
A Review of the Relationship Between Insulin and Bone Health



Abstract
1. Introduction
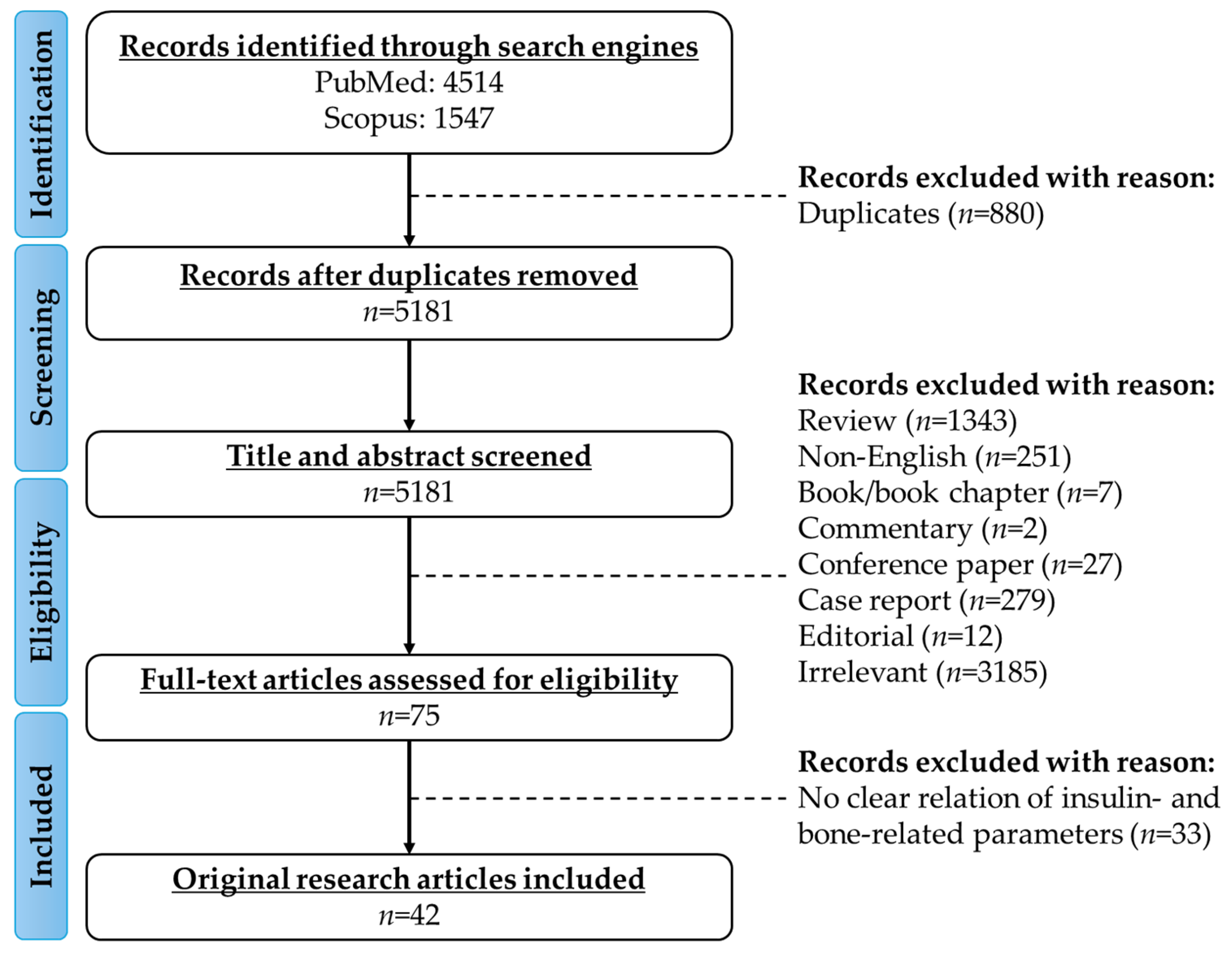
2. Literature Search
3. Evidence from In Vivo Studies
3.1. Effects of Circulating Insulin Levels on Bone Health

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Animal Model | Findings | Reference |
|---|---|---|
| Rats injected with STZ | Insulin: ↓; insulin receptor: ↔; femur length: ↓; tibia length: ↓; bone weight (femur, tibia, proximal tibia): ↓; ALP: ↓; TRAP: ↔; CTSK: ↔; calcium: ↓; hydroxyproline: ↓; deoxypyridinoline: ↔; BV/TV: ↓; trabecular bone surface: ↓; Tb.Th: ↓; Tb.N: ↓; Ob.N: ↓; Oc.N: ↔; BMP-2: ↔; Dlx5: ↓; Runx2: ↓; OSX: ↓; OCN: ↓; COL1: ↓; Wnt3a: ↔; LRP5: ↔; Akt: ↔; p-Akt: ↓; GSK3β: ↔; p-GSK3β: ↓; β-catenin: ↓; p-β-catenin: ↓; Sost: ↑; DKK1: ↑ | [12] |
| Mice injected with STZ | Glucose: ↑; CTX: ↑; P1NP: ↓; stiffness: ↓; force: ↓; femoral length: ↔; modulus: ↔; bending strength: ↓; toughness: ↔ Cortical parameters: Ct.Ar: ↓; Ct.Th: ↓; mineral density: ↔ Trabecular parameters: BV/TV: ↓; Tb.N: ↓; Tb.Th: ↓; mineral density: ↓ | [13] |
| Rats injected with STZ and fed high trans-fat diet with 2% cholesterol | Insufficiency of β-cells; glucose: ↑; insulin: ↔; tibial length: ↔; tibial width: ↔; stiffness: ↔; load: ↔; strength: ↓ Cortical parameters: BMC: ↔; BMD: ↔; Ct.Th: ↓; periosteal perimeter: ↔; endosteal perimeter: ↑ Trabecular parameters: BMD: ↔; BV/TV: ↓; bone surface: ↓; Tb.N: ↓; Tb.Th: ↔; Tb.Sp: ↑; SMI: ↔; Conn.D: ↓ | [14] |
| Rats with metabolic syndrome induced by high-carbohydrate high-fat diet | Insulin: ↑; glucose: ↑; calcium: ↔; load: ↓; displacement: ↔; stiffness: ↔; stress: ↔; strain: ↑; elasticity: ↔; Ob.S: ↓; Oc.S: ↔; ES: ↑; OS: ↓; OV: ↓; sLS: ↔; dLS: ↔; MS: ↔; MAR: ↔; BFR: ↔ Cortical parameters: Ct.Th: ↔; Ct.Ar: ↔; Tt.Ar: ↓ Trabecular parameters: BV/TV: ↓; Tb.N: ↓; Tb.Sp: ↑; Tb.Th: ↔; SMI: ↑; Conn.D: ↓ | [16] |
| C57BL/6 mice fed high-fat diet | Fasting glucose: ↑; fasting insulin: ↑; glucose intolerance: ↑; leptin: ↑; tibia length: ↓; whole body BMD: ↓; Runx2: ↓; COL1: ↓ Cortical parameters: Ct.Th: ↔; Ct.Ar: ↔ Trabecular parameters: BV/TV: ↓ | [17] |
| C57BL/6 mice injected with low-dose STZ and fed hypercaloric diet | Glucose: ↑; insulin: ↓; HOMA-β cell function: ↓; alveolar bone loss: ↑; alveolar RANKL: ↑; load: ↓; force: ↓; stiffness: ↓; elastic modulus: ↓; stress: ↓ Cortical parameter: Ct.Th: ↓ Trabecular parameter: BV/TV: ↓; Tb.Th: ↓; Tb.Sp: ↑ | [18] |
| KK-Ay diabetic mice | Insulin: ↑; glucose: ↑; OCN: ↑; BSP: ↑; COL1: ↑; osteonectin: ↑; ALP: ↓; Foxo1: ↑; Runx2: ↑; OSX: ↑; TRAP: ↑; V-ATPase: ↑ Cortical parameters: BMD: ↑; Ct.Th: ↑ Trabecular parameters: BMD: ↓; BV/TV: ↓; Tb.Th: ↓; Tb.N: ↔; Tb.Sp: ↑ | [19] |
| Zucker diabetic fatty rats fed high-fat, high-carbohydrate diet | Non-enzymatic glycation: ↑; mineral density ratio: ↓; elastic modulus: ↔; toughness: ↓; maximum stress: ↓ Cortical parameters: Porosity: ↑ | [20] |
| Zucker diabetic fatty rats with subcritical femur defects | Glucose: ↑; HbA1c: ↑; calcium: ↑; phosphate: ↑; P1NP: ↓; CTX: ↑; TRAP: ↑; PTH: ↔; force: ↓; work to failure: ↓; BFR: ↓; MS: ↓; MAR: ↓ Cortical parameters: BV/TV: ↓; BMD: ↓; Tb.N: ↓; Tb.Th: ↓ Trabecular parameters: BV/TV: ↓; BMD: ↓; Ct.Th: ↓ | [21] |
| Goto-Kakizaki rats | Insulin: ↑; OCN: ↓; ALP: ↑; CTX: ↑; TRAP: ↓; calcium: ↔; phosphate: ↓; BMD: ↓; Ob.S: ↓; Oc.S: ↔; MS: ↓; MAR: ↓; BFR: ↓; load: ↓; stiffness: ↓ Cortical parameters: Ct.Ar: ↓ Trabecular parameters: BV/TV: ↓; Tb.N: ↓; Tb.Th: ↓; Tb.Sp: ↑; Conn.D: ↓ | [22] |
| Tsumura Suzuki Obese Diabetes mice | Glucose: ↑; insulin: ↑; glucose intolerance: ↑; OCN: ↔; TRAP: ↑; BMC: ↓; BMD: ↓; force: ↔; stiffness: ↔; work to failure: ↔; stress: ↓; elastic modulus: ↔; toughness: ↔ Cortical parameters: Ct.V: ↔; Tt.V: ↓; Ct.V/Tt.V: ↑; porosity: ↔; Ct.Th: ↑ Trabecular parameters: BV/TV: ↑; Tb.Th: ↑; Tb.N: ↑; Tb.Sp: ↓ | [23] |
| Nagoya Shibata Yasuda mice | Glucose: ↑; insulin: ↔; glucose intolerance: ↑; femur length: ↓; BMD: ↓; BMC: ↓; OCN: ↓; TRAP: ↓; force: ↓; stiffness: ↔; work to failure: ↔; stress: ↔; elastic modulus: ↓; toughness: ↓ Cortical parameters: Ct.Ar: ↓; Ct.Th: ↔ Trabecular parameters: Tb.Ar: ↔ | [24] |
3.2. Effects of Insulin Treatment on Bone Health
3.3. Effects of Insulin Receptor Silencing on Bone Health
4. Evidence from Human Studies
4.1. Effects of Circulating Insulin Levels on Bone Health
4.2. Effects of Insulin Resistance on Bone Health
4.3. Effects of Insulin Treatment on Bone Health
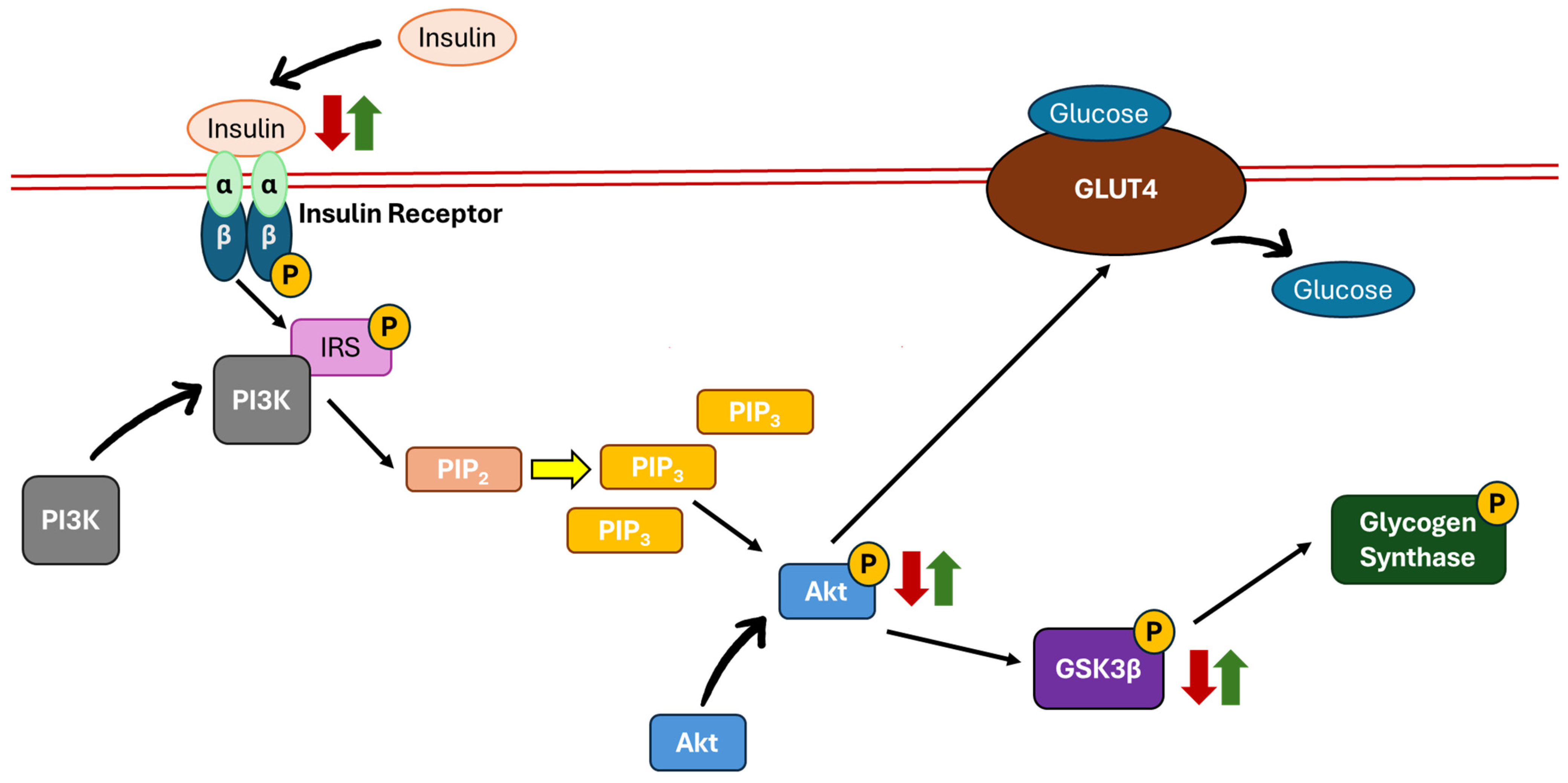
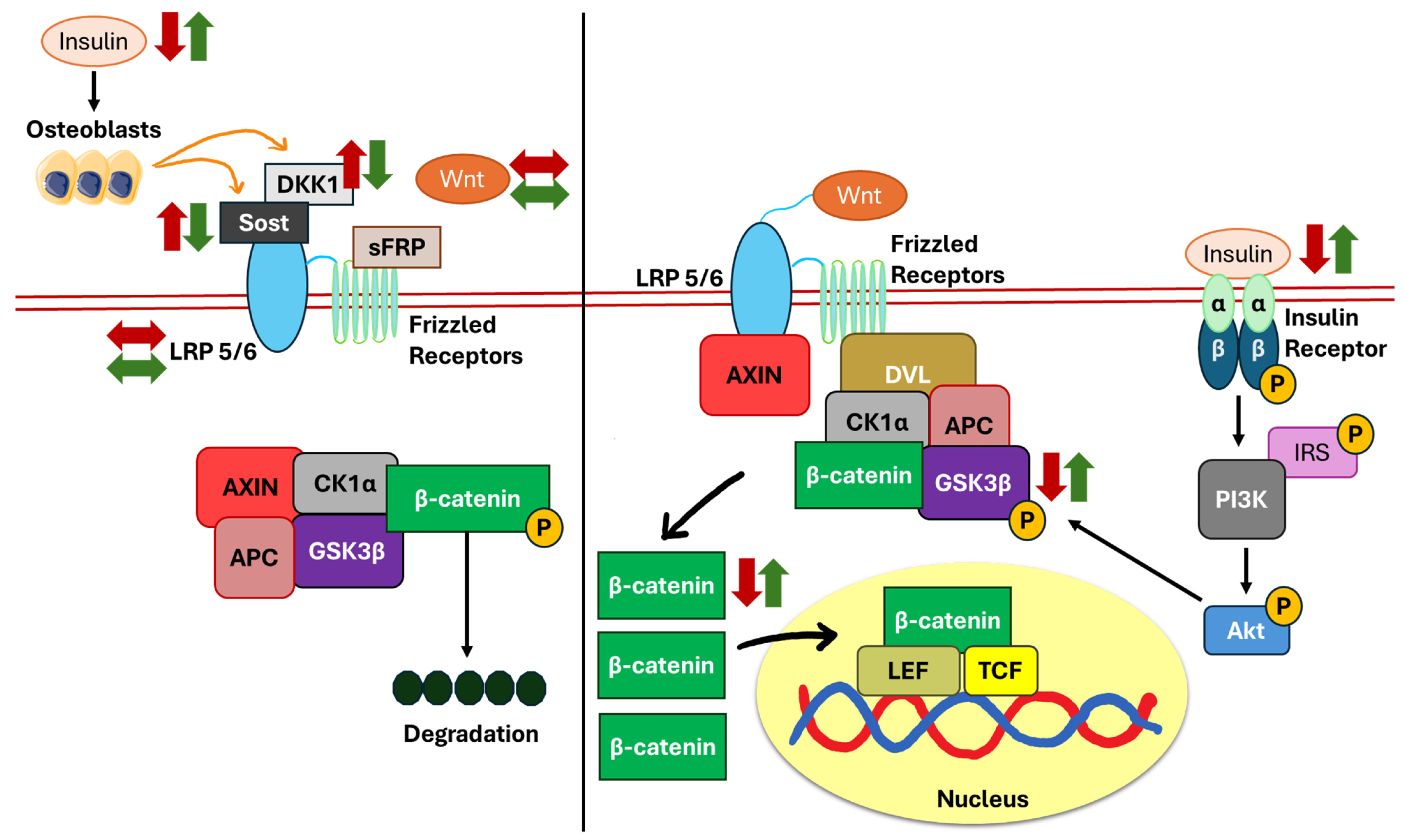
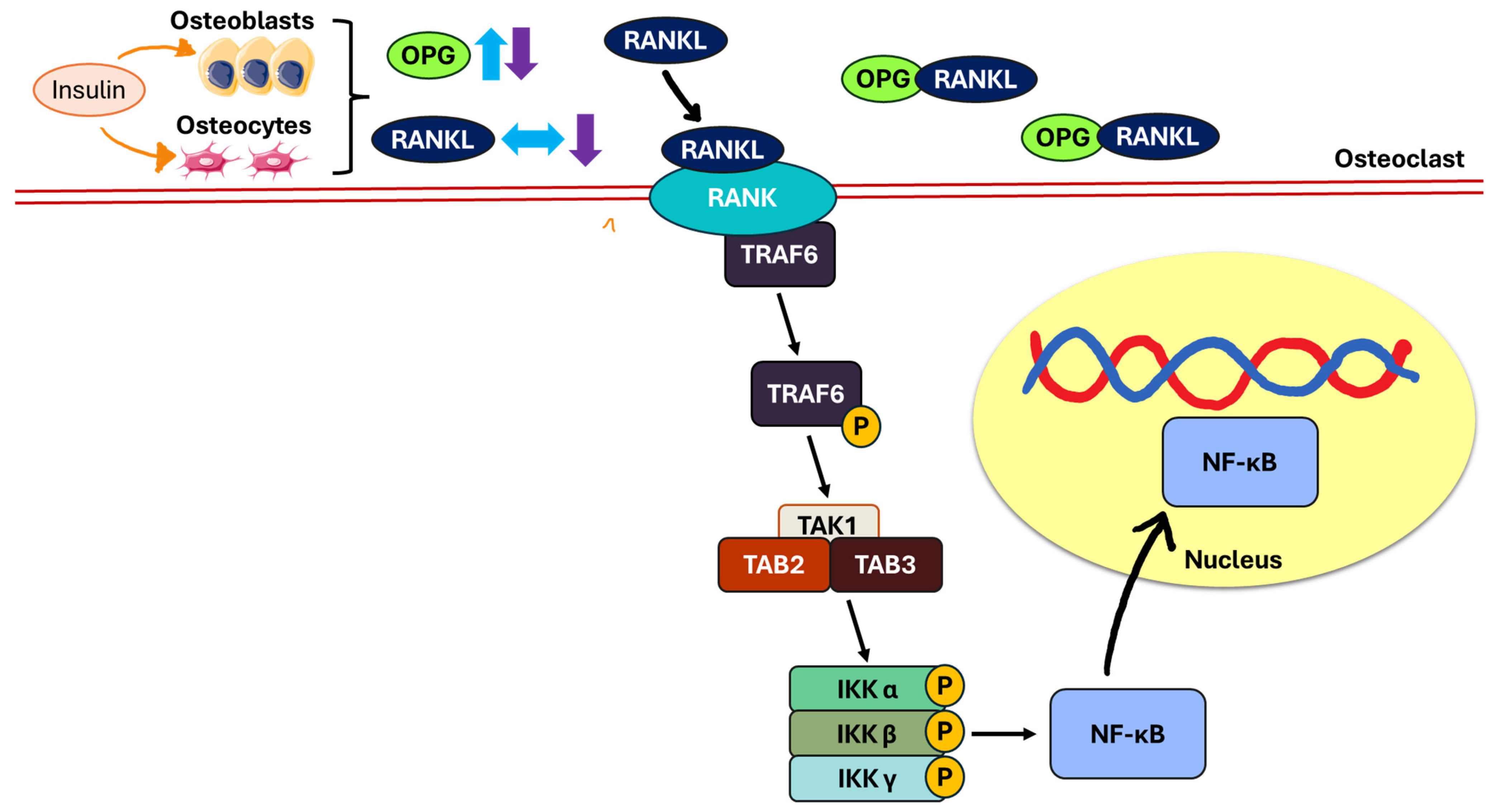
5. The Mechanistic Pathway of Insulin
5.1. Phosphatidylinositol 3-Kinase (PI3K)/Protein Kinase B (Akt)/Glycogen Synthase Kinase 3-Beta (GSK3β) Pathway
5.2. Wnt/β-Catenin Pathway
5.3. Receptor Activator of Nuclear Factor Kappa B (RANK)/RANKL/OPG Pathway
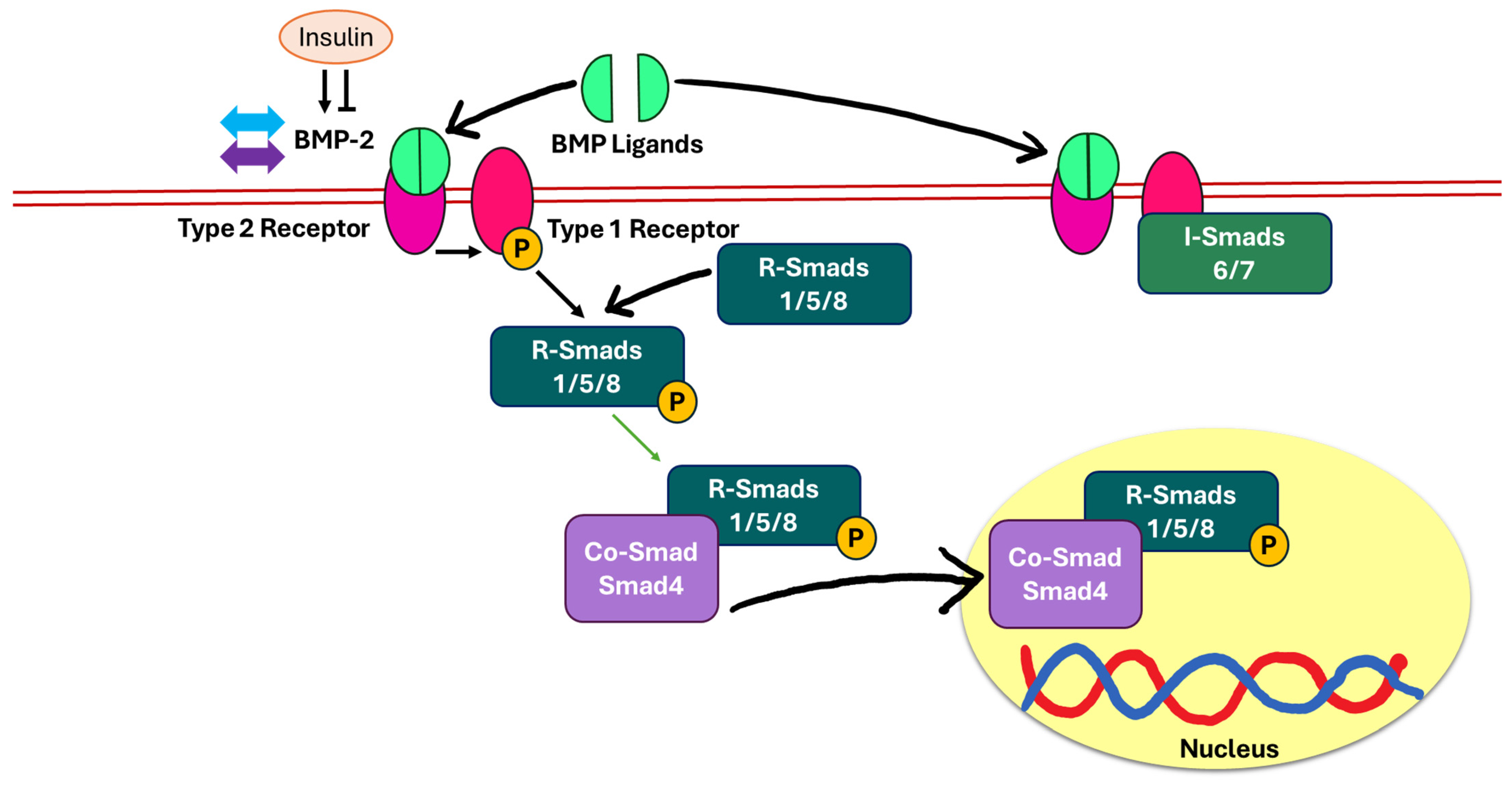
5.4. Bone Morphogenic Protein-2 (BMP-2)/Suppressor of Mothers Against Decapentaplegic (Smad)-Dependent Pathway
6. Sarcopenia, Insulin Resistance, and Bone Health
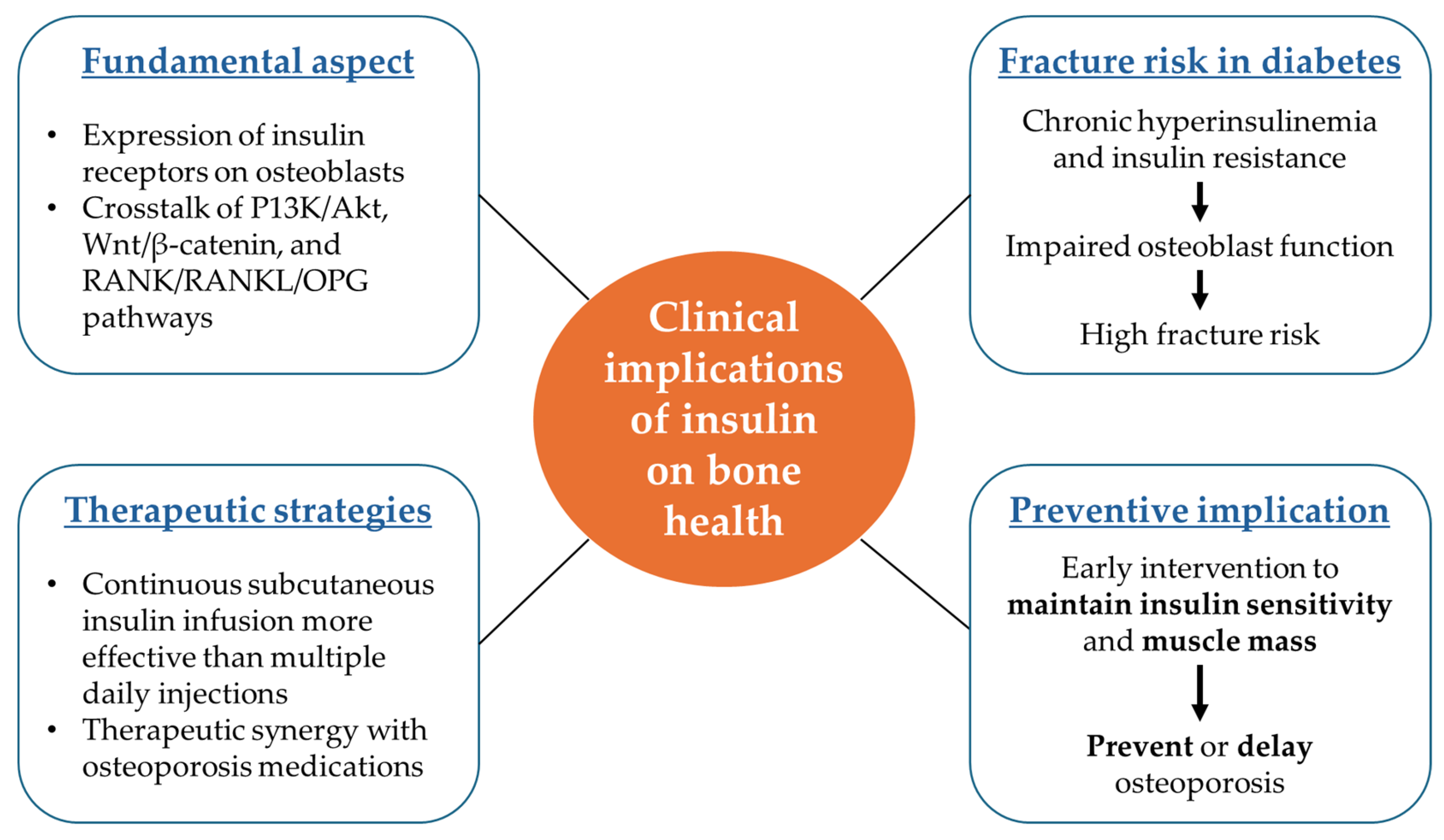
7. Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| ALP | Alkaline phosphatase |
| APC | Adenomatous polyposis coli |
| AXIN | Axis inhibition protein |
| BA/TA | Bone area/total area |
| BFR | Bone formation rate |
| BMAD | Bone mineral apparent density |
| BMC | Bone mineral content |
| BMD | Bone mineral density |
| BMI | Body mass index |
| BMP-2 | Bone morphogenic protein-2 |
| BMPRI | BMP type I receptor |
| BMPRII | BMP type II receptor |
| BSP | Bone sialoprotein |
| BV/TV | Bone volume/total volume |
| CI | Confidence interval |
| CK1α | Casein kinase alpha |
| COL1 | Type I collagen |
| Conn.D | Connectivity density |
| Co-Smad | Common-mediator Smad |
| CTSK | Cathepsin K |
| Ct.Ar | Cortical area |
| Ct.Ar/Tt.Ar | Cortical area fraction |
| Ct.Th | Cortical thickness |
| CTX | Carboxyl-terminal cross-linked telopeptide of type 1 collagen |
| DKK1 | Dickkopf-related protein 1 |
| dLS | Double-labelled surface |
| Dlx5 | Distal-less homeobox 5 |
| DVL | Protein dishevelled |
| ES | Eroded surface |
| Foxo1 | Forkhead box protein O1 |
| GLUT4 | Glucose transporter 4 |
| GSK3β | Glycogen synthase kinase 3-beta |
| HOMA-IR | Homeostatic model assessment of insulin resistance |
| HOMA-β | Homeostatic model assessment of β-cell function |
| HR | Hazard ratio |
| IKK | Inhibitor of nuclear factor-kappa B |
| IRS | Insulin receptor substrate |
| IRβ | Insulin receptor β |
| I-Smads | Inhibitory Smads |
| KK-Ay | Yellow Kuo Kondo |
| LEF | Lymphoid enhancer-binding factor |
| LRP5 | Low-density lipoprotein receptor-related protein 5 |
| MAR | Mineral apposition rate |
| METS-IR | Metabolic score for insulin resistance |
| MS | Mineralising surface |
| NF-κB | Nuclear factor-kappa B |
| NHANES | National Health and Nutrition Examination Survey |
| Ob.N | Osteoblast number |
| Ob.S | Osteoblast surface |
| Oc.N | Osteoclast number |
| Oc.S | Osteoclast surface |
| OCN | Osteocalcin |
| OPG | Osteoprotegerin |
| OR | Odds ratio |
| OS | Osteoid surface |
| OSX | Osterix |
| OV | Osteoid volume |
| PIP2 | Phosphorylates phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PI3K | Phosphatidylinositol 3-kinase |
| P1NP | N-terminal propeptide of type 1 collagen |
| p-Akt | Phosphorylated protein kinase B |
| p-β-catenin | Phosphorylated β-catenin |
| p-GSK3β | Phosphorylated glycogen synthase kinase 3-beta |
| QUICKI | Quantitative insulin sensitivity check index |
| RANK | Receptor activator of nuclear factor kappa B |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| R-Smads | Receptor-regulated Smads |
| Runx2 | Runt-related transcription factor 2 |
| sFRP | Secreted Frizzled-related proteins |
| sLS | Single-labelled surface |
| Smad | Suppressor of mothers against decapentaplegic |
| SMI | Structure model index |
| Sost | Sclerostin |
| STZ | Streptozotocin |
| TAB2 | TAK1 binding protein 2 |
| TAB3 | TAK1 binding protein 3 |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| Tb.N | Trabecular number |
| Tb.Sp | Trabecular separation |
| Tb.Th | Trabecular thickness |
| TCF | T-cell-specific factor |
| TGF-β | Transforming growth factor-beta |
| TRAF6 | Tumour necrosis factor receptor-associated factor 6 |
| TRAP | Tartrate-resistant acid phosphatase |
| Tt.Ar | Total cross-sectional area inside the periosteal envelope |
| TyG | Triglyceride–glucose index |
| TyG-BMI | Tyg index with body mass index |
| TyG-WC | Tyg index with waist circumference |
| TyG-WHtR | Tyg index with waist-to-height ratio |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| ucOCN | Uncarboxylated osteocalcin |
| V-ATPase | V-type proton atpase |
| Wnt3a | Wnt ligand 3a |
| β-catenin | Beta-catenin |
References
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 3000605231164548. [Google Scholar] [CrossRef] [PubMed]
- Oghbaei, H.; Fattahi, A.; Hamidian, G.; Sadigh-Eteghad, S.; Ziaee, M.; Mahmoudi, J. A closer look at the role of insulin for the regulation of male reproductive function. Gen. Comp. Endocrinol. 2021, 300, 113643. [Google Scholar] [CrossRef] [PubMed]
- Dakic, T.; Jevdjovic, T.; Lakic, I.; Ruzicic, A.; Jasnic, N.; Djurasevic, S.; Djordjevic, J.; Vujovic, P. The Expression of Insulin in the Central Nervous System: What Have We Learned So Far? Int. J. Mol. Sci. 2023, 24, 6586. [Google Scholar] [CrossRef]
- Sylow, L.; Tokarz, V.L.; Richter, E.A.; Klip, A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021, 33, 758–780. [Google Scholar] [CrossRef]
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone remodeling: An operational process ensuring survival and bone mechanical competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef]
- Smit, A.E.; Meijer, O.C.; Winter, E.M. The multi-faceted nature of age-associated osteoporosis. Bone Rep. 2024, 20, 101750. [Google Scholar] [CrossRef]
- Conte, C.; Epstein, S.; Napoli, N. Insulin resistance and bone: A biological partnership. Acta Diabetol. 2018, 55, 305–314. [Google Scholar] [CrossRef]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Wang, W.; Liu, J. Insulin stimulates osteoblast proliferation and differentiation through ERK and PI3K in MG-63 cells. Cell Biochem. Funct. 2010, 28, 334–341. [Google Scholar] [CrossRef]
- Liu, X.; Chen, F.; Liu, L.; Zhang, Q. Prevalence of osteoporosis in patients with diabetes mellitus: A systematic review and meta-analysis of observational studies. BMC Endocr. Disord. 2023, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Hie, M.; Iitsuka, N.; Otsuka, T.; Tsukamoto, I. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int. J. Mol. Med. 2011, 28, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Kalaitzoglou, E.; Bunn, R.; Uppuganti, S.; Thrailkill, K.; Fowlkes, J. Preserving and restoring bone with continuous insulin infusion therapy in a mouse model of type 1 diabetes. Bone Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bagi, C.M.; Edwards, K.; Berryman, E. Metabolic Syndrome and Bone: Pharmacologically Induced Diabetes has Deleterious Effect on Bone in Growing Obese Rats. Calcif. Tissue Int. 2018, 102, 683–694. [Google Scholar] [CrossRef]
- Wong, S.K.; Chin, K.Y.; Ima-Nirwana, S. Toll-like Receptor as a Molecular Link between Metabolic Syndrome and Inflammation: A Review. Curr. Drug Targets 2019, 20, 1264–1280. [Google Scholar] [CrossRef]
- Wong, S.K.; Chin, K.Y.; Suhaimi, F.H.; Ahmad, F.; Ima-Nirwana, S. Exploring the potential of tocotrienol from Bixa orellana as a single agent targeting metabolic syndrome and bone loss. Bone 2018, 116, 8–21. [Google Scholar] [CrossRef]
- Rendina-Ruedy, E.; Graef, J.L.; Davis, M.R.; Hembree, K.D.; Gimble, J.M.; Clarke, S.L.; Lucas, E.A.; Smith, B.J. Strain differences in the attenuation of bone accrual in a young growing mouse model of insulin resistance. J. Bone Miner. Metab. 2016, 34, 380–394. [Google Scholar] [CrossRef]
- Sólis-Suarez, D.L.; Cifuentes-Mendiola, S.E.; González-Alva, P.; Rodríguez-Hernández, A.P.; Martínez-Dávalos, A.; Llamosas-Hernandez, F.E.; Godínez-Victoria, M.; García-Hernández, A.L. Lipocalin-2 as a fundamental protein in type 2 diabetes and periodontitis in mice. J. Periodontol. 2025, 96, 369–382. [Google Scholar] [CrossRef]
- Fu, C.; Zhang, X.; Ye, F.; Yang, J. High insulin levels in KK-Ay diabetic mice cause increased cortical bone mass and impaired trabecular micro-structure. Int. J. Mol. Sci. 2015, 16, 8213–8226. [Google Scholar] [CrossRef]
- Campbell, G.M.; Tiwari, S.; Picke, A.K.; Hofbauer, C.; Rauner, M.; Morlock, M.M.; Hofbauer, L.C.; Glüer, C.C. Effects of insulin therapy on porosity, non-enzymatic glycation and mechanical competence in the bone of rats with type 2 diabetes mellitus. Bone 2016, 91, 186–193. [Google Scholar] [CrossRef]
- Picke, A.K.; Gordaliza Alaguero, I.; Campbell, G.M.; Glüer, C.C.; Salbach-Hirsch, J.; Rauner, M.; Hofbauer, L.C.; Hofbauer, C. Bone defect regeneration and cortical bone parameters of type 2 diabetic rats are improved by insulin therapy. Bone 2016, 82, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, Y.; Lai, W.; Du, M.; Li, S.; Zhou, L.; Mo, Y.; Wang, P.; Min, Y.; Cui, L. 1,25-Dihydroxy vitamin D3 treatment attenuates osteopenia, and improves bone muscle quality in Goto-Kakizaki type 2 diabetes model rats. Endocrine 2019, 64, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yamashita, T.; Yoneda, M.; Takagi, S.; Miura, T. Characteristics of bone strength and metabolism in type 2 diabetic model Tsumura, Suzuki, Obese Diabetes mice. Bone Rep. 2018, 9, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Miura, T.; Yamashita, T.; Yoneda, M.; Takagi, S. Characteristics of Bone Strength and Metabolism in Type 2 Diabetic Model Nagoya Shibata Yasuda Mice. Biol. Pharm. Bull. 2018, 41, 1567–1573. [Google Scholar] [CrossRef]
- Bortolin, R.H.; Freire Neto, F.P.; Arcaro, C.A.; Bezerra, J.F.; da Silva, F.S.; Ururahy, M.A.; Souza, K.S.; Lima, V.M.; Luchessi, A.D.; Lima, F.P.; et al. Anabolic Effect of Insulin Therapy on the Bone: Osteoprotegerin and Osteocalcin Up-Regulation in Streptozotocin-Induced Diabetic Rats. Basic Clin. Pharmacol. Toxicol. 2017, 120, 227–234. [Google Scholar] [CrossRef]
- Cirano, F.R.; Molez, A.M.; Ribeiro, F.V.; Tenenbaum, H.C.; Casati, M.Z.; Corrêa, M.G.; Pimentel, S.P. Resveratrol and insulin association reduced alveolar bone loss and produced an antioxidant effect in diabetic rats. J. Periodontol. 2021, 92, 748–759. [Google Scholar] [CrossRef]
- Shi, P.; Hou, A.; Li, C.; Wu, X.; Jia, S.; Cen, H.; Hu, X.; Gong, H. Continuous subcutaneous insulin infusion ameliorates bone structures and mechanical properties in type 2 diabetic rats by regulating bone remodeling. Bone 2021, 153, 116101. [Google Scholar] [CrossRef]
- Studentsova, V.; Knapp, E.; Loiselle, A.E. Insulin Receptor deletion in S100a4-lineage cells accelerates age-related bone loss. Bone Rep. 2019, 10, 100197. [Google Scholar] [CrossRef]
- Fulzele, K.; Riddle, R.C.; DiGirolamo, D.J.; Cao, X.; Wan, C.; Chen, D.; Faugere, M.C.; Aja, S.; Hussain, M.A.; Brüning, J.C.; et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 2010, 142, 309–319. [Google Scholar] [CrossRef]
- Thrailkill, K.; Bunn, R.C.; Lumpkin, C., Jr.; Wahl, E.; Cockrell, G.; Morris, L.; Kahn, C.R.; Fowlkes, J.; Nyman, J.S. Loss of insulin receptor in osteoprogenitor cells impairs structural strength of bone. J. Diabetes Res. 2014, 2014, 703589. [Google Scholar] [CrossRef]
- Irwin, R.; Lin, H.V.; Motyl, K.J.; McCabe, L.R. Normal bone density obtained in the absence of insulin receptor expression in bone. Endocrinology 2006, 147, 5760–5767. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Sánchez, F.; Usategui-Martín, R.; Ruiz-de Temiño, Á.; Gil, J.; Ruiz-Mambrilla, M.; Fernández-Gómez, J.M.; Dueñas-Laita, A.; Pérez-Castrillón, J.L. Relationship between Insulin Resistance (HOMA-IR), Trabecular Bone Score (TBS), and Three-Dimensional Dual-Energy X-ray Absorptiometry (3D-DXA) in Non-Diabetic Postmenopausal Women. J. Clin. Med. 2020, 9, 1732. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Shi, L.; Zhang, Z. Effect of insulin resistance on gonadotropin and bone mineral density in nondiabetic postmenopausal women. Front. Endocrinol. 2023, 14, 1235102. [Google Scholar] [CrossRef] [PubMed]
- Sayers, A.; Lawlor, D.A.; Sattar, N.; Tobias, J.H. The association between insulin levels and cortical bone: Findings from a cross-sectional analysis of pQCT parameters in adolescents. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 610–618. [Google Scholar] [CrossRef]
- Srikanthan, P.; Crandall, C.J.; Miller-Martinez, D.; Seeman, T.E.; Greendale, G.A.; Binkley, N.; Karlamangla, A.S. Insulin resistance and bone strength: Findings from the study of midlife in the United States. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 796–803. [Google Scholar] [CrossRef]
- Torres-Costoso, A.; Pozuelo-Carrascosa, D.P.; Álvarez-Bueno, C.; Ferri-Morales, A.; Miota Ibarra, J.; Notario-Pacheco, B.; Martínez-Vizcaíno, V. Insulin and bone health in young adults: The mediator role of lean mass. PLoS ONE 2017, 12, e0173874. [Google Scholar] [CrossRef]
- Wong, S.K.; Mohamad, N.V.; Jayusman, P.A.; Ibrahim, N. A Review on the Crosstalk between Insulin and Wnt/β-Catenin Signalling for Bone Health. Int. J. Mol. Sci. 2023, 24, 12441. [Google Scholar] [CrossRef]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef]
- Minh, H.V.; Tien, H.A.; Sinh, C.T.; Thang, D.C.; Chen, C.H.; Tay, J.C.; Siddique, S.; Wang, T.D.; Sogunuru, G.P.; Chia, Y.C.; et al. Assessment of preferred methods to measure insulin resistance in Asian patients with hypertension. J. Clin. Hypertens. (Greenwich Conn.) 2021, 23, 529–537. [Google Scholar] [CrossRef]
- Sung, K.C.; Reaven, G.M.; Kim, S.H. Utility of homeostasis model assessment of beta-cell function in predicting diabetes in 12,924 healthy Koreans. Diabetes Care 2010, 33, 200–202. [Google Scholar] [CrossRef]
- Katz, A.; Nambi, S.S.; Mather, K.; Baron, A.D.; Follmann, D.A.; Sullivan, G.; Quon, M.J. Quantitative insulin sensitivity check index: A simple, accurate method for assessing insulin sensitivity in humans. J. Clin. Endocrinol. Metab. 2000, 85, 2402–2410. [Google Scholar] [CrossRef] [PubMed]
- Dang, K.; Wang, X.; Hu, J.; Zhang, Y.; Cheng, L.; Qi, X.; Liu, L.; Ming, Z.; Tao, X.; Li, Y. The association between triglyceride-glucose index and its combination with obesity indicators and cardiovascular disease: NHANES 2003-2018. Cardiovasc. Diabetol. 2024, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Jung, D.; Lee, Y.; Park, B. The Metabolic Score for Insulin Resistance (METS-IR) as a Predictor of Incident Ischemic Heart Disease: A Longitudinal Study among Korean without Diabetes. J. Pers. Med. 2021, 11, 742. [Google Scholar] [CrossRef] [PubMed]
- Karimi, F.; Ranjbar Omrani, G.; Dabbaghmanesh, M.H. Insulin resistance and bone health in adolescents. Arch. Osteoporos. 2021, 16, 66. [Google Scholar] [CrossRef]
- Fu, Y.H.; Liu, W.J.; Lee, C.L.; Wang, J.S. Associations of insulin resistance and insulin secretion with bone mineral density and osteoporosis in a general population. Front. Endocrinol. 2022, 13, 971960. [Google Scholar] [CrossRef]
- Zhuo, M.; Chen, Z.; Zhong, M.L.; Lei, F.; Qin, J.J.; Liu, S.; Liu, Y.M.; Sun, T.; Zhang, X.J.; Zhu, L.; et al. Association of insulin resistance with bone mineral density in a nationwide health check-up population in China. Bone 2023, 170, 116703. [Google Scholar] [CrossRef]
- Sheu, A.; Blank, R.D.; Tran, T.; Bliuc, D.; Greenfield, J.R.; White, C.P.; Center, J.R. Associations of Type 2 Diabetes, Body Composition, and Insulin Resistance with Bone Parameters: The Dubbo Osteoporosis Epidemiology Study. JBMR Plus 2023, 7, e10780. [Google Scholar] [CrossRef]
- Guo, H.; Wang, C.; Jiang, B.; Ge, S.; Cai, J.; Zhou, Y.; Ying, R.; Zha, K.; Zhou, J.; Wang, N.; et al. Association of Insulin Resistance and β-cell Function With Bone Turnover Biomarkers in Dysglycemia Patients. Front. Endocrinol. 2021, 12, 554604. [Google Scholar] [CrossRef]
- Shieh, A.; Greendale, G.A.; Cauley, J.A.; Srikanthan, P.; Karlamangla, A.S. Longitudinal associations of insulin resistance with change in bone mineral density in midlife women. JCI Insight 2022, 7, e162085. [Google Scholar] [CrossRef]
- Yang, J.; Hong, N.; Shim, J.S.; Rhee, Y.; Kim, H.C. Association of Insulin Resistance with Lower Bone Volume and Strength Index of the Proximal Femur in Nondiabetic Postmenopausal Women. J. Bone Metab. 2018, 25, 123–132. [Google Scholar] [CrossRef]
- Mesinovic, J.; McMillan, L.B.; Shore-Lorenti, C.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sex-specific associations between insulin resistance and bone parameters in overweight and obese older adults. Clin. Endocrinol. 2019, 90, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, L.; Shao, X. Association Analysis of Insulin Resistance and Osteoporosis Risk in Chinese Patients with T2DM. Ther. Clin. Risk Manag. 2021, 17, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Chen, S.; Han, H.; Jin, J.; Li, Z. Association between triglyceride glucose index and total bone mineral density: A cross-sectional study from NHANES 2011-2018. Sci. Rep. 2024, 14, 4208. [Google Scholar] [CrossRef] [PubMed]
- Cherif, R.; Mahjoub, F.; Sahli, H.; Cheour, E.; Vico, L.; Sakly, M.; Attia, N. Positive Association of Obesity and Insulin Resistance With Bone Mineral Density in Tunisian Postmenopausal Women. J. Clin. Densitom. Off. J. Int. Soc. Clin. Densitom. 2018, 21, 163–171. [Google Scholar] [CrossRef]
- Gu, P.; Pu, B.; Xin, Q.; Yue, D.; Luo, L.; Tao, J.; Li, H.; Chen, M.; Hu, M.; Hu, X.; et al. The metabolic score of insulin resistance is positively correlated with bone mineral density in postmenopausal patients with type 2 diabetes mellitus. Sci. Rep. 2023, 13, 8796. [Google Scholar] [CrossRef]
- Seeman, E.; Delmas, P.D. Bone quality—The material and structural basis of bone strength and fragility. N. Engl. J. Med. 2006, 354, 2250–2261. [Google Scholar] [CrossRef]
- Roomi, A.B.; AL-Salih, R.; Ali, S.A. The effect insulin therapy and metformin on osteoporosis in diabetic postmenopausal Iraqi women. Indian J. Public Health 2019, 10, 1479. [Google Scholar]
- Liu, D.; Bai, J.J.; Yao, J.J.; Wang, Y.B.; Chen, T.; Xing, Q.; Bai, R. Association of Insulin Glargine Treatment with Bone Mineral Density in Patients with Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 1909–1917. [Google Scholar] [CrossRef]
- Ivaska, K.K.; Heliövaara, M.K.; Ebeling, P.; Bucci, M.; Huovinen, V.; Väänänen, H.K.; Nuutila, P.; Koistinen, H.A. The effects of acute hyperinsulinemia on bone metabolism. Endocr. Connect. 2015, 4, 155–162. [Google Scholar] [CrossRef]
- Ruppert, K.; Cauley, J.; Lian, Y.; Zgibor, J.C.; Derby, C.; Solomon, D.H. The effect of insulin on bone mineral density among women with type 2 diabetes: A SWAN Pharmacoepidemiology study. Osteoporos. Int. A J. Establ. Result Coop. Between Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 2018, 29, 347–354. [Google Scholar] [CrossRef]
- Losada-Grande, E.; Hawley, S.; Soldevila, B.; Martinez-Laguna, D.; Nogues, X.; Diez-Perez, A.; Puig-Domingo, M.; Mauricio, D.; Prieto-Alhambra, D. Insulin use and excess fracture risk in patients with type 2 diabetes: A propensity-matched cohort analysis. Sci. Rep. 2017, 7, 3781. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Peterson, J.; Rizza, R.; Khosla, S. Effects of physiological variations in circulating insulin levels on bone turnover in humans. J. Clin. Endocrinol. Metab. 2011, 96, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Stage, T.B.; Christensen, M.H.; Jørgensen, N.R.; Beck-Nielsen, H.; Brøsen, K.; Gram, J.; Frost, M. Effects of metformin, rosiglitazone and insulin on bone metabolism in patients with type 2 diabetes. Bone 2018, 112, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too much of a good thing is bad. BMC Med. 2020, 18, 224. [Google Scholar] [CrossRef]
- Tong, C.; Wu, Y.; Zhang, L.; Yu, Y. Insulin resistance, autophagy and apoptosis in patients with polycystic ovary syndrome: Association with PI3K signaling pathway. Front. Endocrinol. 2022, 13, 1091147. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Carrillo-López, N.; Martínez-Arias, L.; Fernández-Villabrille, S.; Ruiz-Torres, M.P.; Dusso, A.; Cannata-Andía, J.B.; Naves-Díaz, M.; Panizo, S. Role of the RANK/RANKL/OPG and Wnt/β-Catenin Systems in CKD Bone and Cardiovascular Disorders. Calcif. Tissue Int. 2021, 108, 439–451. [Google Scholar] [CrossRef]
- Kamiya, N.; Kobayashi, T.; Mochida, Y.; Yu, P.B.; Yamauchi, M.; Kronenberg, H.M.; Mishina, Y. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2010, 25, 200–210. [Google Scholar] [CrossRef]
- De Leon-Oliva, D.; Barrena-Blázquez, S.; Jiménez-Álvarez, L.; Fraile-Martinez, O.; García-Montero, C.; López-González, L.; Torres-Carranza, D.; García-Puente, L.M.; Carranza, S.T.; Álvarez-Mon, M.; et al. The RANK-RANKL-OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina 2023, 59, 1752. [Google Scholar] [CrossRef]
- Ayyasamy, R.; Fan, S.; Czernik, P.; Lecka-Czernik, B.; Chattopadhyay, S.; Chakravarti, R. 14-3-3ζ suppresses RANKL signaling by destabilizing TRAF6. J. Biol. Chem. 2024, 300, 107487. [Google Scholar] [CrossRef]
- Deng, T.; Hu, B.; Wang, X.; Ding, S.; Lin, L.; Yan, Y.; Peng, X.; Zheng, X.; Liao, M.; Jin, Y.; et al. TRAF6 autophagic degradation by avibirnavirus VP3 inhibits antiviral innate immunity via blocking NFKB/NF-κB activation. Autophagy 2022, 18, 2781–2798. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, Q. BMP Signaling: Lighting up the Way for Embryonic Dorsoventral Patterning. Front. Cell Dev. Biol. 2021, 9, 799772. [Google Scholar] [CrossRef] [PubMed]
- Manzari-Tavakoli, A.; Babajani, A.; Farjoo, M.H.; Hajinasrollah, M.; Bahrami, S.; Niknejad, H. The Cross-Talks Among Bone Morphogenetic Protein (BMP) Signaling and Other Prominent Pathways Involved in Neural Differentiation. Front. Mol. Neurosci. 2022, 15, 827275. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.L.; Chen, Z.H.; Teng, Y.Y.; Liu, S.Y.; Jia, Y.; Zhang, K.W.; Sun, Z.L.; Wu, J.J.; Yuan, Z.D.; Feng, Y.; et al. The Smad Dependent TGF-β and BMP Signaling Pathway in Bone Remodeling and Therapies. Front. Mol. Biosci. 2021, 8, 593310. [Google Scholar] [CrossRef]
- Jeong, J.S.; Lee, W.K.; Moon, Y.S.; Kim, N.R. Early changes in retinal structure and BMP2 expression in the retina and crystalline lens of streptozotocin-induced diabetic pigs. Lab. Anim. Res. 2017, 33, 216–222. [Google Scholar] [CrossRef]
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.D.; Sun, P.Y.; Davies, K.J.A.; Grune, T. Sarcopenia—Molecular mechanisms and open questions. Ageing Res. Rev. 2021, 65, 101200. [Google Scholar] [CrossRef]
- Liu, Z.-j.; Zhu, C.-f. Causal relationship between insulin resistance and sarcopenia. Diabetol. Metab. Syndr. 2023, 15, 46. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.; Dong, M.; Wen, S.; Zhou, L.; Yuan, X. The Association Between Sarcopenia and Diabetes: From Pathophysiology Mechanism to Therapeutic Strategy. Diabetes Metab. Syndr. Obes. Targets Ther. 2023, 16, 1541–1554. [Google Scholar] [CrossRef]
- Das, C.; Das, P.P.; Kambhampati, S.B.S. Sarcopenia and Osteoporosis. Indian J. Orthop. 2023, 57, 33–41. [Google Scholar] [CrossRef]
- Zhi, X.; Chen, Q.; Song, S.; Gu, Z.; Wei, W.; Chen, H.; Chen, X.; Weng, W.; Zhou, Q.; Cui, J.; et al. Myostatin Promotes Osteoclastogenesis by Regulating Ccdc50 Gene Expression and RANKL-Induced NF-κB and MAPK Pathways. Front. Pharmacol. 2020, 11, 565163. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Zeng, X.; Yi, X.; Yang, Y.; Hu, J.; Yin, R.; Chen, X. The Function of Myostatin in Ameliorating Bone Metabolism Abnormalities in Individuals with Type 2 Diabetes Mellitus by Exercise. Curr. Issues Mol. Biol. 2025, 47, 158. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Chin, K.-Y.; Suhaimi, F.H.; Ahmad, F.; Jamil, N.A.; Ima-Nirwana, S. Osteoporosis is associated with metabolic syndrome induced by high-carbohydrate high-fat diet in a rat model. Biomed. Pharmacother. 2018, 98, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Karimi Fard, M.; Aminorroaya, A.; Kachuei, A.; Salamat, M.R.; Hadi Alijanvand, M.; Aminorroaya Yamini, S.; Karimifar, M.; Feizi, A.; Amini, M. Alendronate improves fasting plasma glucose and insulin sensitivity, and decreases insulin resistance in prediabetic osteopenic postmenopausal women: A randomized triple-blind clinical trial. J. Diabetes Investig. 2019, 10, 731–737. [Google Scholar] [CrossRef]
- Maugeri, D.; Panebianco, P.; Rosso, D.; Calanna, A.; Speciale, S.; Santangelo, A.; Rizza, I.; Motta, M.; Lentini, A.; Malaguarnera, M. Alendronate reduces the daily consumption of insulin (DCI) in patients with senile type I diabetes and osteoporosis. Arch. Gerontol. Geriatr. 2002, 34, 117–122. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Wu, Z.; Li, W.; Jiang, K.; Lin, Z.; Qian, C.; Wu, M.; Xia, Y.; Li, N.; Zhang, H.; Xiao, H.; et al. Regulation of bone homeostasis: Signaling pathways and therapeutic targets. MedComm 2024, 5, e657. [Google Scholar] [CrossRef]
- Hu, L.; Chen, W.; Qian, A.; Li, Y.-P. Wnt/β-catenin signaling components and mechanisms in bone formation, homeostasis, and disease. Bone Res. 2024, 12, 39. [Google Scholar] [CrossRef]
| Type of Animal Model | Treatment/Intervention (Dose, Route and Duration) | Findings | Reference |
|---|---|---|---|
| STZ-induced diabetic rats | Neutral protamine Hagedorn insulin (2.5 IU twice daily, subcutaneous injection, 8 weeks) | Calcium: ↑; ionised calcium: ↔; phosphorus: ↔; ALP: ↓; Tb.Sp: ↓; trabecular width: ↑; trabecular bone area: ↑; load: ↔; stiffness: ↔; stress: ↔; strain: ↔; Young’s modulus: ↑; RANKL: ↔; OPG: ↑; OCN: ↑ | [25] |
| STZ induced diabetic rats | Neutral protamine Hagedorn (100 IU twice daily, subcutaneous injection, 30 days) | Glucose: ↓; alveolar bone loss: ↓; OPG: ↑; RANKL: ↔ | [26] |
| STZ-induced diabetic mice | Humulin R insulin (0.25 IU/day, osmotic minipump, 4 weeks) | Glucose: ↓; CTX: ↓; P1NP: ↑; stiffness: ↑; peak force: ↑; femoral length: ↑; modulus: ↔; bending strength: ↔; toughness: ↔ Cortical parameters: Ct.Ar: ↑; Ct.Th: ↑; mineral density: ↔ Trabecular parameters: BV/TV: ↑; Tb.N: ↑; Tb.Th: ↑; mineral density: ↑ | [13] |
| STZ-induced diabetic rats | Insulin from bovine pancreas (1.6 IU/day, continuous subcutaneous infusion, 4 weeks) | Insulin: ↑; insulin receptor: ↔; femur length: ↑; tibia length: ↑; bone weight (femur, tibia, proximal tibia): ↑; ALP: ↑; TRAP: ↔; CTSK: ↔; calcium: ↑; hydroxyproline: ↑; deoxypyridinoline: ↔; BV/TV: ↑; trabecular bone surface: ↑; Tb.Th: ↑; Tb.N: ↑; Ob.N: ↑; Oc.N: ↔; BMP-2: ↔; Dlx5: ↑; Runx2: ↑; OSX: ↑; OCN: ↑; COL1: ↑; Wnt3a: ↔; LRP5: ↔; Akt: ↔; p-Akt: ↑; GSK3β: ↔; p-GSK3β: ↑; β-catenin: ↑; p-β-catenin: ↑; Sost: ↓; DKK1: ↓ | [12] |
| Zucker diabetic fatty rats fed high-fat, high-carbohydrate diet | Insulin glargine (0.5–13.0 IU/ daily, subcutaneous injection, 12 weeks) | Non-enzymatic glycation: ↔; tissue mineral density ratio: ↔; elastic modulus: ↔; toughness: ↔; stress: ↔ Cortical parameters: Porosity: ↔ | [20] |
| Zucker diabetic fatty rats with subcritical femur defects | Insulin glargine (0.5–13.0 IU/ daily, subcutaneous injection, 12 weeks) | Glucose: ↓; HbA1c: ↓; calcium: ↔; phosphate: ↔; P1NP: ↑; CTX: ↔; TRAP: ↑; PTH: ↑; force: ↔; work to failure: ↔; BFR: ↑ Cortical parameters: BV/TV: ↑; BMD: ↔; Ct.Th: ↔ Trabecular parameters: BV/TV: ↔; BMD: ↔; Tb.N: ↔; Tb.Th: ↔ | [21] |
| STZ- and high-fat-diet-induced diabetic rats | Insulin (2.5 IU/day, continuous subcutaneous infusion, 4 weeks) | Glucose: ↓; OCN: ↑; TRAP: ↔; CTX: ↓; BMD: ↔; load: ↔; elasticity: ↔; toughness: ↑; matrix mineralisation: ↓ Trabecular parameters: BV/TV: ↑; Tb.N: ↑; Tb.Th: ↑; Tb.Sp: ↓; indentation modulus: ↔; hardness: ↔; surface grain size: ↔; roughness: ↔; OPG: ↑; RANKL: ↓ Cortical parameters: Indentation modulus: ↔; hardness: ↔; surface grain size: ↔; roughness: ↓; OPG: ↑; RANKL: ↓ | [27] |
| Insulin (2.5 IU/day, continuous subcutaneous infusion, 8 weeks) | Glucose: ↔; OCN: ↑; TRAP: ↓; CTX: ↓; BMD: ↔; load: ↑; elasticity: ↑; toughness: ↑; matrix mineralisation: ↓ Trabecular parameters: BV/TV: ↑; Tb.N: ↑; Tb.Th: ↑; Tb.Sp: ↓; indentation modulus: ↔; hardness: ↑; surface grain size: ↓; roughness: ↔; OPG: ↑; RANKL: ↓ Cortical parameters: Indentation modulus: ↑; hardness: ↑; surface grain size: ↓; roughness: ↓; OPG: ↑; RANKL: ↓ |
| Type of Animal Model | Findings | Reference |
|---|---|---|
| Mice with IRβ deletion (15 weeks) | Cortical parameters: BV: ↔; BV/TV: ↔; Ct.Th: ↔; mineral density: ↔ Trabecular parameters: BV/TV: ↔; Tb.N: ↔; Tb.Sp: ↔; Tb.Th: ↔; ultimate torque: ↔; torsional rigidity: ↔ | [28] |
| Mice with IRβ deletion (48 weeks) | Cortical parameters: BV: ↔; BV/TV: ↔; Ct.Th: ↔; mineral density: ↔ Trabecular parameters: BV/TV: ↓; Tb.N: ↔; Tb.Sp: ↔; Tb.Th: ↔; ultimate torque: ↔; torsional rigidity: ↓ | |
| Mice lacking insulin receptors in osteoblasts | Insulin: ↓; glucose: ↑; BV/TV: ↓; Tb.N: ↓; Tb.Th: ↓; Tb.Sp: ↑; Ob.N: ↓; BFR: ↔; Oc.N: ↔; erosion depth: ↓; OCN: ↓; CTX: ↓; OPG: ↑ | [29] |
| Female mice with osteoprogenitor-selective ablation of insulin receptors | Femur length: ↓; femur slenderness: ↑; force: ↓; stiffness: ↓; elasticity: ↔; binding strength: ↔; OCN: ↔; CTX: ↔ Cortical parameters: Ct.Th: ↓; Ct.Ar: ↓; mineral density: ↓ Trabecular parameters: BV/TV: ↔; Tb.Th: ↔; Tb.N: ↔; Tb.Sp: ↔; SMI: ↓; mineral density: ↔ | [30] |
| Male mice with osteoprogenitor-selective ablation of insulin receptors | Femur length: ↓; femur slenderness: ↑; force: ↓; stiffness: ↔; elasticity: ↔; binding strength: ↔; OCN: ↔; CTX: ↓ Cortical parameters: Ct.Th: ↓; Ct.Ar: ↓; mineral density: ↓ Trabecular parameters: BV/TV: ↓; Tb.Th: ↓; Tb.N: ↔; Tb.Sp: ↔; SMI: ↑; mineral density: ↔; binding strength: ↔; OCN: ↔; CTX: ↑ | |
| Transgenic insulin receptor knockout mice | Tibial length: ↔; pyridinoline: ↔; Runx2: ↔; OCN: ↔ Cortical parameters: BMC: ↔; BMD: ↔; volume fraction: ↔; Ct.Th: ↔; inner perimeter: ↓; medullary area: ↓; BA/TA: ↑ Trabecular parameters: BMC: ↔; BMD: ↔; volume fraction: ↔; Tb.Th: ↔; Tb.Sp: ↔; Conn.D: ↔ | [31] |
| Type of Study | Subject Characteristics | Findings | References |
|---|---|---|---|
| Cross-sectional study | Non-diabetic postmenopausal women with suspected or diagnosed osteoporosis (n = 381; aged 62 ± 9 years old) | Women with higher insulin level had higher hip BMD and volumetric BMD. | [32] |
| Cross-sectional study | Non-diabetic postmenopausal women (n = 437; aged 51–56 years) | Fasting insulin level was positively associated with BMD (β = 0.033). | [33] |
| Cross-sectional study | Boys (n = 1344; age = 15.47 ± 0.3 years old) and girls (n = 1440; aged 15.48 ± 0.3 years old) | Insulin level was negatively correlated with cortical BMD and periosteal circumference. | [34] |
| Cross-sectional study | Participants in biomarker project of Midlife in the United States Study (n = 717; aged 56.8 ± 11.3 years old) | A higher level of fasting insulin was associated with lower femoral neck (effect size = −0.099; 95% CI −0.19, −0.01) and lumbar spine BMD (effect size = −0.129; 95% CI −0.23, −0.03). A higher level of fasting insulin was associated with lower compression (effect size = −0.121; 95% CI −0.19, −0.05), bending (effect size = −0.180; 95% CI −0.27, −0.09), and impact (effect size = −0.158; 95% CI −0.25, −0.07) strength. | [35] |
| Cross-sectional study | Young men (n = 113; aged 19.8 ± 2.3 years old) and women (n = 353; aged 19.4 ± 2.2 years old) | Insulin level was positively associated with total body BMC, mediated by lean mass. | [36] |
| Type of Study | Subject Characteristics | Findings | Reference |
|---|---|---|---|
| Cross-sectional study | Iranian boys and girls (n = 423; aged 13.93 ± 2.64 years old) | HOMA-IR was correlated with low lumbar spine BMC (β = −1.1; p = 0.008), BMD (β = −0.01; p = 0.011), BMAD (β = −0.002; p = 0.029), and z-score (β = −0.105; p = 0.009); low femoral neck BMC (β = −0.06; p = 0.004) and BMD (β = −0.010; p = 0.010); and low whole-body BMD (β = −0.005; p = 0.029) and z-score (β = −0.076; 0.036). QUICKI was associated high lumbar spine BMC (β = 37.21; p = 0.0001), BMD (β = 0.277; p = 0.007), BMAD (β = 0.062; p = 0.026), and z-score (β = 2.63; p = 0.009); and high femoral neck BMC (β = 1.297; p = 0.013). | [44] |
| Cross-sectional study | Participants in the NHANES (n = 5292; aged ≥ 18 years old) | Subjects with HOMA-IR ≥ 2 and HOMA-β < 100 had a higher risk of osteoporosis (OR = 1.070; 95% CI 0.656, 1.744) as compared to subjects with HOMA-IR < 2 and HOMA-β < 100. Subjects with HOMA-IR ≥ 2 and HOMA-β ≥ 100 had a higher risk of osteoporosis (OR = 1.256; 95% CI 0.625, 2.526) as compared to subjects with HOMA-IR < 2 and HOMA-β < 100. | [45] |
| Longitudinal study | Chinese adults without low bone mass or osteoporosis (n = 8770; aged ≥ 18 years old) | TyG index was negatively associated with bone mass (HR = 1.56; 95% CI: 1.25, 1.93), osteoporosis (HR = 1.66; 95% CI: 1.06, 2.59), and both (HR = 1.55; 95% CI: 1.27, 1.88). | [46] |
| Cross-sectional study | Participants in biomarker project of Midlife in the United States Study (n = 717; aged 56.8 ± 11.3 years old) | HOMA-IR was negatively associated with femoral neck compression (effect size = −0.091; 95% CI: −0.153, −0.030), bending (effect size = −0.141; 95% CI: −0.222, −0.060), and impact (effect size = −0.141; 95% CI: −0.222, −0.048) strength. HOMA-IR was negatively associated with lumbar spine BMD (effect size = −0.087; 95% CI: −0.171, −0.002). | [35] |
| Cross-sectional study | Men and women (n = 525; aged ≥ 60 years old) | Insulin resistance was associated with low CTX (estimate = −24.8%; 95% CI: −38.9, −7.5) in men. T2DM was associated with low bone turnover marker levels in men [CTX (estimate = −34.7%; 95% CI: −48.1, −17.8)] and women [OCN (estimate = −31.9%; 95% CI: −41.8, −20.4), P1NP (estimate = −26.0%; 95% CI: −38.7, −10.8), and CTX (estimate = −30.9%; 95% CI: −46.2, −11.3)]. | [47] |
| Cross-sectional study | Subjects with dysglycaemia (n = 5277; aged ≥ 18 years old) | HOMA-IR was negatively associated with CTX (β = −0.044; 95% CI −0.053, −0.035), P1NP (β = −7.340; 95% CI −9.130, −5.550), and OCN (β = −2.885; 95% CI −3.357, −2.412). HOMA-β was positively associated with CTX (β = 0.022; 95% CI 0.014, 0.031), P1NP (β = 6.951; 95% CI 5.300, 8.602), and OCN (β = 1.361; 95% CI 0.921, 1.800). | [48] |
| Cohort study | Premenopausal (n = 861; aged 45.44 ± 2.51 years old), menopausal transition (n = 571; aged 50.71 ± 2.48 years old), and postmenopausal (n = 693; aged 55.11 ± 3.35 years old) women | Slower BMD loss was observed when HOMA-IR level was <2.82, while faster BMD loss was evident when HOMA-IR level was ≥2.82 in all groups. | [49] |
| Cross-sectional study | Non-diabetic postmenopausal women (n = 1008; aged ≥ 50 years old) | HOMA-IR was associated with low total bone volume at the femoral neck (β = −0.12), intertrochanter (β = −0.43), and total proximal femur (β = −0.76); low cortical volume at the femoral neck (β = −0.05); high cortical BMD at the femoral neck (β = 23.0), intertrochanter (β = 6.8), and total proximal femur (β = 12.2); and low femoral neck strength indices [estimated cortical depth (β = −0.011)], compressive strength index (β = −0.013), section modulus (β = −0.017) | [50] |
| Cross-sectional study | Overweight and obese men and women (n = 79; aged 62.8 ± 7.9 years old) | HOMA-IR was associated with low proximal radius cortical BMD in women (β = −4.79; 95% CI −8.66, −0.92). There was no association between HOMA-IR and bone parameters in men. | [51] |
| Cross-sectional study | T2DM patients (n = 234; aged 57.5 ± 10.8 years old) | HOMA-IR was associated with higher risk of osteoporosis in females (OR = 2.63; 95% CI 1.15, 5.99). | [52] |
| Cross-sectional study | Men and women from United States (n = 5456; aged 30.33 ± 13.55 years old) | TyG (β = 0.0124; 95% CI 0.0006, 0.0242), TyG-WC (β = 0.0001; 95% CI 0.0001, 0.0001), TyG-WHtR (β = 0.0116; 95% CI 0.0076, 0.0156), and TyG-BMI (β = 0.0004; 95% CI 0.0003, 0.0004) were associated with high total BMD. | [53] |
| Cohort study | Postmenopausal women (n = 81; aged 58.40 ± 6.08 years old) | Higher left femur and total hip BMD was observed in the insulin resistant than the non-insulin resistant group. | [54] |
| Cross-sectional study | Non-diabetic postmenopausal women (n = 437; aged 51–56 years old) | HOMA-IR (β = 0.139), HOMA-β (β = 0.137), and MET-IR (β = 0.145) were positively associated with femoral neck BMD. HOMA-IR (β = 0.131), HOMA-β (β = 0.134), and MET-IR (β = 0.138) were positively associated with femoral neck T-score. | [33] |
| Cross-sectional study | Non-diabetic postmenopausal women with suspected or diagnosed osteoporosis (n = 381; mean aged 62 ± 9 years old) | Women with higher HOMA-IR values had higher hip BMD and volumetric BMD. No association was found between HOMA-IR and trabecular bone score. | [32] |
| Retrospective study | Postmenopausal T2DM patients with (n = 91; aged 68.74 ± 7.89 years old) and without osteoporosis (n = 119; aged 61.94 ± 7.33 years old) | METS-IR was positively associated with lumbar spine (β = 0.006), femoral neck (β = 0.005), and hip (β = 0.005) BMD. | [55] |
| Type of Study | Subject Characteristics | Findings | Reference |
|---|---|---|---|
| Cohort study | Healthy postmenopausal women, postmenopausal women with osteoporosis, T2DM postmenopausal women with insulin treatment once daily, and T2DM postmenopausal women with metformin (500 mg) treatment twice daily (n = 200; aged 50–73 years old) | The levels of BMD were higher and OCN and CTX were lower in T2DM women taking insulin compared to postmenopausal women with osteoporosis. | [57] |
| Retrospective study | Non-diabetic controls (n = 30; aged 42.34 ± 6.71 years old), T2DM subjects receiving oral glucose-lowering medication (n = 25; aged 44.44 ± 8.13 years old), and T2DM subjects receiving oral glucose-lowering medication with insulin glargine injection (n = 25; aged 42.60 ± 9.25 years old) | T2DM subjects receiving oral glucose-lowering medication with insulin glargine injection had higher lumbar vertebrae and spine BMD and serum calcium levels but lower serum phosphate levels as compared to those taking oral glucose-lowering medication only. | [58] |
| Cohort study | Cohort 1: Healthy adults (n = 8; age: 37–63 years) | CTX: ↓; TRAP: ↔; ucOCN: ↔; OCN: ↔; P1NP: ↔. | [59] |
| Cohort 2: Healthy young men (n = 12; age = 18–34 years) | CTX: ↓; TRAP: ↔; ucOCN: ↓; OCN: ↔; P1NP: ↔. | ||
| Cohort 3: Healthy elderly women (n = 13; age = 69–79 years) | CTX: ↔; TRAP: ↓; ucOCN: ↓; OCN: ↓; P1NP: ↔. | ||
| Cohort study | Women with T2DM using insulin (n = 55; aged 53.9 ± 5.7 years old) and not using insulin (n = 55; aged 53.3 ± 4.9 years old) | Insulin users experienced a greater loss in BMD at the femoral neck than non-insulin users, while BMD at spine and total hip were not affected. | [60] |
| Cohort study | T2DM patients using insulin (n = 2979; aged 61.7 ± 11.9 years old) and not using insulin (n = 14,895; aged 61.8 ± 11.9 years old) | Major fracture rates were higher in insulin users than non-insulin users. An association was confirmed between insulin use and fracture risk (subhazard ratio = 1.38; 95% CI 1.06, 1.80) | [61] |
| Cohort study | Diabetic (n = 7; aged 62.0 ± 1.8 years old) and non-diabetic (n = 7; aged 57.9 ± 4.0 years old) subjects | P1NP, OCN, ucOCN, CTX, and OPG levels were not significantly different at the end of low-, intermediate- and high-dose insulin treatment. | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindran, S.; Wong, S.K.; Mohamad, N.-V.; Chin, K.-Y. A Review of the Relationship Between Insulin and Bone Health. Biomedicines 2025, 13, 1504. https://doi.org/10.3390/biomedicines13061504
Ravindran S, Wong SK, Mohamad N-V, Chin K-Y. A Review of the Relationship Between Insulin and Bone Health. Biomedicines. 2025; 13(6):1504. https://doi.org/10.3390/biomedicines13061504
Chicago/Turabian StyleRavindran, Sivasree, Sok Kuan Wong, Nur-Vaizura Mohamad, and Kok-Yong Chin. 2025. "A Review of the Relationship Between Insulin and Bone Health" Biomedicines 13, no. 6: 1504. https://doi.org/10.3390/biomedicines13061504
APA StyleRavindran, S., Wong, S. K., Mohamad, N.-V., & Chin, K.-Y. (2025). A Review of the Relationship Between Insulin and Bone Health. Biomedicines, 13(6), 1504. https://doi.org/10.3390/biomedicines13061504