; Takemura, H.; Yamakita, S.; Matsuda, M.; Sawa, T.; Amaya, F. Endoplasmic Reticulum Stress in the Dorsal Root Ganglion Contributes to the Development of Pain Hypersensitivity after Nerve Injury. Neuroscience 2018, 394, 288–299. [Google Scholar] [CrossRef]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef]
- Fan, H.; Tang, H.B.; Kang, J.; Shan, L.; Song, H.; Zhu, K.; Wang, J.; Ju, G.; Wang, Y.Z. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience 2015, 311, 362–373. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Wang, Z.; Ding, M.; Li, X.; Guo, J.; Han, G.; Zhao, P. Dexmedetomidine Alleviated Endoplasmic Reticulum Stress via Inducing ER-phagy in the Spinal Cord of Neuropathic Pain Model. Front. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Ha, D.P.; Zhao, H.; Carlos, A.J.; Wei, S.; Pun, T.K.; Wu, K.; Zandi, E.; Kelly, K.; Lee, A.S. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4245–E4254. [Google Scholar] [CrossRef]
- Chérasse, Y.; Maurin, A.C.; Chaveroux, C.; Jousse, C.; Carraro, V.; Parry, L.; Deval, C.; Chambon, C.; Fafournoux, P.; Bruhat, A. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007, 35, 5954–5965. [Google Scholar] [CrossRef]
- Dong, L.; Guarino, B.B.; Jordan-Sciutto, K.L.; Winkelstein, B.A. Activating transcription factor 4, a mediator of the integrated stress response, is increased in the dorsal root ganglia following painful facet joint distraction. Neuroscience 2011, 193, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, Y.; Jin, H.; Kokubun, H.; Aoe, T. Pharmacological Chaperones Attenuate the Development of Opioid Tolerance. Int. J. Mol. Sci. 2020, 21, 7536. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Nagata, K. ER Stress Proteins in Autoimmune and Inflammatory Diseases. Front. Immunol. 2012, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Denk, F.; Bennett, D.L.; McMahon, S.B. Nerve Growth Factor and Pain Mechanisms. Annu. Rev. Neurosci. 2017, 40, 307–325. [Google Scholar] [CrossRef]
- Indo, Y. NGF-dependent neurons and neurobiology of emotions and feelings: Lessons from congenital insensitivity to pain with anhidrosis. Neurosci. Biobehav. Rev. 2018, 87, 1–16. [Google Scholar] [CrossRef]
- Li, N.; Guo, S.; Wang, Q.; Duan, G.; Sun, J.; Liu, Y.; Zhang, J.; Wang, C.; Zhu, C.; Liu, J.; et al. Heterogeneity of clinical features and mutation analysis of NTRK1 in Han Chinese patients with congenital insensitivity to pain with anhidrosis. J. Pain Res. 2019, 12, 453–465. [Google Scholar] [CrossRef]
- Hirose, M.; Kuroda, Y.; Murata, E. NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract. 2016, 16, 175–182. [Google Scholar] [CrossRef]
- Hefti, F.F.; Rosenthal, A.; Walicke, P.A.; Wyatt, S.; Vergara, G.; Shelton, D.L.; Davies, A.M. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol. Sci. 2006, 27, 85–91. [Google Scholar] [CrossRef]
- Gneo, L.; Ruggeri, P.; Cappabianca, L.; Farina, A.R.; Di Ianni, N.; Mackay, A.R. TRAIL induces pro-apoptotic crosstalk between the TRAIL-receptor signaling pathway and TrkAIII in SH-SY5Y cells, unveiling a potential therapeutic “Achilles heel” for the TrkAIII oncoprotein in neuroblastoma. Oncotarget 2016, 7, 80820–80841. [Google Scholar] [CrossRef]
- Sandgren, J.; Andersson, R.; Rada-Iglesias, A.; Enroth, S.; Akerstrom, G.; Dumanski, J.P.; Komorowski, J.; Westin, G.; Wadelius, C. Integrative epigenomic and genomic analysis of malignant pheochromocytoma. Exp. Mol. Med. 2010, 42, 484–502. [Google Scholar] [CrossRef]
- Hedborg, F.; Holmgren, L.; Sandstedt, B.; Ohlsson, R. The cell type-specific IGF2 expression during early human development correlates to the pattern of overgrowth and neoplasia in the Beckwith-Wiedemann syndrome. Am. J. Pathol. 1994, 145, 802–817. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Formulation | Administration | Dose Rationale |
|---|---|---|---|
| GW441756 (NTRK1i; Selleck #S2789) | 100 μM in saline + 1% DMSO | i.t. daily, days 10–14 | Dose-response pilot [28] |
| IGF2 siRNA (Tsingke Biotechnology) | 10–100 μM in 5% glucose + PEI (1:5 w/w) | i.t. single dose (day 10) | Prior efficacy in neuropathic pain [51] |
| IGF2 siRNA (Tsingke Biotechnology) | 10 μM in 5% glucose + PEI (1:5 w/w) | i.t. daily, days 10–14 | Prior efficacy in neuropathic pain [51] |
| Vehicle controls | Saline + 1% DMSO or PEI/glucose | Matched volumes/timing | N/A |
| Gene | Forward Primer Sequence (5′ to 3′) | Reverse Primer Sequence (5′ to 3′) |
|---|---|---|
| GAPDH | GAAGGTCGGTGTGAACGGAT | CCCATTTGATGTTAGCGGGAT |
| NTRK1 | AGGAGGATTTGTGTGGTGTGTAT | GAGTCATTGGGCATCTGGATCTT |
| IGF2 | GGGAAGTCGATGTTGGTGCT | AAGCAGCACTCTTCCACGAT |
| GRP78 | GGTTGGCGGATCTACTCGAATTC | AAGAGGACACACATCAAGCAGAA |
| XBP1 | CCCAGAACATCTTCCCATGGATT | CAGAGAAAGGGAGGCTGGTAAG |
| CHOP | TTCATACACCACCACACCTGAAA | TAGGGATGCAGGGTCAAGAGTAG |
| ATF6 | AGCAAGATTCCAGGAGAGTGAAA | TGACATGGAGGTGGAGGGATATA |
| PERK | TTGGAAGGTCATGGCGTTTAGTA | TGGCCTCTGTACATCCCTAAGTA |
| IRE1α | TCAAGGCGATGATCTCAGACTTT | GTTGCCCTCAGAGATGACATAGT |
| Antibodies | Source | Identifier | Dilution | Applications |
|---|---|---|---|---|
| Anti-TrkA antibody | ABclonal | A15618 | 1:1000,1:50 | WB,IF |
| Anti-IGF2 antibody | CST | Ab9574 | 1:2000 | WB |
| Anti-IGF2 antibody | ABclonal | A2086 | 1:50 | IF |
| Anti-GRP78 antibody | Affinity | Cat# AF5366 | 1:1000,1:50 | WB,IF |
| Anti-β-actin antibody | Proteintech | Cat# 81115-1-RR | 1:5000 | WB |
| Anti-p-PERK antibody | Affinity | Cat# DF7576 | 1:100 | WB |
| Anti-PERK antibody | ABclonal | Cat# A21255 | 1:1000 | WB |
| Anti-p-IRE1α antibody | Affinity | Cat# AF7150 | 1:100 | WB |
| Anti-IRE1α antibody | Proteintech | Cat# 27528-1-AP | 1:1000 | WB |
| Anti-p-eIF antibody | Affinity | Cat# AF3087 | 1:100 | WB |
| Anti-eIF antibody | Proteintech | Cat# 11170-1-AP | 1:1000 | WB |
| Anti-XBP1 antibody | Proteintech | Cat# 24868-1-AP | 1:1000 | WB |
| Anti-CHOP antibody | ABclonal | A20987 | 1:1000 | WB |
| Anti-ATF6 antibody | Affinity | Cat# DF6009 | 1:1000 | WB |
| Anti-NeuN antibody | Abcam | Ab104224 | 1:200 | IF |
| Anti-GFAP antibody | CST | Cat# 3670 | 1:200 | IF |
| Anti-IBA1 antibody | Abcam | Ab5076 | 1:200 | IF |
| oraLite594 anti-rabbit IgG | Proteintech | Cat# SA00013-8 | 1:200 | IF |
| oraLite488 anti-mouse IgG | Proteintech | Cat# SA00013-5 | 1:200 | IF |
| FITC affinipure anti-goat IgG | Proteintech | Cat# SA00003-3 | 1:200 | IF |
| Anti-rabbit IgG HRP | Abbkine | Cat# A21020 | 1:5000 | WB |
| Anti-goat IgG HRP | ABclonal | Cat# AS031 | 1:5000 | WB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhang, K.; Zhang, W.; Jiao, B.; Cao, X.; Yu, S.; Zhang, M.; Zhang, X. Unidirectional Crosstalk Between NTRK1 and IGF2 Drives ER Stress in Chronic Pain. Biomedicines 2025, 13, 1632. https://doi.org/10.3390/biomedicines13071632
Zhang C, Zhang K, Zhang W, Jiao B, Cao X, Yu S, Zhang M, Zhang X. Unidirectional Crosstalk Between NTRK1 and IGF2 Drives ER Stress in Chronic Pain. Biomedicines. 2025; 13(7):1632. https://doi.org/10.3390/biomedicines13071632
Chicago/Turabian StyleZhang, Caixia, Kaiwen Zhang, Wencui Zhang, Bo Jiao, Xueqin Cao, Shangchen Yu, Mi Zhang, and Xianwei Zhang. 2025. "Unidirectional Crosstalk Between NTRK1 and IGF2 Drives ER Stress in Chronic Pain" Biomedicines 13, no. 7: 1632. https://doi.org/10.3390/biomedicines13071632
APA StyleZhang, C., Zhang, K., Zhang, W., Jiao, B., Cao, X., Yu, S., Zhang, M., & Zhang, X. (2025). Unidirectional Crosstalk Between NTRK1 and IGF2 Drives ER Stress in Chronic Pain. Biomedicines, 13(7), 1632. https://doi.org/10.3390/biomedicines13071632