
Identification Exploring the Mechanism and Clinical Validation of Mitochondrial Dynamics-Related Genes in Membranous Nephropathy Based on Mendelian Randomization Study and Bioinformatics Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Differential Expression Analysis
2.3. Weighted Gene Co-Expression Network (WGCNA) Analysis
2.4. Identification and Functional Analysis of MD-DEGs
2.5. Selection of Single Nucleotide Polymorphisms (SNPs)
2.6. Mendelian Randomization (MR) Analysis
2.7. Identification and Analysis of Key Genes
2.8. Construction of Nomogram
2.9. Analysis of Correlation and Functional Resemblance Among Key Genes
2.10. Chromosome and Subcellular Localization Analysis
2.11. Gene Set Enrichment Analysis (GSEA)
2.12. Immune Cell Infiltration and Immune Factor Correlation
2.13. Construction of Regulatory Networks
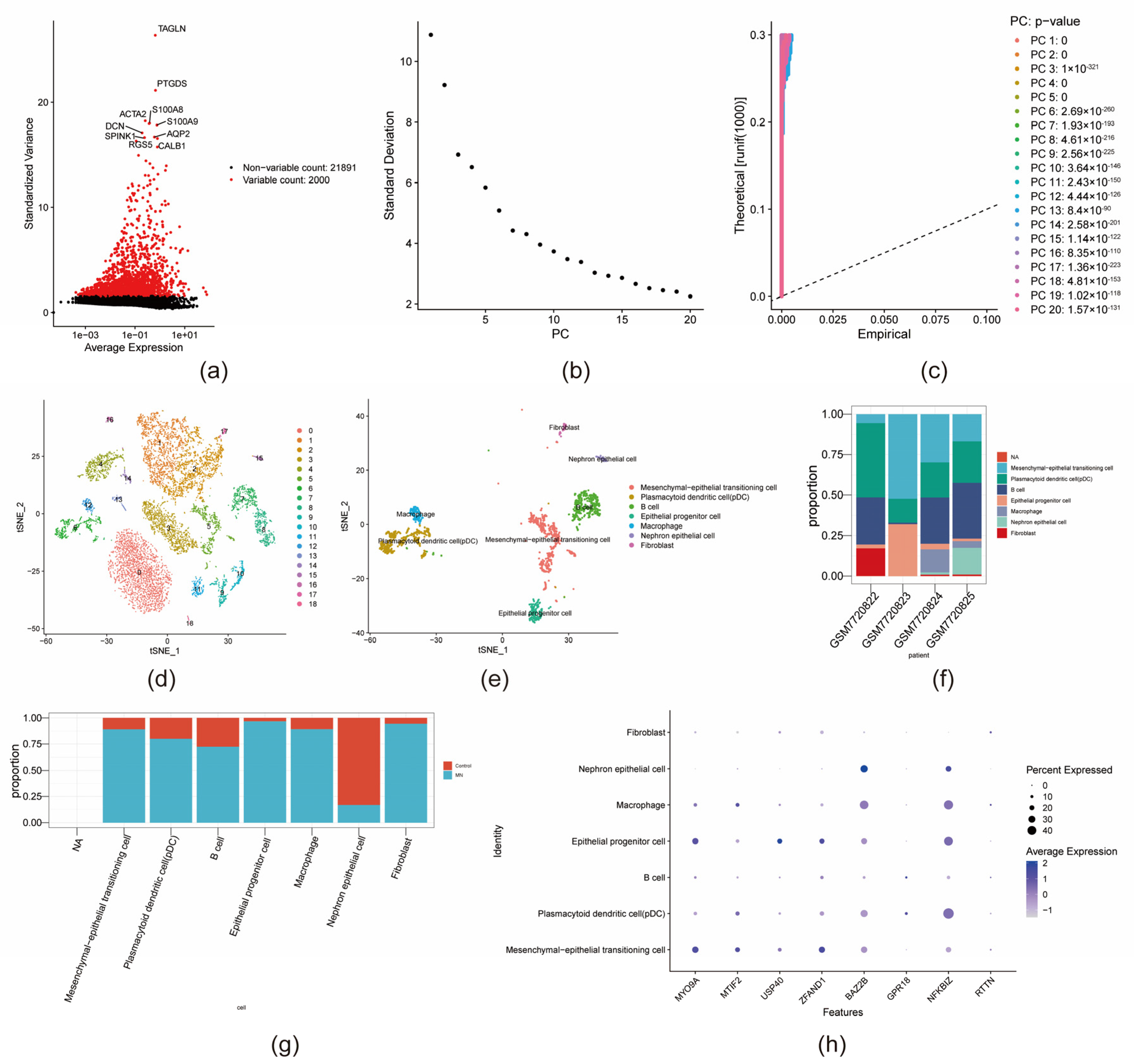
2.14. Single-Cell Analysis
2.15. Cellular Communication and Pseudotime Analysis
2.16. Reverse-Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.17. Statistical Analysis
3. Results
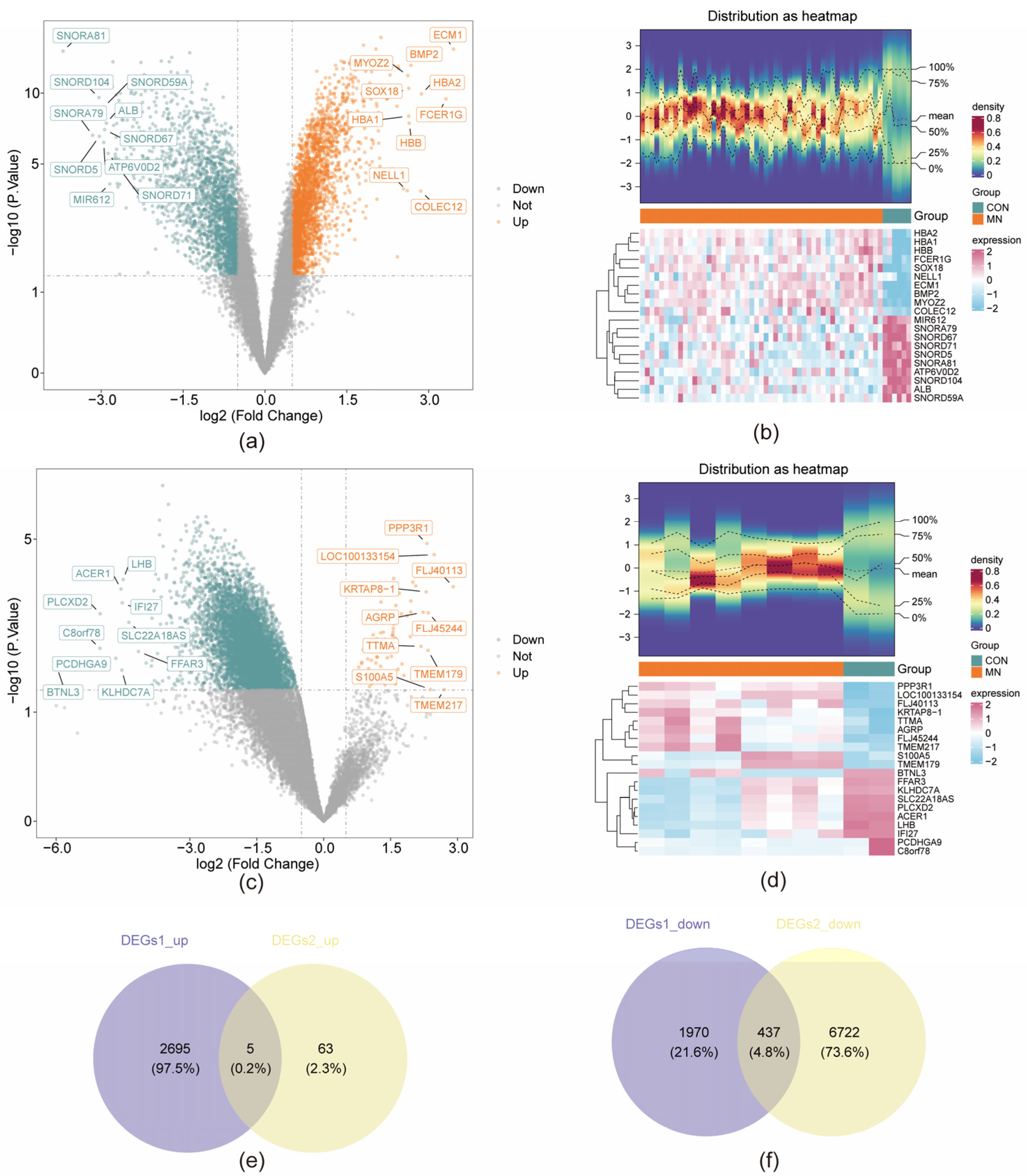
3.1. Identification of MD-DEGs
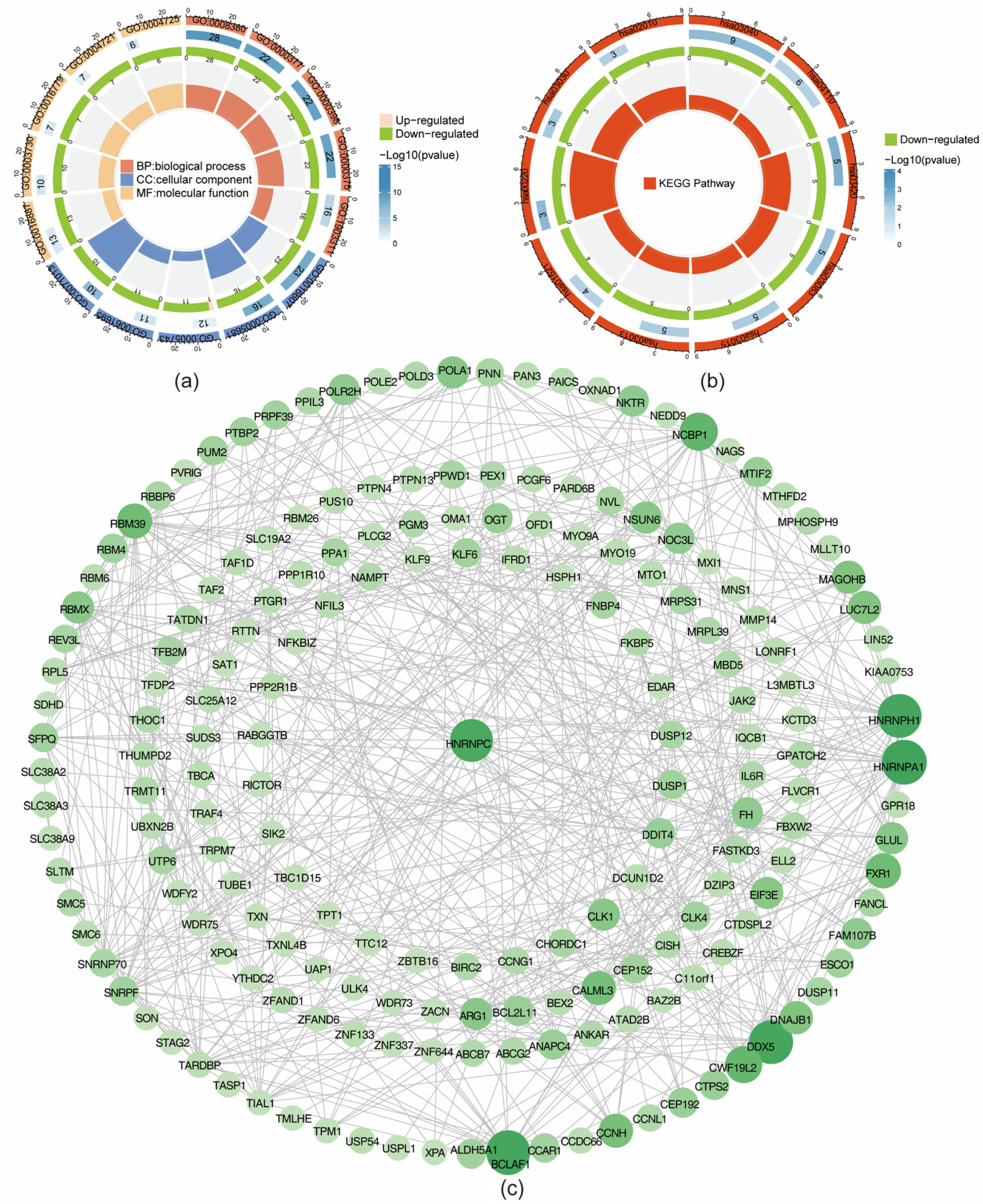
3.2. MD-DEGs Enriched Pathways and Protein Interactions Analysis
3.3. MR Screening of Candidate Genes
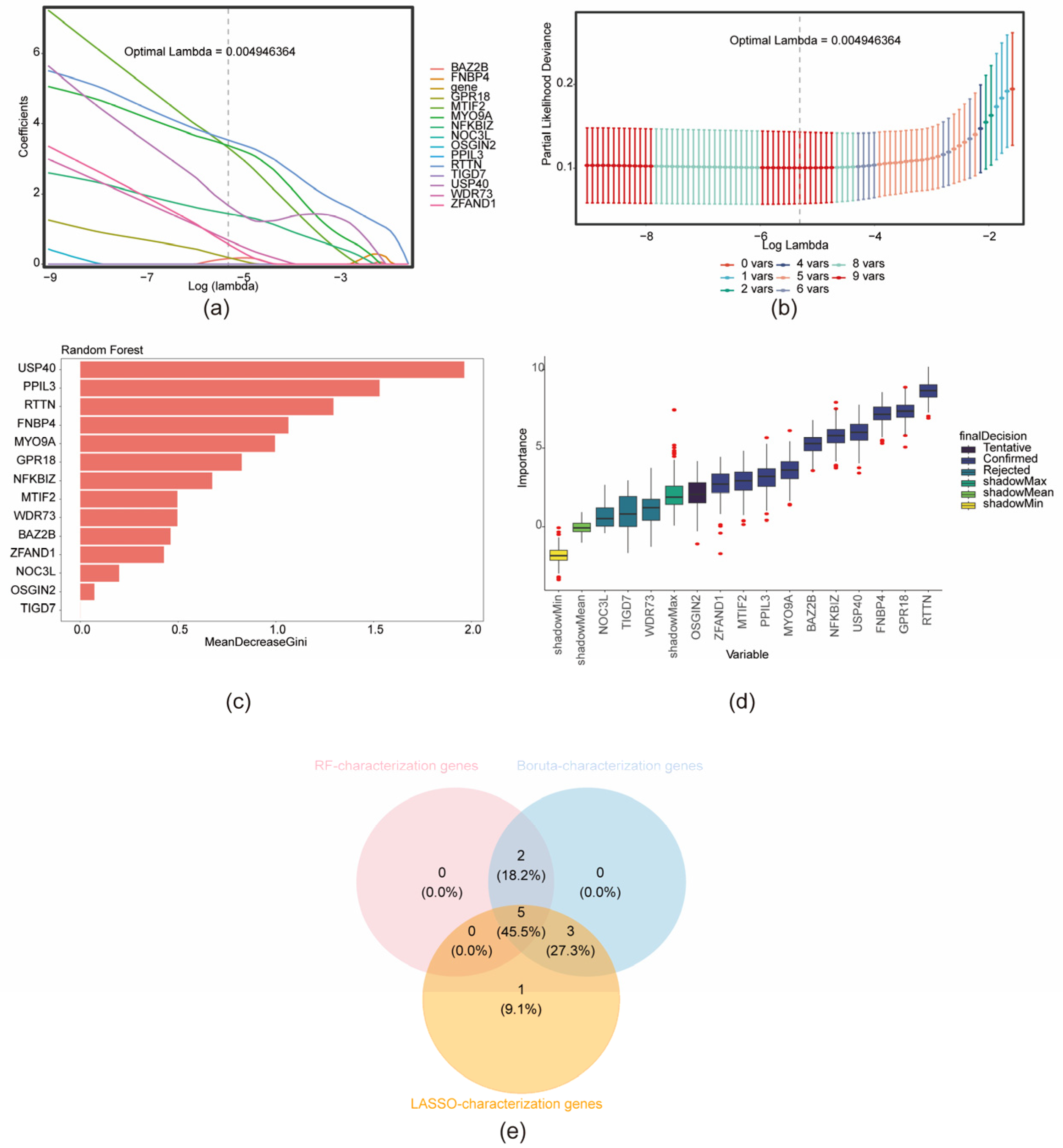
3.4. Identification of Key Genes and Construction of Nomogram
3.5. Subcellular and Chromosomal Localization and Functional Enrichment Analysis
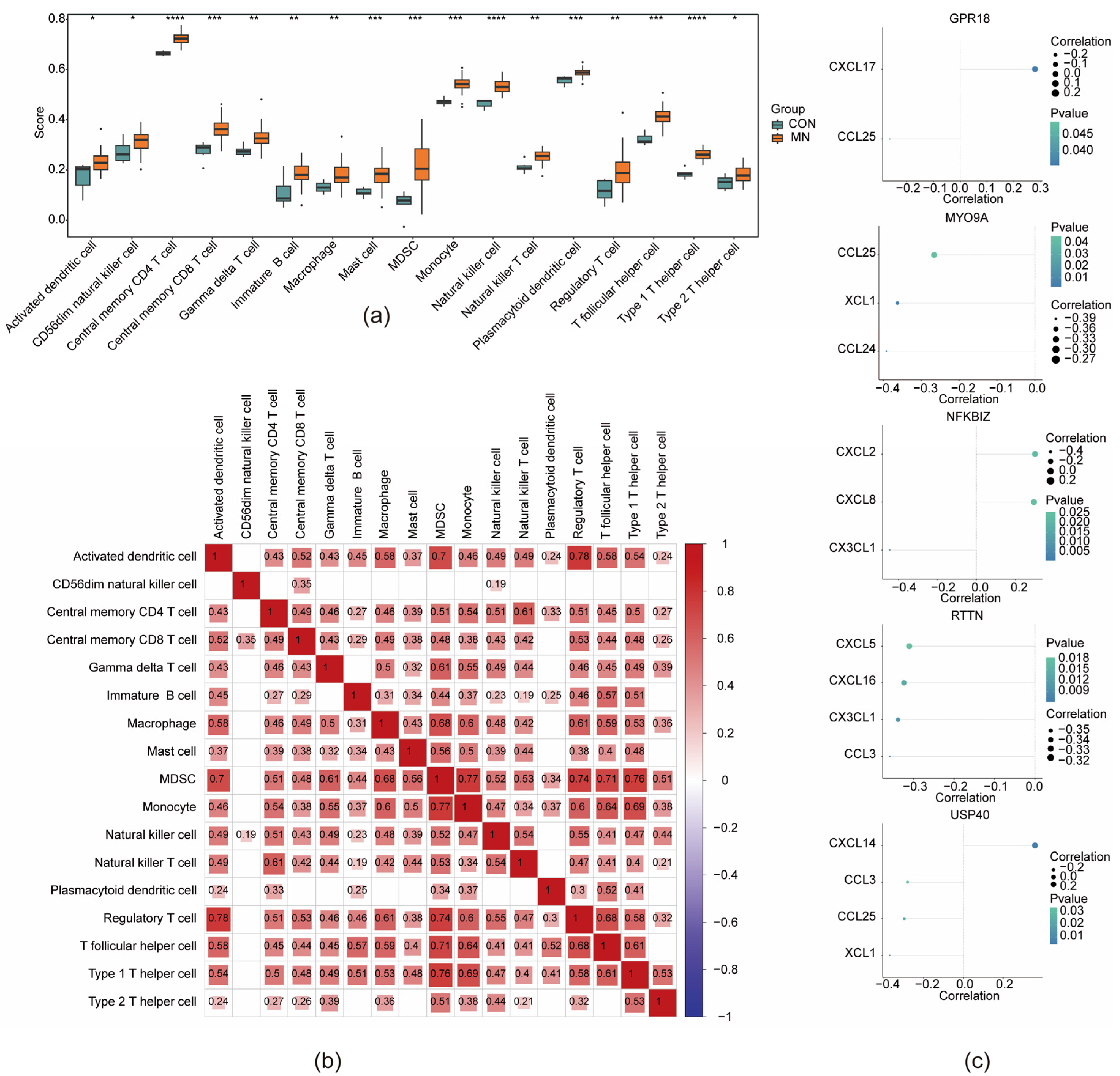
3.6. Immune Microenvironment in MN
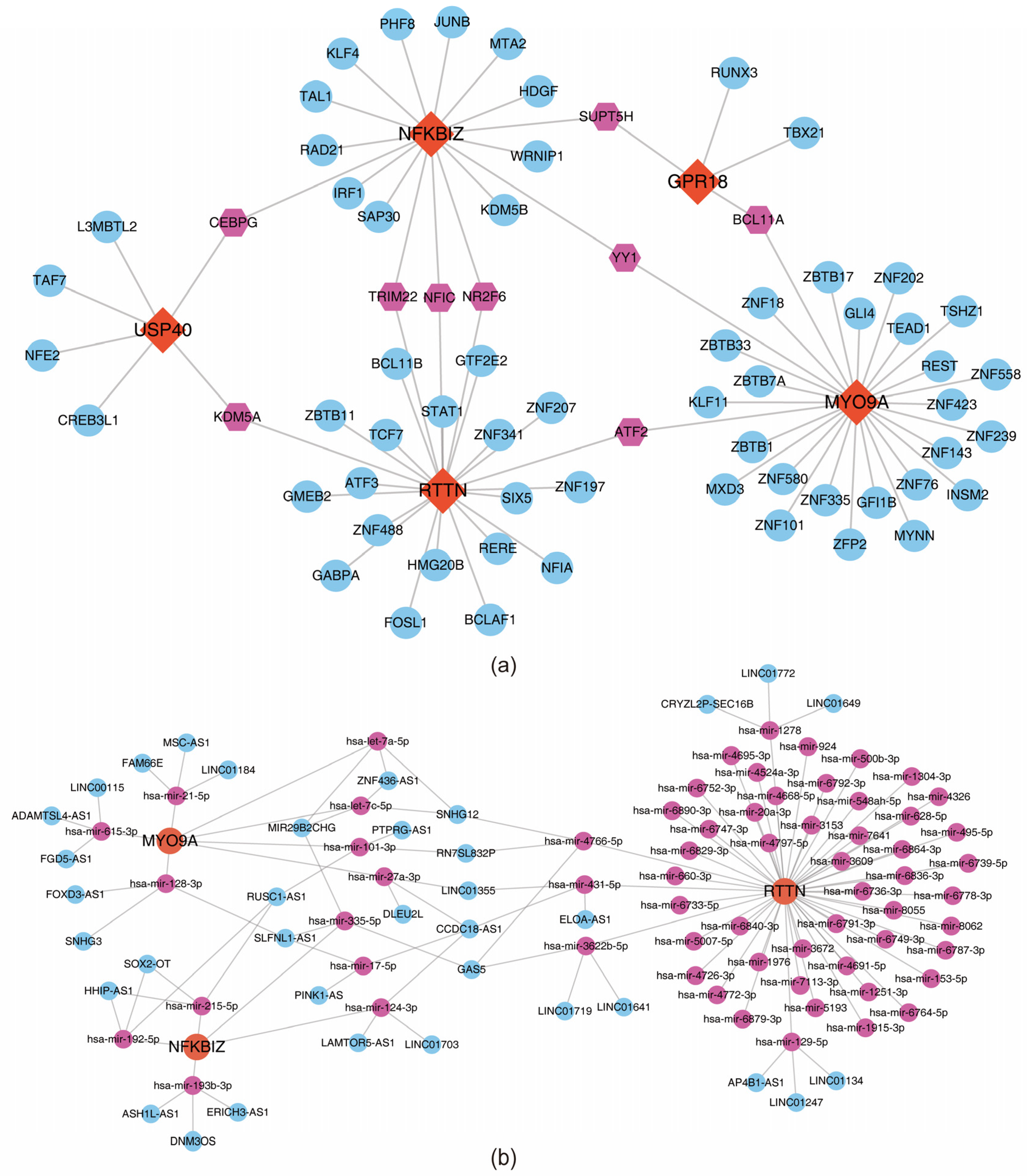
3.7. Acquisition of Regulatory Relationships
3.8. Identification of Key Cell
3.9. Communication Network and Pseudotime Analysis
3.10. Validation of the Expression of Key Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MN | Membranous Nephropathy |
| MD | Mitochondrial Dynamics |
| MDGs | Mitochondrial Dynamics-Related Genes |
| scRNA-seq | Single-Cell RNA Sequencing |
| AUC | Area Under the Curve |
| RT-qPCR | Reverse-Transcription Quantitative PCR |
| GEO | Gene Expression Omnibus |
| DEGs | Differentially Expressed Genes |
| WGCNA | Weighted Gene Co-expression Network Analysis |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | Protein–Protein Interaction |
| SNPs | Single Nucleotide Polymorphisms |
| MR | Mendelian Randomization |
| IVW | Inverse Variance Weighted |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| RF | Random Forest |
| ROC | Receiver Operating Characteristic |
| PCA | Principal Component Analysis |
| MET | Mesenchymal–Epithelial Transition |
| EMT | Epithelial–Mesenchymal Transition |
| TF | Transcription Factor |
| miRNA | MicroRNA |
| lncRNA | Long Non-coding RNA |
| Tfh | Follicular Helper T cells |
| Tfr | Follicular Regulatory T cells |
| PMN | Primary Membranous Nephropathy |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| PD-1 | Programmed Cell Death Protein 1 |
| IL-21 | Interleukin-21 |
| PKR1 | Pre-Kinase Receptor 1 |
References
- Avasare, R.; Andeen, N.; Beck, L. Novel Antigens and Clinical Updates in Membranous Nephropathy. Annu. Rev. Med. 2024, 75, 219–332. [Google Scholar] [CrossRef] [PubMed]
- Prunotto, M.; Nachman, P.H.; Gillespie, B.S.; Beck, L.H., Jr.; Thompson, A.M.; Hu, A.H.; Stafford, E.A.; Tarnoff, J.M.; Rovin, B.H. Designing clinical trials for the treatment of membranous nephropathy in the anti-PLA2R era: Results of a Nephcure membranous nephropathy workshop. Kidney Int. 2025, 107, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Storrar, J.; Gill-Taylor, T.; Chinnadurai, R.; Chrysochou, C.; Poulikakos, D.; Rainone, F.; Ritchie, J.; Lamerton, E.; Kalra, P.A.; Sinha, S. A low rate of end-stage kidney disease in membranous nephropathy: A single centre study over 2 decades. PLoS ONE 2022, 17, e0276053. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B. Visualizing Mitochondrial Form and Function within the Cell. Trends Mol. Med. 2020, 26, 58–70. [Google Scholar] [CrossRef]
- Li, H.; Zhou, L.; Zhou, W.; Zhang, X.; Shang, J.; Feng, X.; Yu, L.; Fan, J.; Ren, J.; Zhang, R.; et al. Decoding the mitochondrial connection: Development and validation of biomarkers for classifying and treating systemic lupus erythematosus through bioinformatics and machine learning. BMC Rheumatol. 2023, 7, 44. [Google Scholar] [CrossRef]
- Wang, H.; Lv, D.; Jiang, S.; Hou, Q.; Zhang, L.; Li, S.; Zhu, X.; Xu, X.; Wen, J.; Zeng, C.; et al. Complement induces podocyte pyroptosis in membranous nephropathy by mediating mitochondrial dysfunction. Cell Death Dis. 2022, 13, 281. [Google Scholar] [CrossRef]
- Debiec, H.; Ronco, P. Immunopathogenesis of membranous nephropathy: An update. Semin. Immunopathol. 2014, 36, 381–397. [Google Scholar] [CrossRef]
- Zhang, D.; Ji, P.; Sun, R.; Zhou, H.; Huang, L.; Kong, L.; Li, W.; Li, W. Ginsenoside Rg1 attenuates LPS-induced chronic renal injury by inhibiting NOX4-NLRP3 signaling in mice. Biomed. Pharmacother. 2022, 150, 112936. [Google Scholar] [CrossRef]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Wei, Q.J.; Xu, H.; Guan, N.; Ren, Y.L.; Liu, X.Y.; Wu, G.H.; Zhu, S.N. Overproduction of Mitochondrial Fission Proteins in Membranous Nephropathy in Children. Kidney Blood Press. Res. 2018, 43, 1927–1934. [Google Scholar] [CrossRef]
- Simoneau, J.; Dumontier, S.; Gosselin, R.; Scott, M.S. Current RNA-seq methodology reporting limits reproducibility. Brief. Bioinform. 2021, 22, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Mwambi, H.; Konig, I.R. Mendelian Randomization versus Path Models: Making Causal Inferences in Genetic Epidemiology. Hum. Hered. 2015, 79, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Birney, E. Mendelian Randomization. Cold Spring Harb. Perspect. Med. 2022, 12, a041302. [Google Scholar] [CrossRef] [PubMed]
- Sekula, P.; Del Greco, M.F.; Pattaro, C.; Kottgen, A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef]
- Yang, K.; Ding, X.; Liu, J.; Liu, S.; Liu, Q.; Li, J.; Zhang, P. Two-sample mendelian randomization reveals a causal association between membranous nephropathy and lung cancer. Commun. Biol. 2023, 6, 887. [Google Scholar] [CrossRef]
- Han, M.; Wang, Y.; Huang, X.; Li, P.; Shan, W.; Gu, H.; Wang, H.; Zhang, Q.; Bao, K. Prediction of biomarkers associated with membranous nephropathy: Bioinformatic analysis and experimental validation. Int. Immunopharmacol. 2024, 126, 111266. [Google Scholar] [CrossRef]
- Dong, Z.; Geng, Y.; Zhang, P.; Tang, J.; Cao, Z.; Zheng, H.; Guo, J.; Zhang, C.; Liu, B.; Liu, W.J. Identification of molecular mechanism and key biomarkers in membranous nephropathy by bioinformatics analysis. Am. J. Transl. Res. 2022, 14, 5833–5847. [Google Scholar]
- Shi, M.; Wang, Y.; Zhang, H.; Ling, Z.; Chen, X.; Wang, C.; Liu, J.; Ma, Y. Single-cell RNA sequencing shows the immune cell landscape in the kidneys of patients with idiopathic membranous nephropathy. Front. Immunol. 2023, 14, 1203062. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Liu, H.; Yang, C.; Yu, J.; Zhao, W.; Guo, J.; Zhou, B.; Jiang, N. MTFR2-dependent mitochondrial fission promotes HCC progression. J. Transl. Med. 2024, 22, 73. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Ito, K.; Murphy, D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Burgess, S.; Scott, R.A.; Timpson, N.J.; Davey Smith, G.; Thompson, S.G.; Consortium, E.-I. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, F.; Yin, Y. Applying logistic LASSO regression for the diagnosis of atypical Crohn’s disease. Sci. Rep. 2022, 12, 11340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huettmann, F.; Zhang, X.; Liu, S.; Sun, P.; Yu, Z.; Mi, C. The use of classification and regression algorithms using the random forests method with presence-only data to model species’ distribution. MethodsX 2019, 6, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Nunez, E.; Steyerberg, E.W.; Nunez, J. Regression modeling strategies. Rev. Esp. Cardiol. 2011, 64, 501–507. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, H.; Hao, B.; Zhan, W.; Yan, T.; Zhang, J.; Wang, S.; Hu, H.; Zhang, T. Insights into a Machine Learning-Based Palmitoylation-Related Gene Model for Predicting the Prognosis and Treatment Response of Breast Cancer Patients. Technol. Cancer Res. Treat. 2024, 23, 15330338241263434. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Yu, G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. Methods Mol. Biol. 2020, 2117, 207–215. [Google Scholar] [CrossRef]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, H.; Shen, H. Identification of Critical Biomarkers and Immune Infiltration in Rheumatoid Arthritis Based on WGCNA and LASSO Algorithm. Front. Immunol. 2022, 13, 925695. [Google Scholar] [CrossRef]
- Gharehhasani, B.S.; Rezaei, M.; Naghipour, A.; Sayad, N.; Mostafaei, S.; Alimohammadi, E. The most important variables associated with death due to COVID-19 disease, based on three data mining models Decision Tree, AdaBoost, and Support Vector Machine: A cross-sectional study. Health Sci. Rep. 2024, 7, e2266. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Lu, X.; Wang, X.; Wu, H.; Miao, X. From single-cell to spatial transcriptomics: Decoding the glioma stem cell niche and its clinical implications. Front. Immunol. 2024, 15, 1475235. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Grande, M.T.; López-Novoa, J.M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 2009, 5, 319–328. [Google Scholar] [CrossRef]
- de Haij, S.; Bakker, A.C.; van der Geest, R.N.; Haegeman, G.; Vanden Berghe, W.; Aarbiou, J.; Daha, M.R.; van Kooten, C. NF-kappaB mediated IL-6 production by renal epithelial cells is regulated by c-jun NH2-terminal kinase. J. Am. Soc. Nephrol. 2005, 16, 1603–1611. [Google Scholar] [CrossRef]
- Thelen, S.; Abouhamed, M.; Ciarimboli, G.; Edemir, B.; Bahler, M. Rho GAP myosin IXa is a regulator of kidney tubule function. Am. J. Physiol. Renal Physiol. 2015, 309, F501–F513. [Google Scholar] [CrossRef]
- Li, Q.; Gulati, A.; Lemaire, M.; Nottoli, T.; Bale, A.; Tufro, A. Rho-GTPase Activating Protein myosin MYO9A identified as a novel candidate gene for monogenic focal segmental glomerulosclerosis. Kidney Int. 2021, 99, 1102–1117. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, M.; Wei, T.; Zhang, X.; Shi, X.; Tang, H.; Ding, M.; Li, J.; Zhang, S.; Guo, W. NFKBIZ regulates NFkappaB signaling pathway to mediate tumorigenesis and metastasis of hepatocellular carcinoma by direct interaction with TRIM16. Cell Mol. Life Sci. 2024, 81, 167. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, J.; Dai, C.; Chen, M.X.; Fan, J.; Humphreys, B.D.; Fulton, D.J.R.; Kleven, D.T.; Fan, X.; Dong, Z.; et al. Pik3c3 expression profiling in the mouse kidney and its role in proximal tubule cell physiology. Am. J. Physiol. Cell Physiol. 2024, 327, C1094–C1110. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cai, H.; Zhao, Q.; Xing, T.; Li, J.; Wang, N. Epinephrine evokes renalase secretion via α-adrenoceptor/NF-κB pathways in renal proximal tubular epithelial cells. Kidney Blood Press. Res. 2014, 39, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lu, R.; Li, H.; Zhou, Y.; Zhang, P.; Bai, L.; Chen, D.; Chen, J.; Li, J.; Yu, P.; et al. Zhen-wu-tang ameliorates membranous nephropathy rats through inhibiting NF-kappaB pathway and NLRP3 inflammasome. Phytomedicine 2019, 59, 152913. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ping, J.; Guo, F.; Song, J.; Luo, M.; Chen, L. PLA2G12B Mediates Arachidonic Acid Metabolism through Activation of the NF-kappaB Pathway to Promote Membrane Nephropathy. Kidney Blood Press. Res. 2023, 48, 652–665. [Google Scholar] [CrossRef]
- Imran Naseer, M.; Abdulrahman Abdulkareem, A.; Yousef Muthaffar, O.; Chaudhary, A.G. Exome sequencing reveled a compound heterozygous mutations in RTTN gene causing developmental delay and primary microcephaly. Saudi J. Biol. Sci. 2021, 28, 2824–2829. [Google Scholar] [CrossRef]
- Chen, L.; Chou, C.L.; Knepper, M.A. Targeted Single-Cell RNA-seq Identifies Minority Cell Types of Kidney Distal Nephron. J. Am. Soc. Nephrol. 2021, 32, 886–896. [Google Scholar] [CrossRef]
- Takagi, H.; Nishibori, Y.; Katayama, K.; Katada, T.; Takahashi, S.; Kiuchi, Z.; Takahashi, S.I.; Kamei, H.; Kawakami, H.; Akimoto, Y.; et al. USP40 gene knockdown disrupts glomerular permeability in zebrafish. Am. J. Physiol. Renal Physiol. 2017, 312, F702–F715. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukuhara, D.; Kimura, T.; Fukutomi, T.; Tanaka, E.; Mikami, N.; Hada, I.; Takematsu, H.; Nishibori, Y.; Akimoto, Y.; et al. USP40 deubiquitinates HINT1 and stabilizes p53 in podocyte damage. Biochem. Biophys. Res. Commun. 2022, 614, 198–206. [Google Scholar] [CrossRef]
- Liu, W.J.; Li, Z.H.; Chen, X.C.; Zhao, X.L.; Zhong, Z.; Yang, C.; Wu, H.L.; An, N.; Li, W.Y.; Liu, H.F. Blockage of the lysosome-dependent autophagic pathway contributes to complement membrane attack complex-induced podocyte injury in idiopathic membranous nephropathy. Sci. Rep. 2017, 7, 8643. [Google Scholar] [CrossRef]
- Rood, I.M.; Merchant, M.L.; Wilkey, D.W.; Zhang, T.; Zabrouskov, V.; van der Vlag, J.; Dijkman, H.B.; Willemsen, B.K.; Wetzels, J.F.; Klein, J.B.; et al. Increased expression of lysosome membrane protein 2 in glomeruli of patients with idiopathic membranous nephropathy. Proteomics 2015, 15, 3722–3730. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, H.; Hu, Y.; Dai, H.; Zhao, Q.; Zheng, Y.; Liu, W.; Rui, H.; Liu, B. B cell dysregulation and depletion therapy in primary membranous nephropathy: Prospects and potential challenges. Int. Immunopharmacol. 2024, 140, 112769. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tayier, D.; Yang, S.; Zhang, X.; Lu, C. Follicular Helper T Cells and Follicular Regulatory T Cells Involved in Immune Disorders of Idiopathic Membranous Nephropathy. Indian. J. Pediatr. 2024, 91, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Ma, X.; Ji, A.; Geng, K.; Feng, H.; Li, L.; Zhang, A.; Cheng, Z. Development of a novel (18)F-labeled small molecule probe for PET imaging of mesenchymal epithelial transition receptor expression. Eur. J. Nucl. Med. Mol. Imaging 2024, 51, 656–668. [Google Scholar] [CrossRef]
- Price, K.L.; Kolatsi-Joannou, M.; Mari, C.; Long, D.A.; Winyard, P.J.D. Lithium induces mesenchymal-epithelial differentiation during human kidney development by activation of the Wnt signalling system. Cell Death Discov. 2018, 4, 13. [Google Scholar] [CrossRef]
- Arora, H.; Boulberdaa, M.; Qureshi, R.; Bitirim, V.; Messadeq, N.; Dolle, P.; Nebigil, C.G. Prokineticin receptor 1 is required for mesenchymal-epithelial transition in kidney development. FASEB J. 2016, 30, 2733–2740. [Google Scholar] [CrossRef]
- Al-Rashidi, R.R.; Noraldeen, S.A.M.; Kareem, A.K.; Mahmoud, A.K.; Kadhum, W.R.; Ramírez-Coronel, A.A.; Iswanto, A.H.; Obaid, R.F.; Jalil, A.T.; Mustafa, Y.F.; et al. Malignant function of nuclear factor-kappaB axis in prostate cancer: Molecular interactions and regulation by non-coding RNAs. Pharmacol. Res. 2023, 194, 106775. [Google Scholar] [CrossRef]
- Chou, E.J.; Tang, T.K. Human Microcephaly Protein RTTN Is Required for Proper Mitotic Progression and Correct Spindle Position. Cells 2021, 10, 1441. [Google Scholar] [CrossRef]
- Miao, J.; Li, L.; Shaheen, N.; Wei, J.; Jacko, A.M.; Sundd, P.; Taleb, S.J.; Mallampalli, R.K.; Zhao, Y.; Zhao, J. The deubiquitinase USP40 preserves endothelial integrity by targeting the heat shock protein HSP90β. Exp. Mol. Med. 2024, 56, 395–407. [Google Scholar] [CrossRef]
- Li, X.; Zhao, X.; Xie, L.; Song, X.; Song, X. Identification of four snoRNAs (SNORD16, SNORA73B, SCARNA4, and SNORD49B) as novel non-invasive biomarkers for diagnosis of breast cancer. Cancer Cell Int. 2024, 24, 55. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Q.; Li, N.; Qiu, H.; Zhao, M.; Jiang, C.; Wan, C. Identification Exploring the Mechanism and Clinical Validation of Mitochondrial Dynamics-Related Genes in Membranous Nephropathy Based on Mendelian Randomization Study and Bioinformatics Analysis. Biomedicines 2025, 13, 1489. https://doi.org/10.3390/biomedicines13061489
Shao Q, Li N, Qiu H, Zhao M, Jiang C, Wan C. Identification Exploring the Mechanism and Clinical Validation of Mitochondrial Dynamics-Related Genes in Membranous Nephropathy Based on Mendelian Randomization Study and Bioinformatics Analysis. Biomedicines. 2025; 13(6):1489. https://doi.org/10.3390/biomedicines13061489
Chicago/Turabian StyleShao, Qiuyuan, Nan Li, Huimin Qiu, Min Zhao, Chunming Jiang, and Cheng Wan. 2025. "Identification Exploring the Mechanism and Clinical Validation of Mitochondrial Dynamics-Related Genes in Membranous Nephropathy Based on Mendelian Randomization Study and Bioinformatics Analysis" Biomedicines 13, no. 6: 1489. https://doi.org/10.3390/biomedicines13061489
APA StyleShao, Q., Li, N., Qiu, H., Zhao, M., Jiang, C., & Wan, C. (2025). Identification Exploring the Mechanism and Clinical Validation of Mitochondrial Dynamics-Related Genes in Membranous Nephropathy Based on Mendelian Randomization Study and Bioinformatics Analysis. Biomedicines, 13(6), 1489. https://doi.org/10.3390/biomedicines13061489