
The Overlapping Biology of Sepsis and Cancer and Therapeutic Implications



Abstract
1. Introduction
2. Pathophysiological Overlap Between Sepsis and Cancer and Therapeutic Implications
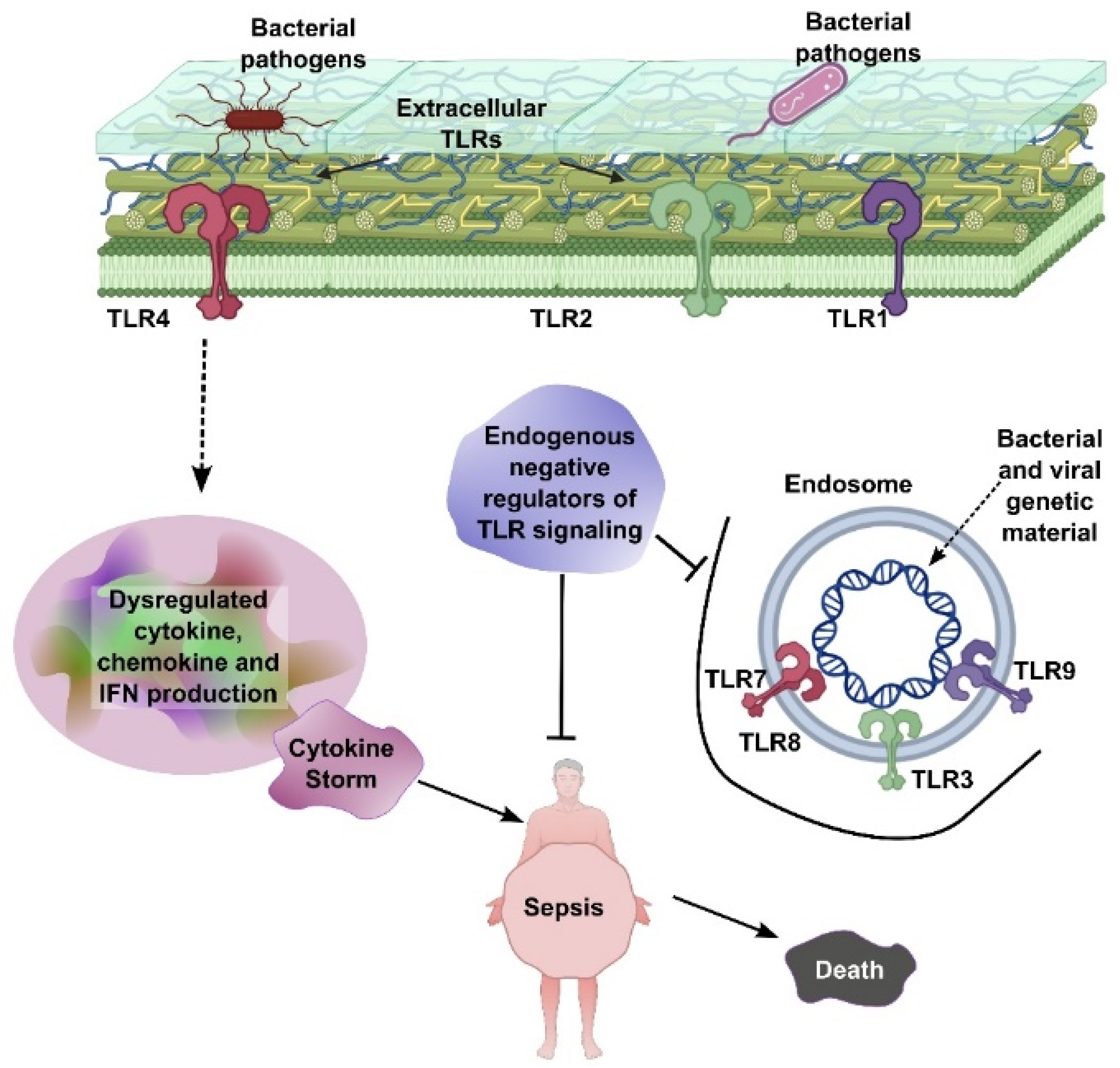
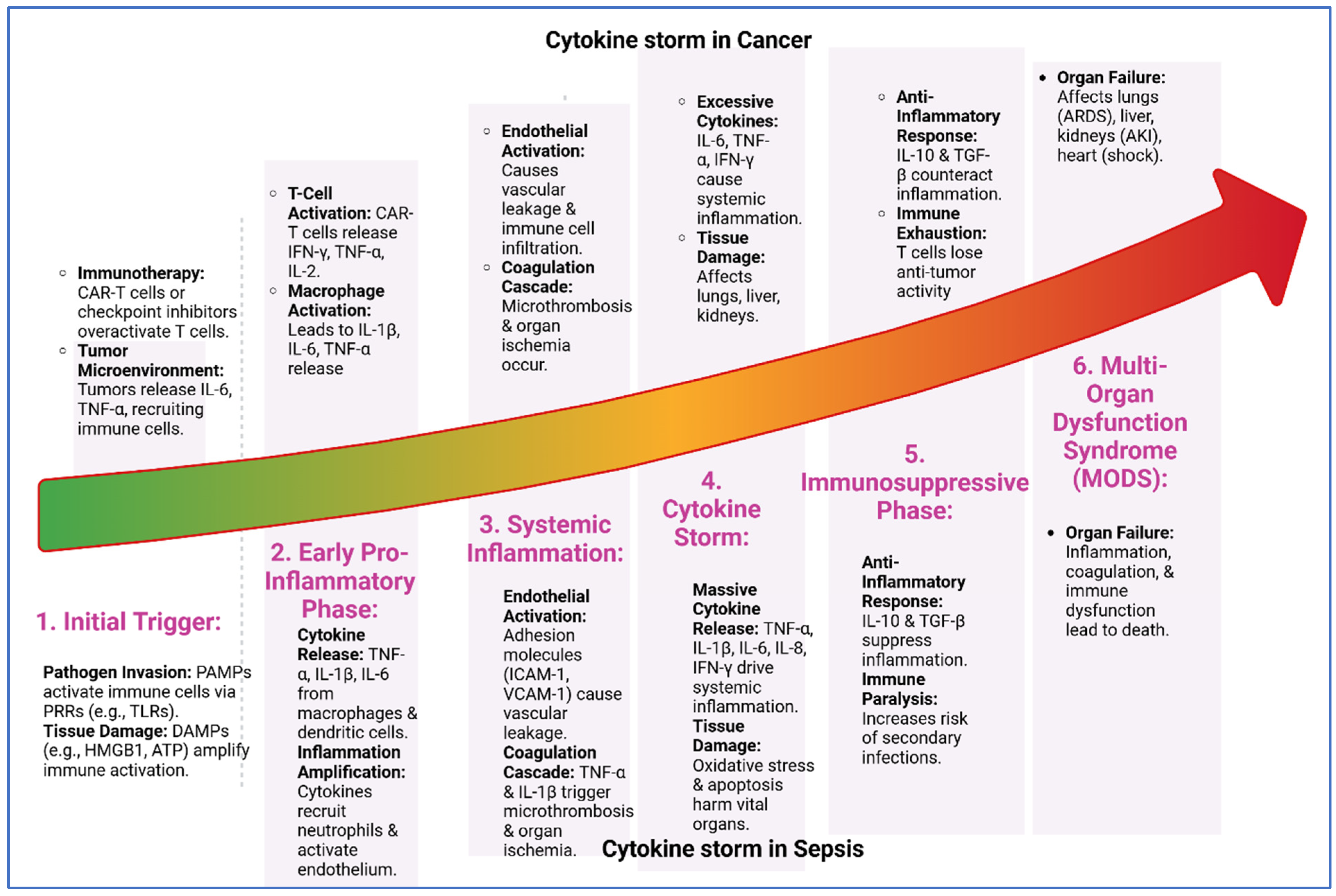
2.1. Cytokine Storm in Sepsis
2.2. Cytokines in Cancer
3. Immunosuppression in Sepsis and Cancer and Therapeutic Implications
3.1. Immune Paralysis in Sepsis
3.2. Immunosuppression in Cancer
4. Metabolic Alterations
4.1. The Warburg Effect on Cancer
4.2. Metabolic Shifts in Sepsis
4.3. Shared Metabolic Pathways and Therapeutic Implications
4.4. Glutamine Metabolism
4.5. Lipid Metabolism
5. Clinical Implications of the Sepsis–Cancer Connection
5.1. Impact of Sepsis on Tumor Microenvironment
5.2. Inflammatory Cytokines and Reactive Oxygen Species (ROS)
6. Long-Term Consequences of Sepsis in Cancer Patients
7. Therapeutic Opportunities
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- La Via, L.; Sangiorgio, G.; Stefani, S.; Marino, A.; Nunnari, G.; Cocuzza, S.; La Mantia, I.; Cacopardo, B.; Stracquadanio, S.; Spampinato, S.; et al. The Global Burden of Sepsis and Septic Shock. Epidemiologia 2024, 5, 456–478. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Pan, S.; Fang, X.; Wang, S.; Zou, X.; Shu, H.; Yang, X.; Xu, J.; Shang, Y. Association of cancers with the occurrence and 28-day mortality of sepsis: A mendelian randomization and mediator analysis. Sci. Rep. 2025, 15, 5600. [Google Scholar] [CrossRef]
- Gudiol, C.; Albasanz-Puig, A.; Cuervo, G.; Carratala, J. Understanding and Managing Sepsis in Patients with Cancer in the Era of Antimicrobial Resistance. Front. Med. 2021, 8, 636547. [Google Scholar] [CrossRef]
- Staudinger, T.; Pene, F. Current insights into severe sepsis in cancer patients. Rev. Bras. Ter. Intensiv. 2014, 26, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Said, S.A.; de Hullu, J.A.; van der Aa, M.A.; Walraven, J.E.W.; Bekkers, R.L.M.; Slangen, B.F.M.; Pickkers, P.; van Altena, A.M. Impact of Sepsis on the Oncologic Outcomes of Advanced Epithelial Ovarian Cancer Patients: A Multicenter Observational Study. Cancers 2023, 15, 4642. [Google Scholar] [CrossRef]
- Hensley, M.K.; Donnelly, J.P.; Carlton, E.F.; Prescott, H.C. Epidemiology and Outcomes of Cancer-Related Versus Non-Cancer-Related Sepsis Hospitalizations. Crit. Care Med. 2019, 47, 1310–1316. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Schulte, W.; Bernhagen, J.; Bucala, R. Cytokines in sepsis: Potent immunoregulators and potential therapeutic targets—An updated view. Mediat. Inflamm. 2013, 2013, 165974. [Google Scholar] [CrossRef] [PubMed]
- Damas, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; De Groote, D.; Franchimont, P.; Lamy, M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann. Surg. 1992, 215, 356–362. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, X.; Zhang, X.; Li, Z.; Wang, L.; Sun, Y.; Liu, Z.; Ma, X. Elevated levels of plasma TNF-alpha are associated with microvascular endothelial dysfunction in patients with sepsis through activating the NF-kappaB and p38 mitogen-activated protein kinase in endothelial cells. Shock 2014, 41, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Neff, T.A.; Guo, R.F.; Bernacki, K.D.; Laudes, I.J.; Sarma, J.V.; Lambris, J.D.; Ward, P.A. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J. Immunol. 2003, 170, 503–507. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320. [Google Scholar] [CrossRef]
- Raskova, M.; Lacina, L.; Kejik, Z.; Venhauerova, A.; Skalickova, M.; Kolar, M.; Jakubek, M.; Rosel, D.; Smetana, K., Jr.; Brabek, J. The Role of IL-6 in Cancer Cell Invasiveness and Metastasis-Overview and Therapeutic Opportunities. Cells 2022, 11, 3698. [Google Scholar] [CrossRef]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef]
- Yan, B.; Wang, H.; Rabbani, Z.N.; Zhao, Y.; Li, W.; Yuan, Y.; Li, F.; Dewhirst, M.W.; Li, C.Y. Tumor necrosis factor-alpha is a potent endogenous mutagen that promotes cellular transformation. Cancer Res. 2006, 66, 11565–11570. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, X.Y.; Pan, R.L.; Zhang, X.T.; Si, X.Y.; Chen, M.J.; Wang, M.Z.; Zhang, L. Tocilizumab for Advanced Non-Small-Cell Lung Cancer With Concomitant Cachexia: An Observational Study. J. Cachexia Sarcopenia Muscle 2024, 15, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Tavaci, T.; Halici, Z.; Cadirci, E.; Ozkaraca, M.; Kasali, K. The impact of tocilizumab treatment on the severity of inflammation and survival rates in sepsis is significantly influence by the timing of administration. Inflammopharmacology 2025, 33, 1393–1405. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Bilal, J.; Berlinberg, A.; Riaz, I.B.; Faridi, W.; Bhattacharjee, S.; Ortega, G.; Murad, M.H.; Wang, Z.; Prokop, L.J.; Alhifany, A.A.; et al. Risk of Infections and Cancer in Patients With Rheumatologic Diseases Receiving Interleukin Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e1913102. [Google Scholar] [CrossRef]
- Nakamori, Y.; Park, E.J.; Shimaoka, M. Immune Deregulation in Sepsis and Septic Shock: Reversing Immune Paralysis by Targeting PD-1/PD-L1 Pathway. Front. Immunol. 2020, 11, 624279. [Google Scholar] [CrossRef]
- Boomer, J.S.; Green, J.M.; Hotchkiss, R.S. The changing immune system in sepsis: Is individualized immuno-modulatory therapy the answer? Virulence 2014, 5, 45–56. [Google Scholar] [CrossRef]
- Jensen, I.J.; Sjaastad, F.V.; Griffith, T.S.; Badovinac, V.P. Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J. Immunol. 2018, 200, 1543–1553. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Morales, S.; Aranda-Uribe, I.S.; Perez-Amado, C.J.; Ramirez-Bello, J.; Hidalgo-Miranda, A. Mechanisms of Immunosuppressive Tumor Evasion: Focus on Acute Lymphoblastic Leukemia. Front. Immunol. 2021, 12, 737340. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.P.; Gergich, K.; Lubiniecki, G.M.; de Alwis, D.P.; Chen, C.; Tice, M.A.B.; Rubin, E.H. Pembrolizumab KEYNOTE-001: An adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann. Oncol. 2017, 28, 1388–1398. [Google Scholar] [CrossRef]
- Chang, K.C.; Burnham, C.A.; Compton, S.M.; Rasche, D.P.; Mazuski, R.J.; McDonough, J.S.; Unsinger, J.; Korman, A.J.; Green, J.M.; Hotchkiss, R.S. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit. Care 2013, 17, R85. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Harami-Papp, H.; Pongor, L.S.; Munkacsy, G.; Horvath, G.; Nagy, A.M.; Ambrus, A.; Hauser, P.; Szabo, A.; Tretter, L.; Gyorffy, B. TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget 2016, 7, 67183–67195. [Google Scholar] [CrossRef]
- Ferreira, B.L.; Sousa, M.B.; Leite, G.G.F.; Brunialti, M.K.C.; Nishiduka, E.S.; Tashima, A.K.; van der Poll, T.; Salomao, R. Glucose metabolism is upregulated in the mononuclear cell proteome during sepsis and supports endotoxin-tolerant cell function. Front. Immunol. 2022, 13, 1051514. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, G.; Wang, X.; Liu, D. Metabolic reprogramming consequences of sepsis: Adaptations and contradictions. Cell Mol. Life Sci. 2022, 79, 456. [Google Scholar] [CrossRef] [PubMed]
- Suetrong, B.; Walley, K.R. Lactic Acidosis in Sepsis: It’s Not All Anaerobic: Implications for Diagnosis and Management. Chest 2016, 149, 252–261. [Google Scholar] [CrossRef]
- Fan, Y.; Xue, H.; Li, Z.; Huo, M.; Gao, H.; Guan, X. Exploiting the Achilles’ heel of cancer: Disrupting glutamine metabolism for effective cancer treatment. Front. Pharmacol. 2024, 15, 1345522. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Dorai, T.; Pinto, J.T.; Denton, T.T. Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway. Biology 2023, 12, 1131. [Google Scholar] [CrossRef]
- Fang, L.; Gao, D.; Jiang, Z.; Li, G.; Li, M. Glutamine’s double-edged sword: Fueling tumor growth and offering therapeutic hope. Front. Immunol. 2025, 16, 1578940. [Google Scholar] [CrossRef]
- Najumudeen, A.K.; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; Race, A.M.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef]
- Nachef, M.; Ali, A.K.; Almutairi, S.M.; Lee, S.H. Targeting SLC1A5 and SLC3A2/SLC7A5 as a Potential Strategy to Strengthen Anti-Tumor Immunity in the Tumor Microenvironment. Front. Immunol. 2021, 12, 624324. [Google Scholar] [CrossRef]
- Nan, D.; Yao, W.; Huang, L.; Liu, R.; Chen, X.; Xia, W.; Sheng, H.; Zhang, H.; Liang, X.; Lu, Y. Glutamine and cancer: Metabolism, immune microenvironment, and therapeutic targets. Cell Commun. Signal. 2025, 23, 45. [Google Scholar] [CrossRef]
- Karinch, A.M.; Pan, M.; Lin, C.M.; Strange, R.; Souba, W.W. Glutamine metabolism in sepsis and infection. J. Nutr. 2001, 131, 2535S–2538S, discussion 2550S–2531S. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Tseng, S.-M.; Kuo, T.-C.; Wu, J.-M.; Chen, K.-Y.; Wu, M.-H.; Yang, P.-J.; Lee, P.-C.; Chen, P.-D.; Yeh, S.-L.; et al. The role of glutamine and leucine supplementation in liver metabolic reprogramming during sepsis. Life Sci. 2025, 374, 123708. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Soula, M.; Unlu, G.; Welch, R.; Chudnovskiy, A.; Uygur, B.; Shah, V.; Alwaseem, H.; Bunk, P.; Subramanyam, V.; Yeh, H.W.; et al. Glycosphingolipid synthesis mediates immune evasion in KRAS-driven cancer. Nature 2024, 633, 451–458. [Google Scholar] [CrossRef]
- Van Wyngene, L.; Vanderhaeghen, T.; Timmermans, S.; Vandewalle, J.; Van Looveren, K.; Souffriau, J.; Wallaeys, C.; Eggermont, M.; Ernst, S.; Van Hamme, E.; et al. Hepatic PPARalpha function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol. Med. 2020, 12, e11319. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Jin, Z.; Chai, Y.D.; Hu, S. Fatty Acid Metabolism and Cancer. Adv. Exp. Med. Biol. 2021, 1280, 231–241. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Chen, G.; Wu, K.; Li, H.; Xia, D.; He, T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front. Oncol. 2022, 12, 961637. [Google Scholar] [CrossRef]
- Dong, G.; Lin, X.H.; Liu, H.H.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; Chen, R.X. Intermittent hypoxia alleviates increased VEGF and pro-angiogenic potential in liver cancer cells. Oncol. Lett. 2019, 18, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Mowday, A.M.; Smaill, J.B.; Hermans, I.F.; Patterson, A.V. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells 2021, 10, 1006. [Google Scholar] [CrossRef]
- Jin, F.; Zheng, X.; Yang, Y.; Yao, G.; Ye, L.; Doeppner, T.R.; Hermann, D.M.; Wang, H.; Dai, Y. Impairment of hypoxia-induced angiogenesis by LDL involves a HIF-centered signaling network linking inflammatory TNFalpha and angiogenic VEGF. Aging 2019, 11, 328–349. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, e007530. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Williams, J.C.; Ford, M.L.; Coopersmith, C.M. Cancer and sepsis. Clin. Sci. 2023, 137, 881–893. [Google Scholar] [CrossRef]
- Nates, J.L.; Pene, F.; Darmon, M.; Mokart, D.; Castro, P.; David, S.; Povoa, P.; Russell, L.; Nielsen, N.D.; Gorecki, G.P.; et al. Septic shock in the immunocompromised cancer patient: A narrative review. Crit. Care 2024, 28, 285. [Google Scholar] [CrossRef] [PubMed]
- Mirouse, A.; Vigneron, C.; Llitjos, J.F.; Chiche, J.D.; Mira, J.P.; Mokart, D.; Azoulay, E.; Pene, F. Sepsis and Cancer: An Interplay of Friends and Foes. Am. J. Respir. Crit. Care Med. 2020, 202, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.X.; Akinyemiju, T.; Bartolucci, A.; Wang, H.E.; Waterbor, J.; Griffin, R. A prospective study of cancer survivors and risk of sepsis within the REGARDS cohort. Cancer Epidemiol. 2018, 55, 30–38. [Google Scholar] [CrossRef]
- Verma, D.P.; Tripathi, A.K.; Thakur, A.K. Innovative Strategies and Methodologies in Antimicrobial Peptide Design. J. Funct. Biomater. 2024, 15, 320. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Singh, J.; Trivedi, R.; Ranade, P. Shaping the Future of Antimicrobial Therapy: Harnessing the Power of Antimicrobial Peptides in Biomedical Applications. J. Funct. Biomater. 2023, 14, 539. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Vishwanatha, J.K. Role of Anti-Cancer Peptides as Immunomodulatory Agents: Potential and Design Strategy. Pharmaceutics 2022, 14, 2686. [Google Scholar] [CrossRef]
- Kumar, A.; Tripathi, A.K.; Kathuria, M.; Shree, S.; Tripathi, J.K.; Purshottam, R.K.; Ramachandran, R.; Mitra, K.; Ghosh, J.K. Single Amino Acid Substitutions at Specific Positions of the Heptad Repeat Sequence of Piscidin-1 Yielded Novel Analogs That Show Low Cytotoxicity and In Vitro and In Vivo Antiendotoxin Activity. Antimicrob. Agents Chemother. 2016, 60, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.; Harioudh, M.K.; Verma, N.K.; Saroj, J.; Gupta, A.; Pant, G.; Tripathi, J.K.; Kumar, A.; Kumari, T.; Tripathi, A.K.; et al. Characterization of a Myeloid Differentiation Factor 2-Derived Peptide that Facilitates THP-1 Macrophage-Mediated Phagocytosis of Gram-Negative Bacteria. ACS Infect. Dis. 2024, 10, 845–857. [Google Scholar] [CrossRef]
- Tandon, A.; Harioudh, M.K.; Ishrat, N.; Tripathi, A.K.; Srivastava, S.; Ghosh, J.K. An MD2-derived peptide promotes LPS aggregation, facilitates its internalization in THP-1 cells, and inhibits LPS-induced pro-inflammatory responses. Cell Mol. Life Sci. 2018, 75, 2431–2446. [Google Scholar] [CrossRef]
- Kumari, T.; Verma, D.P.; Kuldeep, J.; Dhanabal, V.B.; Verma, N.K.; Sahai, R.; Tripathi, A.K.; Saroj, J.; Ali, M.; Mitra, K.; et al. 10-Residue MyD88-Peptide Adopts beta-Sheet Structure, Self-Assembles, Binds to Lipopolysaccharides, and Rescues Mice from Endotoxin-Mediated Lung-Infection and Death. ACS Chem. Biol. 2022, 17, 3420–3434. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Desai, P.P.; Tyagi, A.; Lampe, J.B.; Srivastava, Y.; Donkor, M.; Jones, H.P.; Dzyuba, S.V.; Crossley, E.; Williams, N.S.; et al. Short peptides based on the conserved regions of MIEN1 protein exhibit anticancer activity by targeting the MIEN1 signaling pathway. J. Biol. Chem. 2024, 300, 105680. [Google Scholar] [CrossRef] [PubMed]
- Kumar Tripathi, A.; Desai, P.; Vishwanatha, J. Design and Characterization of Inhibitory Peptides (iPeps) Derived from of MIEN 1 Protein Sequence. 2024. Available online: https://unthsc-ir.tdl.org/items/d29a5383-05b1-4837-bc20-db68e1538a9b (accessed on 16 April 2025).
- Desai, P.P.; Narra, K.; James, J.D.; Jones, H.P.; Tripathi, A.K.; Vishwanatha, J.K. Combination of Small Extracellular Vesicle-Derived Annexin A2 Protein and mRNA as a Potential Predictive Biomarker for Chemotherapy Responsiveness in Aggressive Triple-Negative Breast Cancer. Cancers 2022, 15, 212. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Trivedi, R.; Tripathi, A.K.; Nandy, R.R.; Wagner, D.C.; Narra, K.; Chaudhary, P. Higher Expression of Annexin A2 in Metastatic Bladder Urothelial Carcinoma Promotes Migration and Invasion. Cancers 2022, 14, 5664. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Vishwanatha, J.K. Abstract 2184: Short Peptides derived from MIEN1 and their analogs exhibit anti-cancer activity in breast and prostate cancer cells. Cancer Res. 2023, 83, 2184. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Vishwanatha, J.K. Abstract LB034: First-in-class compounds targeting the MIEN1 signaling pathway: A breakthrough approach for breast and prostate cancer therapy. Cancer Res. 2024, 84, LB034. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Kumari, T.; Harioudh, M.K.; Yadav, P.K.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Identification of GXXXXG motif in Chrysophsin-1 and its implication in the design of analogs with cell-selective antimicrobial and anti-endotoxin activities. Sci. Rep. 2017, 7, 3384. [Google Scholar] [CrossRef]
- Saxena, R.; Vekariya, U.K.; Kumar, P.; Tripathi, A.K.; Ghosh, J.K.; Tripathi, R.K. HIV-1 Nef CAWLEAQ motif: A regulator of monocytes invasion through ENO1 modulation. Mol. Cell Biochem. 2018, 447, 151–164. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Kumari, T.; Tandon, A.; Sayeed, M.; Afshan, T.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Selective phenylalanine to proline substitution for improved antimicrobial and anticancer activities of peptides designed on phenylalanine heptad repeat. Acta Biomater. 2017, 57, 170–186. [Google Scholar] [CrossRef]
- Srivastava, S.; Kumar, A.; Tripathi, A.K.; Tandon, A.; Ghosh, J.K. Modulation of anti-endotoxin property of Temporin L by minor amino acid substitution in identified phenylalanine zipper sequence. Biochem. J. 2016, 473, 4045–4062. [Google Scholar] [CrossRef]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, T.Y.; Wang, Y.L.; Lee, E.P.; Lin, J.J.; Hsiao, Y.W.; Jaing, T.H.; Yang, C.P.; Hung, I.J. Outcome of Tocilizumab Treatment in Febrile Neutropenic Children with Severe Sepsis/Septic Shock in a Single-Center Retrospective Case Series. Cancers 2024, 16, 1512. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, A.; Ritter, H.L.; Dueck, A.; Nguyen, P.L.; Nikcevich, D.A.; Luyun, R.F.; Mattar, B.I.; Loprinzi, C.L. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 2010, 68, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Ljung, T.; Karlen, P.; Schmidt, D.; Hellstrom, P.M.; Lapidus, A.; Janczewska, I.; Sjoqvist, U.; Lofberg, R. Infliximab in inflammatory bowel disease: Clinical outcome in a population based cohort from Stockholm County. Gut 2004, 53, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Angus, D.C.; Moldawer, L.L.; Crouser, E.D.; Martin, G.S.; Coopersmith, C.M.; Brakenridge, S.; Mayr, F.B.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 Antibody (BMS-936559). Crit. Care Med. 2019, 47, 632–642. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cytokine | Role in Sepsis | Role in Cancer |
|---|---|---|
| TNF-α | -Early pro-inflammatory mediator. -Promotes inflammation, endothelial activation, and organ dysfunction. | -Promotes tumor progression, angiogenesis, and metastasis. -Can induce tumor cell death in high concentrations. |
| IL-1β | -Induces fever, vasodilation, and immune cell recruitment. -Contributes to tissue damage. | -Promotes tumor growth, angiogenesis, and metastasis. -Enhances immunosuppressive microenvironment. |
| IL-6 | -Key mediator of acute-phase response. -Correlates with severity and mortality. | -Promotes tumor growth, survival, and metastasis. -Drives chronic inflammation and immune evasion. |
| IL-8 (CXCL8) | -Chemoattractant for neutrophils. -Contributes to tissue injury. | -Promotes angiogenesis, tumor growth, and metastasis. -Attracts immunosuppressive cells to the tumor microenvironment. |
| IL-10 | -Anti-inflammatory cytokine. -Suppresses pro-inflammatory responses. -Can lead to immunosuppression. | -Promotes immune evasion by suppressing antitumor immunity. -Enhances tumor progression. |
| IL-17 | -Produced by Th17 cells. -Promotes neutrophil recruitment and inflammation. | -Promotes tumor growth, angiogenesis, and metastasis. -Contributes to chronic inflammation. |
| IL-23 | -Promotes Th17 cell differentiation and IL-17 production. -Amplifies inflammation. | -Promotes tumor growth and immune evasion. -Enhances chronic inflammation. |
| IFN-γ | -Activates macrophages and enhances pro-inflammatory responses. -Contributes to tissue damage. | -Can have antitumor effects by activating immune cells. -May promote tumor immune evasion in chronic settings. |
| TGF-β | -Anti-inflammatory cytokine. -Promotes tissue repair and immunosuppression. | -Promotes tumor progression, immune evasion, and metastasis. -Induces epithelial–mesenchymal transition (EMT). |
| VEGF | -Promotes vascular permeability and endothelial dysfunction. | -Drives angiogenesis, supporting tumor growth and metastasis. |
| HMGB1 | -Late-phase mediator of sepsis. -Sustains inflammation and organ damage. | -Promotes tumor growth, metastasis, and immune evasion. -Acts as a damage-associated molecular pattern (DAMP). |
| PD-1/PD-L1 | -Contributes to T-cell exhaustion and immunosuppression in sepsis. | -Key immune checkpoint in cancer. -Promotes immune evasion and tumor progression. |
| G-CSF | -Stimulates neutrophil production and mobilization. | -Promotes tumor growth and metastasis. -Enhances myeloid-derived suppressor cells (MDSCs). |
| MCP-1 (CCL2) | -Recruits monocytes and macrophages to sites of inflammation. | -Recruits tumor-associated macrophages (TAMs), promoting tumor progression and immune evasion. |
| Aspect | Sepsis | Cancer | Clinical Implications |
|---|---|---|---|
| Glucose Metabolism | -Hyperglycemia: Insulin resistance and increased gluconeogenesis. -Warburg Effect: Increased glycolysis. | -Warburg Effect: Aerobic glycolysis for rapid ATP production. -Increased Glucose Uptake: Enhanced by GLUT transporters. | -Targeting Glycolysis: Inhibitors like 2-DG may help in both conditions. -Glucose Control: Tight glucose management improves outcomes in sepsis. |
| Lactate Production | -Lactic Acidosis: Excessive glycolysis leads to lactate accumulation. | -High Lactate Levels: Lactate contributes to tumor microenvironment acidosis. | -Lactate as a Biomarker: High lactate levels correlate with poor prognosis in both conditions. |
| Lipid Metabolism | -Lipolysis: Increased breakdown of fats for energy. -Hyperlipidemia: Elevated free fatty acids. | -Lipid Synthesis: Increased de novo lipogenesis. -Fatty Acid Oxidation: Some cancers rely on fatty acids for energy. | -Lipid-Targeting Therapies: Inhibitors of lipogenesis (e.g., FASN inhibitors) are explored in cancer. |
| Protein Metabolism | -Protein Catabolism: Muscle breakdown for gluconeogenesis. -Negative Nitrogen Balance. | -Increased Protein Synthesis: Supports cell proliferation. -Amino Acid Dependency: Reliance on glutamine. | -Nutritional Support: Glutamine supplementation may benefit both conditions. |
| Glutamine Metabolism | -Glutamine Utilization: Supports immune cell function and energy production. | -Glutamine Addiction: Used for anaplerosis and nucleotide synthesis. | -Glutaminase Inhibitors: CB-839 is being tested in cancer and may have potential in sepsis. |
| Mitochondrial Dysfunction | -Impaired Oxidative Phosphorylation: Reduced ATP production. -ROS Production: Contributes to tissue damage. | -Altered Mitochondrial Function: Dysfunction or upregulation depending on cancer type. -ROS Signaling: Promotes tumor growth. | -Antioxidant Therapies: May help mitigate ROS-induced damage in both conditions. |
| Ketone Body Metabolism | -Increased Ketogenesis: In response to energy demands. | -Ketone Utilization: Some cancers use ketone bodies as an energy source. | -Ketogenic Diets: May benefit cancer patients and potentially sepsis patients. |
| Immune Cell Metabolism | -Metabolic Reprogramming: Immune cells shift to glycolysis. -Immunosuppression: M2 macrophages rely on oxidative metabolism. | -Tumor-Associated Immune Cells: TAMs and Tregs exhibit metabolic changes supporting tumor growth. | -Immunometabolism Targeting: Modulating immune cell metabolism may improve outcomes. |
| Hypoxia Response | -HIF-1α Activation: Promotes glycolysis and angiogenesis. | -HIF-1α Activation: Drives angiogenesis and tumor progression. | -HIF-1α Inhibitors: Potential therapeutic target in both conditions. |
| Insulin Resistance | -Peripheral Insulin Resistance: Reduces glucose uptake in muscle and adipose tissue. | -Altered Insulin Signaling: Some cancers exhibit insulin resistance or upregulate insulin/IGF-1 signaling. | -Insulin Sensitizers: May improve outcomes in sepsis and certain cancers. |
| Acidosis | -Metabolic Acidosis: Due to lactate accumulation and impaired renal function. | -Tumor Microenvironment Acidosis: Results from high lactate production. | -pH Modulation: Alkalinizing agents may help mitigate acidosis in both conditions. |
| Energy Demand | -Increased Energy Demand: To support hypermetabolic state and immune responses. | -Increased Energy Demand: To support rapid cell proliferation and tumor growth. | -Nutritional Support: High-calorie diets may benefit patients in both conditions. |
| Therapy | Indication | Clinical Trial/Approval | Outcome/Status | Reference |
|---|---|---|---|---|
| Tocilizumab | Cancer (CRS) | FDA Approval (2017) | Approved for CAR-T CRS | [91] |
| Tocilizumab | Sepsis (children) | Retrospective study | Reduced mortality, small number of cases | [92] |
| Infliximab | Cancer (NSCLC) | NCT00058264 | No benefit, increased fatigue | [93] |
| Infliximab | Sepsis | Observational | Infection risk, not recommended | [94] |
| Checkpoint Inhibitors | Sepsis | NCT02576457 (Phase 1b) | Safe, immunomodulatory, efficacy unproven | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, A.K.; Srivastava, Y. The Overlapping Biology of Sepsis and Cancer and Therapeutic Implications. Biomedicines 2025, 13, 1280. https://doi.org/10.3390/biomedicines13061280
Tripathi AK, Srivastava Y. The Overlapping Biology of Sepsis and Cancer and Therapeutic Implications. Biomedicines. 2025; 13(6):1280. https://doi.org/10.3390/biomedicines13061280
Chicago/Turabian StyleTripathi, Amit Kumar, and Yogesh Srivastava. 2025. "The Overlapping Biology of Sepsis and Cancer and Therapeutic Implications" Biomedicines 13, no. 6: 1280. https://doi.org/10.3390/biomedicines13061280
APA StyleTripathi, A. K., & Srivastava, Y. (2025). The Overlapping Biology of Sepsis and Cancer and Therapeutic Implications. Biomedicines, 13(6), 1280. https://doi.org/10.3390/biomedicines13061280