
Ketone Body Induction: Insights into Metabolic Disease Management


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
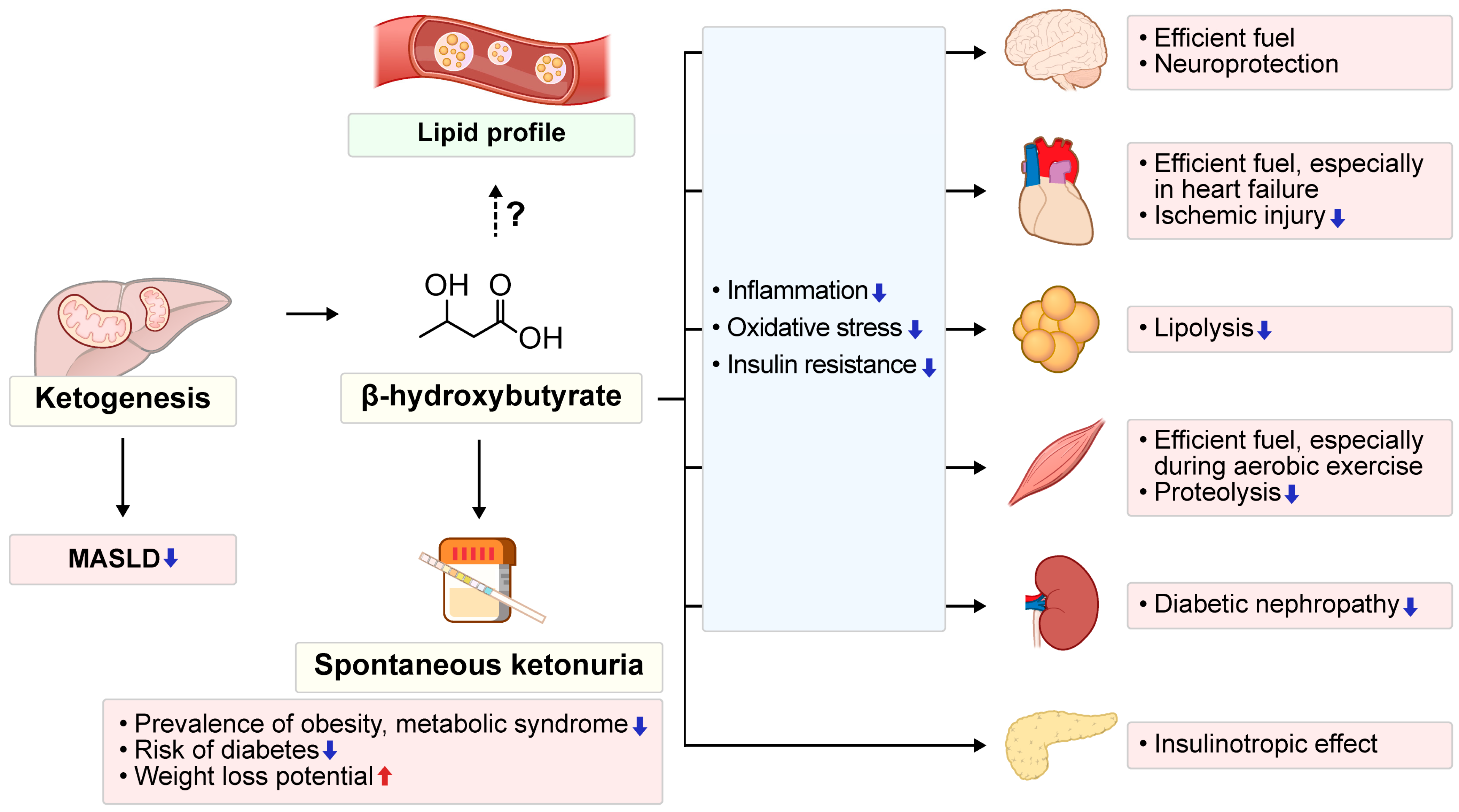
2. Roles of KBs in Metabolic Health
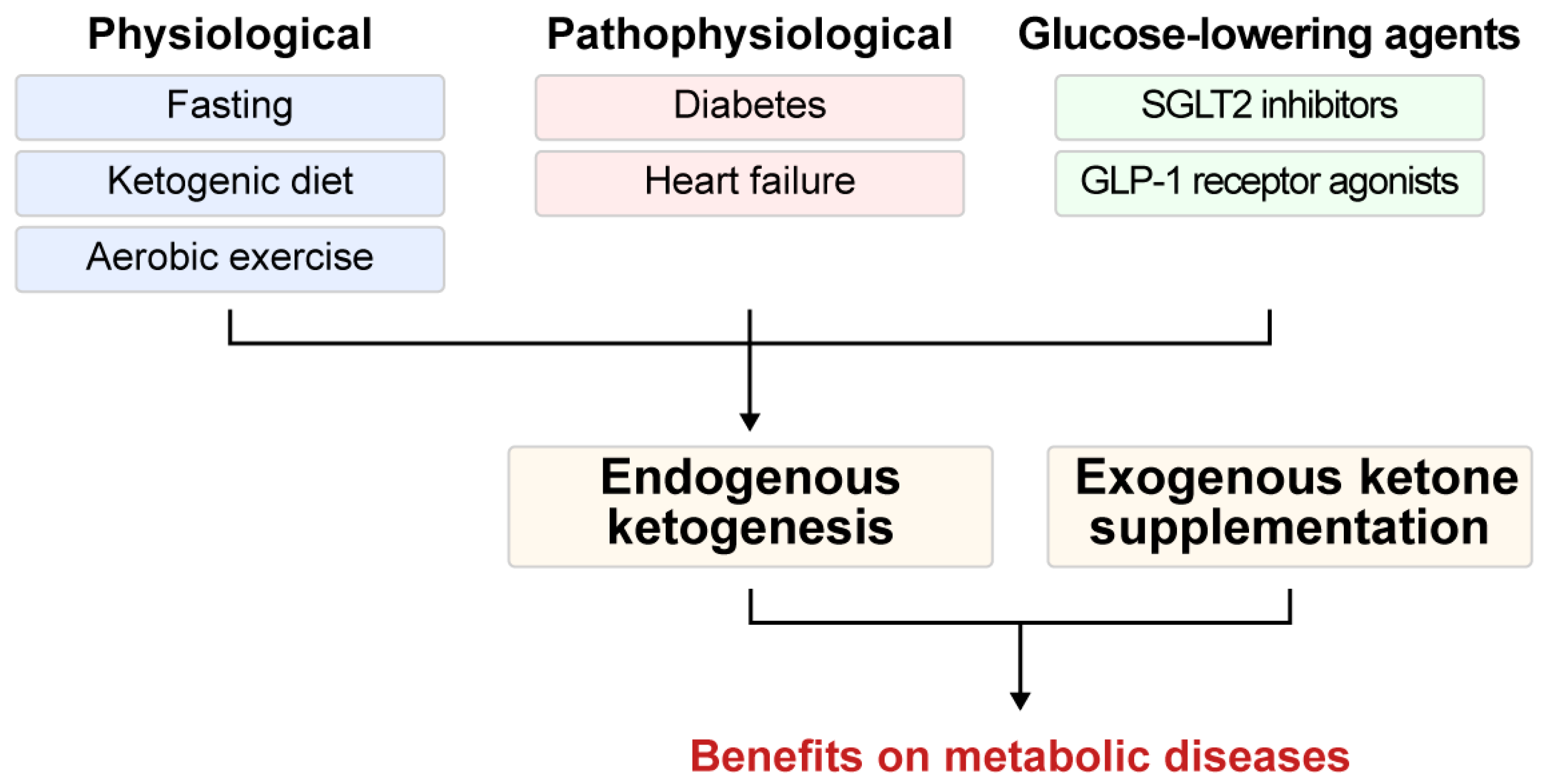
3. Pathways to Ketosis: Endogenous Ketogenesis and Exogenous Ketone Supplementation in Health and Disease
3.1. Endogenous Ketogenesis: Adaptive Functions and Metabolic Implications
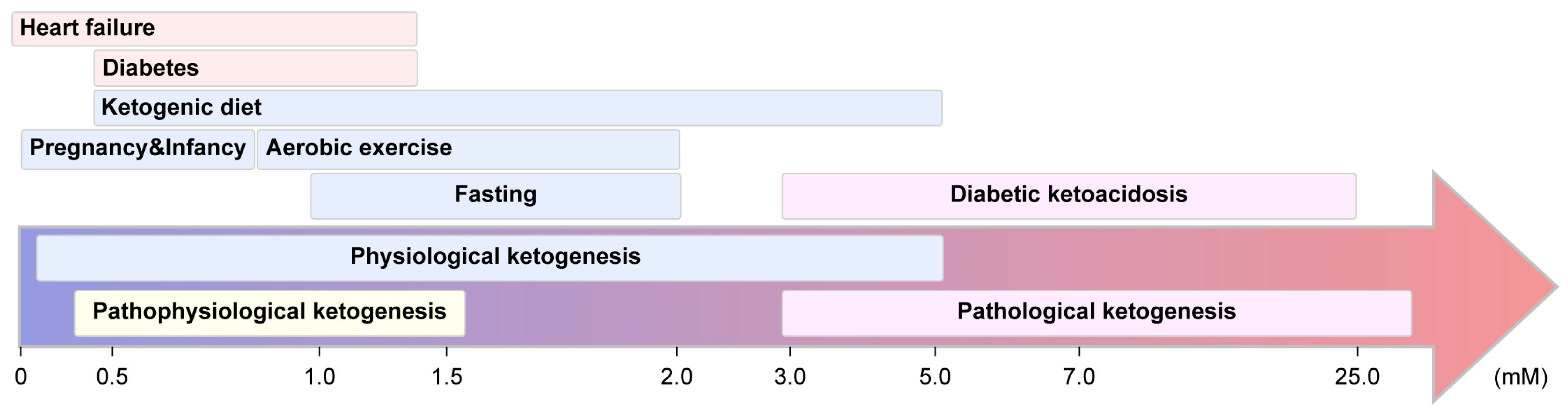
3.1.1. Physiological Ketogenesis
3.1.2. Pathophysiological Ketogenesis
3.1.3. Ketogenesis Induced by Glucose-Lowering Agents
3.1.4. Pathological Ketogenesis
3.2. Exogenous Ketone Supplementation
4. Navigating the Challenges of Ketogenic Strategies: Balancing Health Risks and Therapeutic Benefits in the Management of Metabolic Disorders
4.1. Ketogenesis Induced Lipid Profile Changes and Cardiovascular Risk
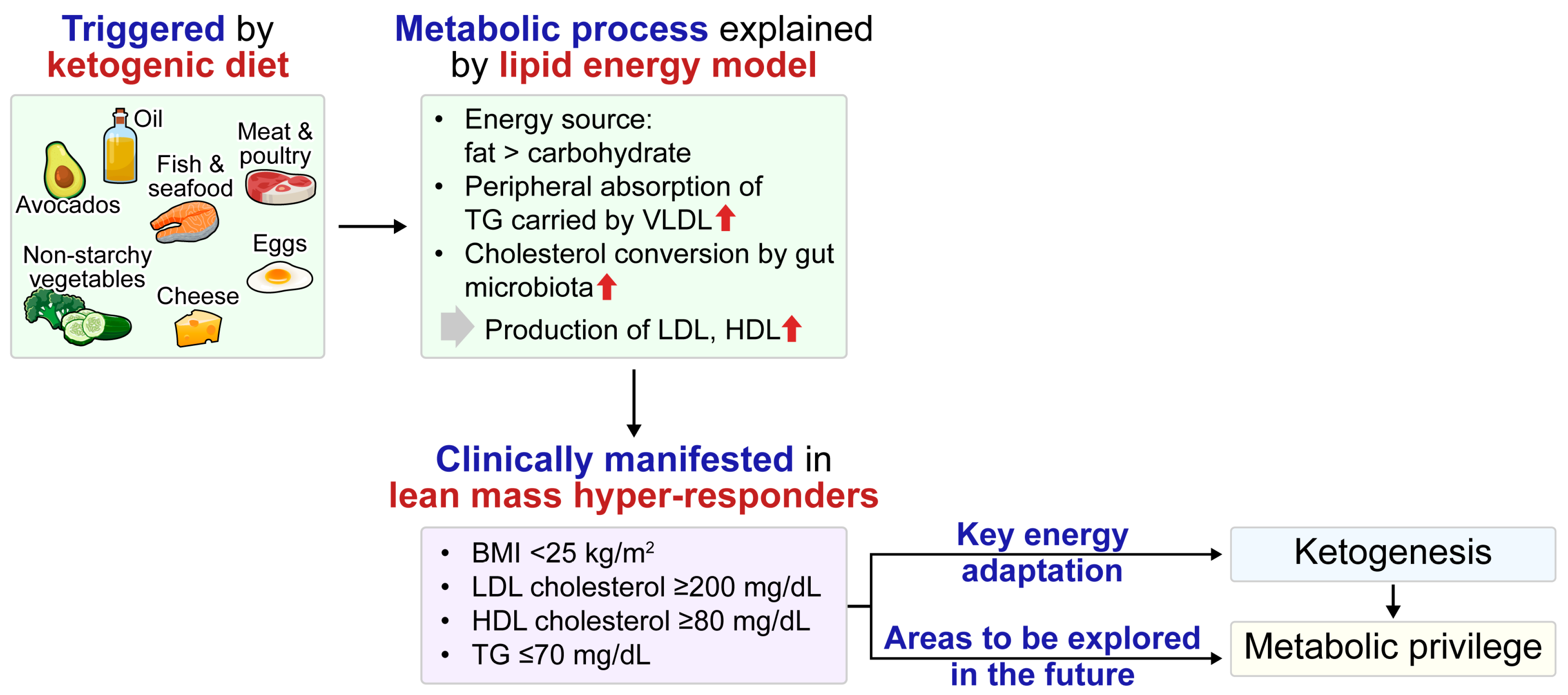
4.2. LMHR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| β-HB | β-Hydroxybutyrate |
| BMI | Body mass index |
| CV | Cardiovascular |
| CVD | Cardiovascular disease |
| DKA | Diabetic ketoacidosis |
| FFA | Free fatty acid |
| FGF21 | Fibroblast growth factor 21 |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-1RA | Glucagon-like peptide-1 receptor agonist |
| HbA1c | Glycated hemoglobin |
| HDL-C | High-density lipoprotein cholesterol |
| KB | Ketone body |
| KD | Ketogenic diet |
| KME | Ketone monoester |
| LCHF | Low-carbohydrate high-fat |
| LDL-C | Low-density lipoprotein cholesterol |
| LEM | Lipid energy model |
| LMHR | Lean mass hyper-responder |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| RCT | Randomized controlled trial |
| SCOT | Succinyl-CoA:3-ketoacid CoA transferase |
| SGLT2 | Sodium-glucose cotransporter 2 |
| TCA | Tricarboxylic acid |
| TG | Triglyceride |
| T2D | Type 2 diabetes |
| VLCKD | Very-low-calorie ketogenic diet |
| VLEKD | Very-low-energy ketogenic diet |
| VLDL | Very-low-density lipoprotein |
References
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Venturini, C.; Mancinelli, L.; Matacchione, G.; Olivieri, F.; Antonicelli, R. The Cardioprotective Effects of Nutritional Ketosis: Mechanisms and Clinical Implications. Nutrients 2024, 16, 4204. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Kim, B.R.; Seo, J.W.; Kim, S.M.; Kim, K.N.; Joo, N.S. The Presence of Urinary Ketones according to Metabolic Status and Obesity. J. Korean Med. Sci. 2020, 35, e273. [Google Scholar] [CrossRef]
- Joo, N.S.; Lee, D.J.; Kim, K.M.; Kim, B.T.; Kim, C.W.; Kim, K.N.; Kim, S.M. Ketonuria after fasting may be related to the metabolic superiority. J. Korean Med. Sci. 2010, 25, 1771–1776. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Xu, D.; Zhou, Y.; Qu, Z.; Yang, Q.; Lv, Q. Low carbohydrate ketogenic diets reduce cardiovascular risk factor levels in obese or overweight patients with T2DM: A meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1092031. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Lee, S.G.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Ferrannini, E.; Lee, Y.H.; Cho, N.H. Spontaneous ketonuria and risk of incident diabetes: A 12 year prospective study. Diabetologia 2019, 62, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Joo, N.S.; Kim, K.M.; Lee, D.J.; Kim, S.M. Different response of body weight change according to ketonuria after fasting in the healthy obese. J. Korean Med. Sci. 2012, 27, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Veneti, S.; Grammatikopoulou, M.G.; Kintiraki, E.; Mintziori, G.; Goulis, D.G. Ketone Bodies in Diabetes Mellitus: Friend or Foe? Nutrients 2023, 15, 4383. [Google Scholar] [CrossRef]
- Makievskaya, C.I.; Popkov, V.A.; Andrianova, N.V.; Liao, X.; Zorov, D.B.; Plotnikov, E.Y. Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 2576. [Google Scholar] [CrossRef]
- Poplawski, M.M.; Mastaitis, J.W.; Isoda, F.; Grosjean, F.; Zheng, F.; Mobbs, C.V. Reversal of diabetic nephropathy by a ketogenic diet. PLoS ONE 2011, 6, e18604. [Google Scholar] [CrossRef]
- Persson, B.; Gentz, J. The pattern of blood lipids, glycerol and ketone bodies during the neonatal period, infancy and childhood. Acta Paediatr. Scand. 1966, 55, 353–362. [Google Scholar] [CrossRef]
- Johnson, R.H.; Walton, J.L.; Krebs, H.A.; Williamson, D.H. Metabolic fuels during and after severe exercise in athletes and non-athletes. Lancet 1969, 2, 452–455. [Google Scholar] [CrossRef]
- Lommi, J.; Koskinen, P.; Näveri, H.; Härkönen, M.; Kupari, M. Heart failure ketosis. J. Intern. Med. 1997, 242, 231–238. [Google Scholar] [CrossRef]
- Bobo, L.; Womeodu, R.J.; Knox, A.L., Jr. Principles of intercultural medicine in an internal medicine program. Am. J. Med. Sci. 1991, 302, 244–248. [Google Scholar] [CrossRef]
- Polin, R.A.; Abman, S.H. Fetal and Neonatal Physiology E-Book; Elsevier Health Sciences: Philadelphia, PA, USA, 2011. [Google Scholar]
- Bronisz, A.; Ozorowski, M.; Hagner-Derengowska, M. Pregnancy Ketonemia and Development of the Fetal Central Nervous System. Int. J. Endocrinol. 2018, 2018, 1242901. [Google Scholar] [CrossRef] [PubMed]
- Knopp, R.H.; Montes, A.; Childs, M.; Li, J.R.; Mabuchi, H. Metabolic adjustments in normal and diabetic pregnancy. Clin. Obstet. Gynecol. 1981, 24, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Frise, C.J.; Mackillop, L.; Joash, K.; Williamson, C. Starvation ketoacidosis in pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 167, 1–7. [Google Scholar] [CrossRef]
- Reichard, G.A., Jr.; Owen, O.E.; Haff, A.C.; Paul, P.; Bortz, W.M. Ketone-body production and oxidation in fasting obese humans. J. Clin. Investig. 1974, 53, 508–515. [Google Scholar] [CrossRef]
- Balasse, E.O.; Féry, F. Ketone body production and disposal: Effects of fasting, diabetes, and exercise. Diabetes Metab. Rev. 1989, 5, 247–270. [Google Scholar] [CrossRef] [PubMed]
- Koeslag, J.H. Post-exercise ketosis and the hormone response to exercise: A review. Med. Sci. Sports Exerc. 1982, 14, 327–334. [Google Scholar] [CrossRef]
- Sherrier, M.; Li, H. The impact of keto-adaptation on exercise performance and the role of metabolic-regulating cytokines. Am. J. Clin. Nutr. 2019, 110, 562–573. [Google Scholar] [CrossRef]
- Fulghum, K.; Salathe, S.F.; Davis, X.; Thyfault, J.P.; Puchalska, P.; Crawford, P.A. Ketone body metabolism and cardiometabolic implications for cognitive health. NPJ Metab. Health Dis. 2024, 2, 29. [Google Scholar] [CrossRef]
- Brehm, B.J.; Seeley, R.J.; Daniels, S.R.; D’Alessio, D.A. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. J. Clin. Endocrinol. Metab. 2003, 88, 1617–1623. [Google Scholar] [CrossRef]
- Yancy, W.S., Jr.; Olsen, M.K.; Guyton, J.R.; Bakst, R.P.; Westman, E.C. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: A randomized, controlled trial. Ann. Intern. Med. 2004, 140, 769–777. [Google Scholar] [CrossRef]
- Moreno, B.; Bellido, D.; Sajoux, I.; Goday, A.; Saavedra, D.; Crujeiras, A.B.; Casanueva, F.F. Comparison of a very low-calorie-ketogenic diet with a standard low-calorie diet in the treatment of obesity. Endocrine 2014, 47, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Hengist, A.; Davies, R.G.; Walhin, J.P.; Buniam, J.; Merrell, L.H.; Rogers, L.; Bradshaw, L.; Moreno-Cabañas, A.; Rogers, P.J.; Brunstrom, J.M.; et al. Ketogenic diet but not free-sugar restriction alters glucose tolerance, lipid metabolism, peripheral tissue phenotype, and gut microbiome: RCT. Cell Rep. Med. 2024, 5, 101667. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.J. Effect of low-carbohydrate diets on cardiometabolic risk, insulin resistance, and metabolic syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 301–307. [Google Scholar] [CrossRef]
- Tinguely, D.; Gross, J.; Kosinski, C. Efficacy of Ketogenic Diets on Type 2 Diabetes: A Systematic Review. Curr. Diabetes Rep. 2021, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Yancy, W.S., Jr.; Foy, M.; Chalecki, A.M.; Vernon, M.C.; Westman, E.C. A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr. Metab. 2005, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Farrés, J.; Pujol, A.; Coma, M.; Ruiz, J.L.; Naval, J.; Mas, J.M.; Molins, A.; Fondevila, J.; Aloy, P. Revealing the molecular relationship between type 2 diabetes and the metabolic changes induced by a very-low-carbohydrate low-fat ketogenic diet. Nutr. Metab. 2010, 7, 88. [Google Scholar] [CrossRef]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- Parry-Strong, A.; Wright-McNaughton, M.; Weatherall, M.; Hall, R.M.; Coppell, K.J.; Barthow, C.; Krebs, J.D. Very low carbohydrate (ketogenic) diets in type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2022, 24, 2431–2442. [Google Scholar] [CrossRef]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: A comprehensive review of the literature. Obes. Rev. 2020, 21, e13024. [Google Scholar] [CrossRef]
- Lundsgaard, A.M.; Holm, J.B.; Sjøberg, K.A.; Bojsen-Møller, K.N.; Myrmel, L.S.; Fjære, E.; Jensen, B.A.H.; Nicolaisen, T.S.; Hingst, J.R.; Hansen, S.L.; et al. Mechanisms Preserving Insulin Action during High Dietary Fat Intake. Cell Metab. 2019, 29, 50–63.e54. [Google Scholar] [CrossRef]
- Ünalp, A.; Ünay, B.; Arhan, E. Editorial: The use of ketogenic diet therapy in the era of individualized therapy. Front. Nutr. 2023, 10, 1272170. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Caprio, M.; Camajani, E.; Verde, L.; Perrini, S.; Cignarelli, A.; Prodam, F.; Gambineri, A.; Isidori, A.M.; Colao, A.; et al. Ketogenic nutritional therapy (KeNuT)-a multi-step dietary model with meal replacements for the management of obesity and its related metabolic disorders: A consensus statement from the working group of the Club of the Italian Society of Endocrinology (SIE)-diet therapies in endocrinology and metabolism. J. Endocrinol. Investig. 2024, 47, 487–500. [Google Scholar]
- Féry, F.; Balasse, E.O. Ketone body production and disposal in diabetic ketosis. A comparison with fasting ketosis. Diabetes 1985, 34, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Cho, Y.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. β-hydroxybutyrate as a biomarker of β-cell function in new-onset type 2 diabetes and its association with treatment response at 6 months. Diabetes Metab. 2023, 49, 101427. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Matsuura, T.R.; Puchalska, P.; Crawford, P.A.; Kelly, D.P. Ketones and the Heart: Metabolic Principles and Therapeutic Implications. Circ. Res. 2023, 132, 882–898. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef]
- Mizuno, Y.; Harada, E.; Nakagawa, H.; Morikawa, Y.; Shono, M.; Kugimiya, F.; Yoshimura, M.; Yasue, H. The diabetic heart utilizes ketone bodies as an energy source. Metabolism 2017, 77, 65–72. [Google Scholar] [CrossRef]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, X.; Li, T.; Zhao, J.; Yang, Y.; Yao, Y.; Wang, L.; Yang, B.; Ren, G.; Tan, Y.; et al. Alternate-Day Ketogenic Diet Feeding Protects against Heart Failure through Preservation of Ketogenesis in the Liver. Oxid. Med. Cell. Longev. 2022, 2022, 4253651. [Google Scholar] [CrossRef]
- Abdul Kadir, A.; Clarke, K.; Evans, R.D. Cardiac ketone body metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165739. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, M.; Kim, S.H.; Kim, S.R.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Cho, J.W.; Lee, Y.H. Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes. Metab. 2019, 21, 801–811. [Google Scholar] [CrossRef]
- Polidori, D.; Iijima, H.; Goda, M.; Maruyama, N.; Inagaki, N.; Crawford, P.A. Intra- and inter-subject variability for increases in serum ketone bodies in patients with type 2 diabetes treated with the sodium glucose co-transporter 2 inhibitor canagliflozin. Diabetes Obes. Metab. 2018, 20, 1321–1326. [Google Scholar] [CrossRef]
- Saucedo-Orozco, H.; Voorrips, S.N.; Yurista, S.R.; de Boer, R.A.; Westenbrink, B.D. SGLT2 Inhibitors and Ketone Metabolism in Heart Failure. J. Lipid Atheroscler. 2022, 11, 1–19. [Google Scholar] [CrossRef]
- Packer, M. Role of ketogenic starvation sensors in mediating the renal protective effects of SGLT2 inhibitors in type 2 diabetes. J. Diabetes Complicat. 2020, 34, 107647. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e406. [Google Scholar] [CrossRef]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies for kidney injury and disease. Adv. Redox Res. 2021, 2, 100009. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Sahebkar, A. Molecular mechanisms by which GLP-1 RA and DPP-4i induce insulin sensitivity. Life Sci. 2019, 234, 116776. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.; Basheer, F.T.; Poojari, P.G.; Thunga, G.; Chandran, V.P.; Acharya, L.D. Adverse drug reactions of GLP-1 agonists: A systematic review of case reports. Diabetes Metab. Syndr. 2022, 16, 102427. [Google Scholar] [CrossRef] [PubMed]
- Alduraibi, R.K.; Alrebdi, Y.M.; Altowayan, Y.F. Euglycemic diabetic ketoacidosis after the initiation of dulaglutide in patient with type 2 diabetes. Medicine 2023, 102, e34027. [Google Scholar] [CrossRef]
- Tan, X.; Pan, X.; Wu, X.; Zheng, S.; Chen, Y.; Liu, D.; Zhang, X. Glucagon-like peptide-1 receptor agonists as add-on therapy to insulin for type 1 diabetes mellitus. Front. Pharmacol. 2023, 14, 975880. [Google Scholar] [CrossRef]
- Cabou, C.; Burcelin, R. GLP-1, the gut-brain, and brain-periphery axes. Rev. Diabet. Stud. 2011, 8, 418–431. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Pfeiffer, A.F.H. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes. Metab. 2021, 23 (Suppl. S3), 5–29. [Google Scholar] [CrossRef]
- Gutzwiller, J.P.; Drewe, J.; Göke, B.; Schmidt, H.; Rohrer, B.; Lareida, J.; Beglinger, C. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am. J. Physiol. 1999, 276, R1541–R1544. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, M.; Mei, M.; Chen, C.; Lv, Y.; Xiang, L.; Li, R. The association between GLP-1 receptor agonist and diabetic ketoacidosis in the FDA adverse event reporting system. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 504–510. [Google Scholar] [CrossRef]
- Edwards, K.; Li, X.; Lingvay, I. Clinical and Safety Outcomes with GLP-1 Receptor Agonists and SGLT2 Inhibitors in Type 1 Diabetes: A Real-World Study. J. Clin. Endocrinol. Metab. 2023, 108, 920–930. [Google Scholar] [CrossRef]
- Olivieri, M.C.; Botelho, L.H. Synergistic inhibition of hepatic ketogenesis in the presence of insulin and a cAMP antagonist. Biochem. Biophys. Res. Commun. 1989, 159, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Kitabchi, A.E.; Wall, B.M. Diabetic ketoacidosis. Med. Clin. N. Am. 1995, 79, 9–37. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.; Agrawal, A.; Morgan, F. Euglycemic Diabetic Ketoacidosis: A Review. Curr. Diabetes Rev. 2017, 13, 315–321. [Google Scholar] [CrossRef]
- Rahimi, L.; Rajpal, A.; Ismail-Beigi, F. Glucocorticoid-Induced Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2020, 13, 1133–1145. [Google Scholar] [CrossRef]
- Sood, N.; Buddhavarapu, V.; Garg, R. GLP-1 receptor agonists causing euglycemic ketoacidosis in patients without diabetes: A brief review. Int. J. Obes. 2025, 49, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Starvation Ketosis and the Kidney. Am. J. Nephrol. 2021, 52, 467–478. [Google Scholar] [CrossRef]
- Sood, N.; Bansal, O.; Garg, R.; Hoskote, A. Euglycemic Ketoacidosis from Semaglutide in a Patient Without Diabetes. JCEM Case Rep. 2024, 2, luae156. [Google Scholar] [CrossRef]
- Umpierrez, G.E.; Watts, N.B.; Phillips, L.S. Clinical utility of beta-hydroxybutyrate determined by reflectance meter in the management of diabetic ketoacidosis. Diabetes Care 1995, 18, 137–138. [Google Scholar] [CrossRef]
- Umpierrez, G.E.; Kelly, J.P.; Navarrete, J.E.; Casals, M.M.; Kitabchi, A.E. Hyperglycemic crises in urban blacks. Arch. Intern. Med. 1997, 157, 669–675. [Google Scholar] [CrossRef]
- McGarry, J.D. Lilly Lecture 1978. New perspectives in the regulation of ketogenesis. Diabetes 1979, 28, 517–523. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Lee, W.Y.; Lee, J.H.; Kwon, H.S.; Lee, J.M.; Kim, S.R.; Moon, S.D.; Song, K.H.; Han, J.H.; Ahn, Y.B.; et al. Clinical characteristics of diabetic ketoacidosis in Korea over the past two decades. Diabet. Med. 2005, 22, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.; Jung, C.H.; Kim, S.W.; Lee, M.J.; Lee, W.J.; Kim, J.H.; Lee, B.W. Clinical characteristics and insulin independence of Koreans with new-onset type 2 diabetes presenting with diabetic ketoacidosis. Diabetes Metab. Res. Rev. 2013, 29, 507–513. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, S.H.; Kim, J.H.; Lee, J.I.; Kim, J.H.; Jang, E.H.; Yoon, K.H.; Lee, K.W.; Song, K.H. Clinical characteristics of Korean patients with new-onset diabetes presenting with diabetic ketoacidosis. Diabetes Res. Clin. Pract. 2009, 85, e8–e11. [Google Scholar] [CrossRef]
- Maldonado, M.; Hampe, C.S.; Gaur, L.K.; D’Amico, S.; Iyer, D.; Hammerle, L.P.; Bolgiano, D.; Rodriguez, L.; Rajan, A.; Lernmark, A.; et al. Ketosis-prone diabetes: Dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis, and clinical outcomes. J. Clin. Endocrinol. Metab. 2003, 88, 5090–5098. [Google Scholar] [CrossRef]
- Lee, T.H. Prevalence of obesity in Korean non-insulin-dependent diabetic patients. Diabetes Res. Clin. Pract. 1996, 32, 71–80. [Google Scholar] [CrossRef]
- Kim, D.J.; Lee, M.S.; Kim, K.W.; Lee, M.K. Insulin secretory dysfunction and insulin resistance in the pathogenesis of korean type 2 diabetes mellitus. Metabolism 2001, 50, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.M.; Pasiakos, S.M.; Howard, E.E. High-fat ketogenic diets and ketone monoester supplements differentially affect substrate metabolism during aerobic exercise. Am. J. Physiol. Cell Physiol. 2023, 325, C1144–C1153. [Google Scholar] [CrossRef]
- Walsh, J.J.; Neudorf, H.; Little, J.P. 14-Day Ketone Supplementation Lowers Glucose and Improves Vascular Function in Obesity: A Randomized Crossover Trial. J. Clin. Endocrinol. Metab. 2021, 106, e1738–e1754. [Google Scholar] [CrossRef]
- Walsh, J.J.; Myette-Côté, É.; Neudorf, H.; Little, J.P. Potential Therapeutic Effects of Exogenous Ketone Supplementation for Type 2 Diabetes: A Review. Curr. Pharm. Des. 2020, 26, 958–969. [Google Scholar] [CrossRef]
- Sansone, M.; Sansone, A.; Borrione, P.; Romanelli, F.; Di Luigi, L.; Sgrò, P. Effects of Ketone Bodies on Endurance Exercise. Curr. Sports Med. Rep. 2018, 17, 444–453. [Google Scholar] [CrossRef]
- Howard, E.E.; Allen, J.T.; Coleman, J.L.; Small, S.D.; Karl, J.P.; O’Fallon, K.S.; Margolis, L.M. Ketone Monoester Plus Carbohydrate Supplementation Does Not Alter Exogenous and Plasma Glucose Oxidation or Metabolic Clearance Rate During Exercise in Men Compared with Carbohydrate Alone. J. Nutr. 2023, 153, 1696–1709. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Morales, J.S.; Castillo-García, A.; Lucia, A. Acute Ketone Supplementation and Exercise Performance: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Sports Physiol. Perform. 2020, 15, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Wang, M.; Ma, Y.; Offermanns, S.; Whim, M.D. The β-Hydroxybutyrate-GPR109A Receptor Regulates Fasting-induced Plasticity in the Mouse Adrenal Medulla. Endocrinology 2022, 163, bqac077. [Google Scholar] [CrossRef]
- Ahmed, N.; Farooq, J.; Siddiqi, H.S.; Meo, S.A.; Kulsoom, B.; Laghari, A.H.; Jamshed, H.; Pasha, F. Impact of Intermittent Fasting on Lipid Profile-A Quasi-Randomized Clinical Trial. Front. Nutr. 2020, 7, 596787. [Google Scholar] [CrossRef] [PubMed]
- Klempel, M.C.; Kroeger, C.M.; Varady, K.A. Alternate day fasting increases LDL particle size independently of dietary fat content in obese humans. Eur. J. Clin. Nutr. 2013, 67, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Han, Y.M.; Ramprasath, T.; Zou, M.H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef]
- Mccafferty, K.J.; Brinker, E.; Graff, E.; Steury, T.D.; Greene, M.W.; Judd, R.L. 146-OR: Loss of Hydroxycarboxylic Acid Receptor 2 (HCA2) Affects Adipose Tissue Homeostasis in a Sex-Specific Manner during Prolonged Fasting. Diabetes 2022, 71, 146-OR. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, D. Effects of aerobic exercise on lipids and lipoproteins. Lipids Health Dis. 2017, 16, 132. [Google Scholar] [CrossRef]
- Dufaux, B.; Order, U.; Müller, R.; Hollmann, W. Delayed effects of prolonged exercise on serum lipoproteins. Metabolism 1986, 35, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Sharman, M.J.; Kraemer, W.J.; Love, D.M.; Avery, N.G.; Gómez, A.L.; Scheett, T.P.; Volek, J.S. A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J. Nutr. 2002, 132, 1879–1885. [Google Scholar] [CrossRef]
- Dashti, H.M.; Mathew, T.C.; Hussein, T.; Asfar, S.K.; Behbahani, A.; Khoursheed, M.A.; Al-Sayer, H.M.; Bo-Abbas, Y.Y.; Al-Zaid, N.S. Long-term effects of a ketogenic diet in obese patients. Exp. Clin. Cardiol. 2004, 9, 200–205. [Google Scholar]
- Qu, X.; Huang, L.; Rong, J. The ketogenic diet has the potential to decrease all-cause mortality without a concomitant increase in cardiovascular-related mortality. Sci. Rep. 2024, 14, 22805. [Google Scholar] [CrossRef] [PubMed]
- Iatan, I.; Huang, K.; Vikulova, D.; Ranjan, S.; Brunham, L.R. Association of a Low-Carbohydrate High-Fat Diet with Plasma Lipid Levels and Cardiovascular Risk. JACC Adv. 2024, 3, 100924. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes and Nutrition Study Group (DNSG) of the European Association for the Study of Diabetes (EASD). Evidence-based European recommendations for the dietary management of diabetes. Diabetologia 2023, 66, 965–985. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 5. Facilitating positive health behaviors and well-being to improve health outcomes: Standards of Care in Diabetes—2025. Diabetes Care 2025, 48, S86–S127. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Feldman, D.; Soto-Mota, A.; Kalayjian, T.; Ludwig, D.S. Elevated LDL Cholesterol with a Carbohydrate-Restricted Diet: Evidence for a “Lean Mass Hyper-Responder” Phenotype. Curr. Dev. Nutr. 2022, 6, nzab144. [Google Scholar] [CrossRef]
- Schmidt, T.; Harmon, D.M.; Kludtke, E.; Mickow, A.; Simha, V.; Kopecky, S. Dramatic elevation of LDL cholesterol from ketogenic-dieting: A Case Series. Am. J. Prev. Cardiol. 2023, 14, 100495. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Flores-Jurado, Y.; Norwitz, N.G.; Feldman, D.; Pereira, M.A.; Danaei, G.; Ludwig, D.S. Increased low-density lipoprotein cholesterol on a low-carbohydrate diet in adults with normal but not high body weight: A meta-analysis. Am. J. Clin. Nutr. 2024, 119, 740–747. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Soto-Mota, A.; Kaplan, B.; Ludwig, D.S.; Budoff, M.; Kontush, A.; Feldman, D. The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets. Metabolites 2022, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Vedamurthy, D.; Burke, F.; Suri, K.; Soffer, D.; Jacoby, D. Clinical vignette-keto diet-induced dyslipidemia and lean mass hyper-responders. J. Clin. Lipidol. 2023, 17, e7–e8. [Google Scholar] [CrossRef]
- Bubeck, A.M.; Urbain, P.; Horn, C.; Jung, A.S.; Ferrari, L.; Ruple, H.K.; Podlesny, D.; Zorn, S.; Laupsa-Borge, J.; Jensen, C.; et al. High-fat diet impact on intestinal cholesterol conversion by the microbiota and serum cholesterol levels. iScience 2023, 26, 107697. [Google Scholar] [CrossRef] [PubMed]
- Cooper, I.D.; Sanchez-Pizarro, C.; Norwitz, N.G.; Feldman, D.; Kyriakidou, Y.; Edwards, K.; Petagine, L.; Elliot, B.T.; Soto-Mota, A. Thyroid markers and body composition predict LDL-cholesterol change in lean healthy women on a ketogenic diet: Experimental support for the lipid energy model. Front. Endocrinol. 2023, 14, 1326768. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.; Manubolu, V.S.; Kinninger, A.; Norwitz, N.G.; Feldman, D.; Wood, T.R.; Fialkow, J.; Cury, R.; Feldman, T.; Nasir, K. Carbohydrate Restriction-Induced Elevations in LDL-Cholesterol and Atherosclerosis: The KETO Trial. JACC Adv. 2024, 3, 101109. [Google Scholar] [CrossRef]
- Zemer, A.; Samaei, S.; Yoel, U.; Biderman, A.; Pincu, Y. Ketogenic diet in clinical populations-a narrative review. Front. Med. 2024, 11, 1432717. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, B.M.; Kim, S.R.; Lee, B.-W. Ketone Body Induction: Insights into Metabolic Disease Management. Biomedicines 2025, 13, 1484. https://doi.org/10.3390/biomedicines13061484
Yoo BM, Kim SR, Lee B-W. Ketone Body Induction: Insights into Metabolic Disease Management. Biomedicines. 2025; 13(6):1484. https://doi.org/10.3390/biomedicines13061484
Chicago/Turabian StyleYoo, Byung Min, So Ra Kim, and Byung-Wan Lee. 2025. "Ketone Body Induction: Insights into Metabolic Disease Management" Biomedicines 13, no. 6: 1484. https://doi.org/10.3390/biomedicines13061484
APA StyleYoo, B. M., Kim, S. R., & Lee, B.-W. (2025). Ketone Body Induction: Insights into Metabolic Disease Management. Biomedicines, 13(6), 1484. https://doi.org/10.3390/biomedicines13061484