
Identification of Biomarkers Co-Associated with Lactylation and Acetylation in Systemic Lupus Erythematosus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Data Processing
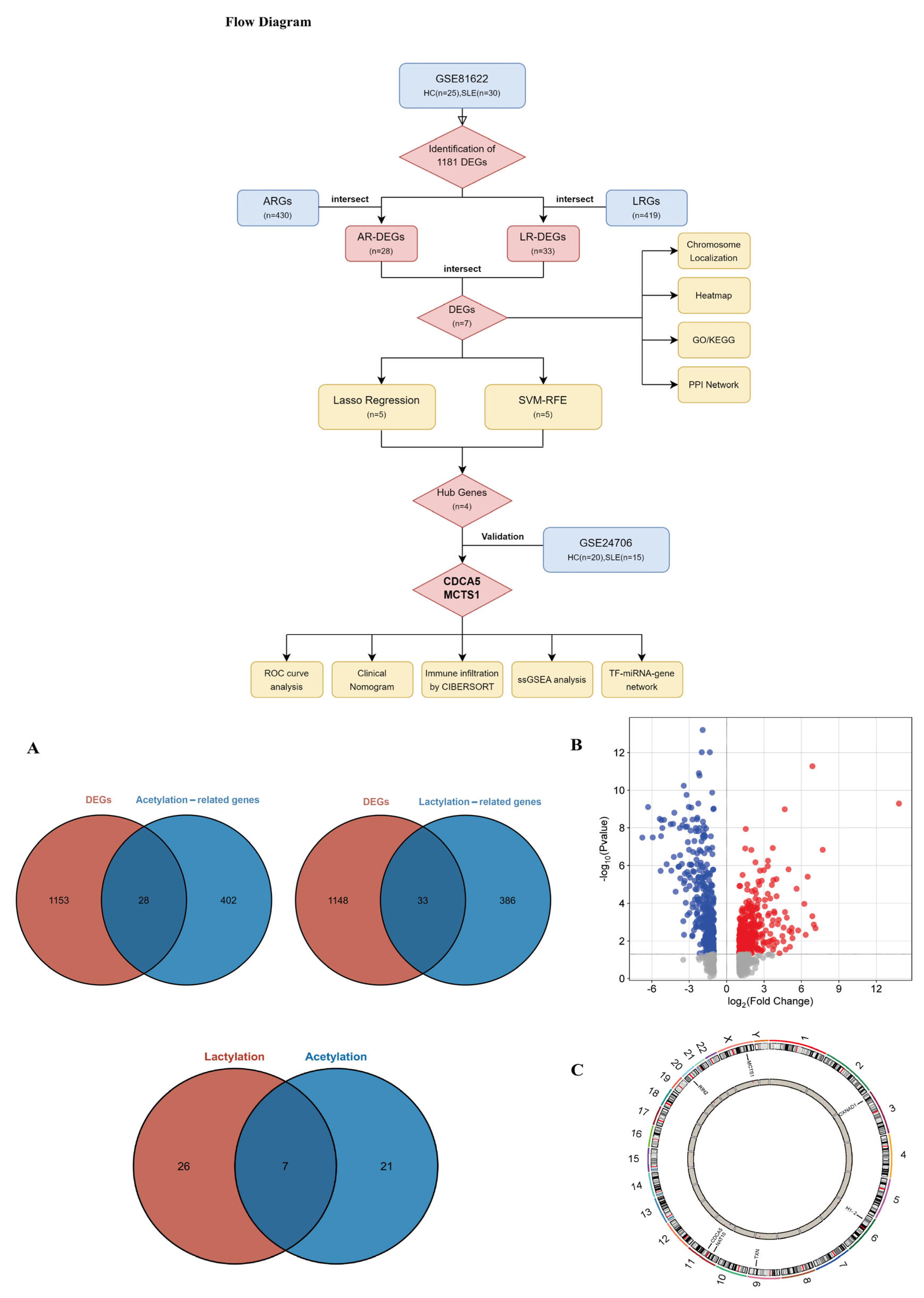
2.2. Indentification of DEGs and Enrichment Analysis
2.3. The Acquisition of Genes Which Are Co-Associated with Lactylation and Acetylation
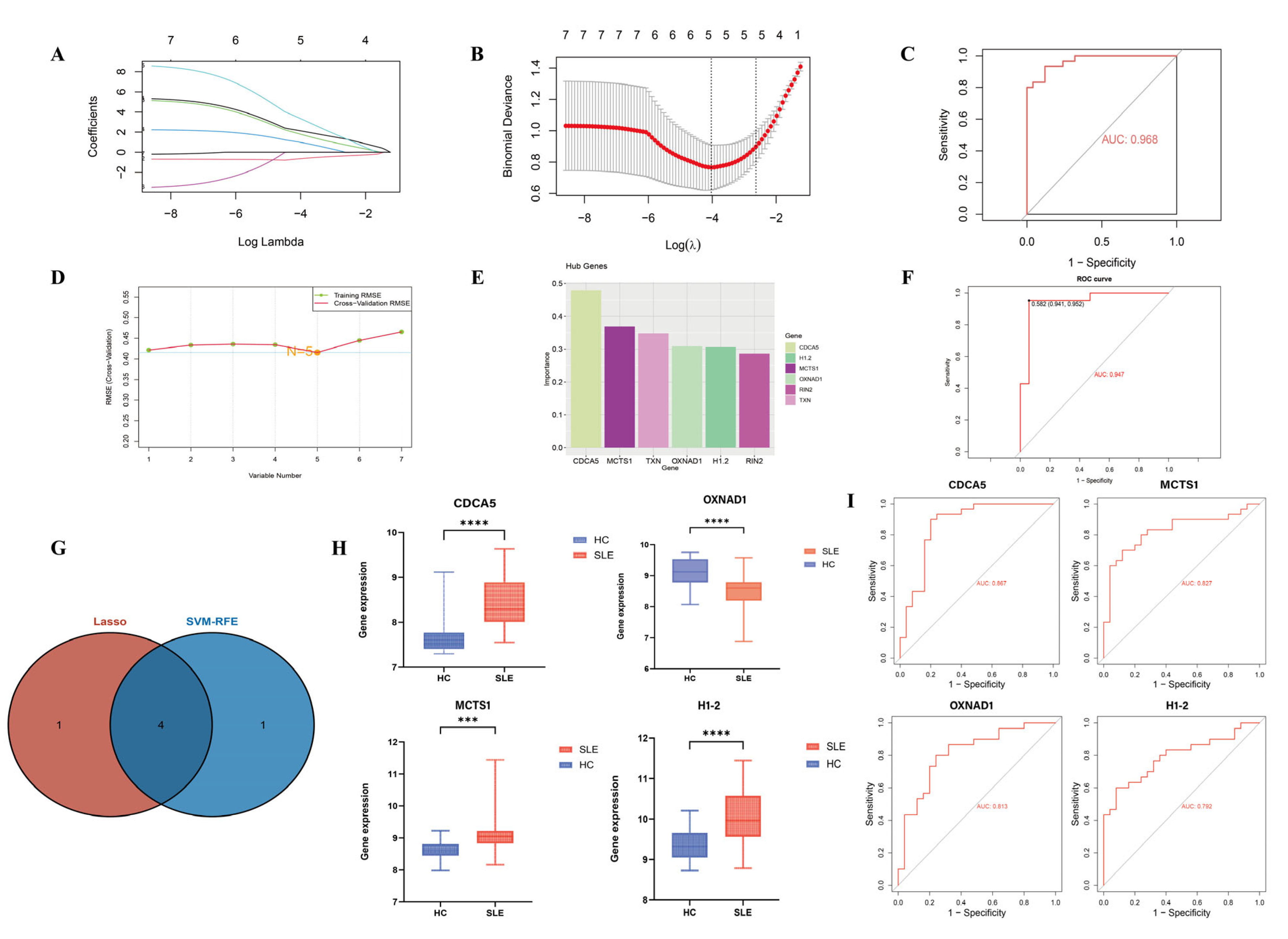
2.4. Machine Learning Algorithms
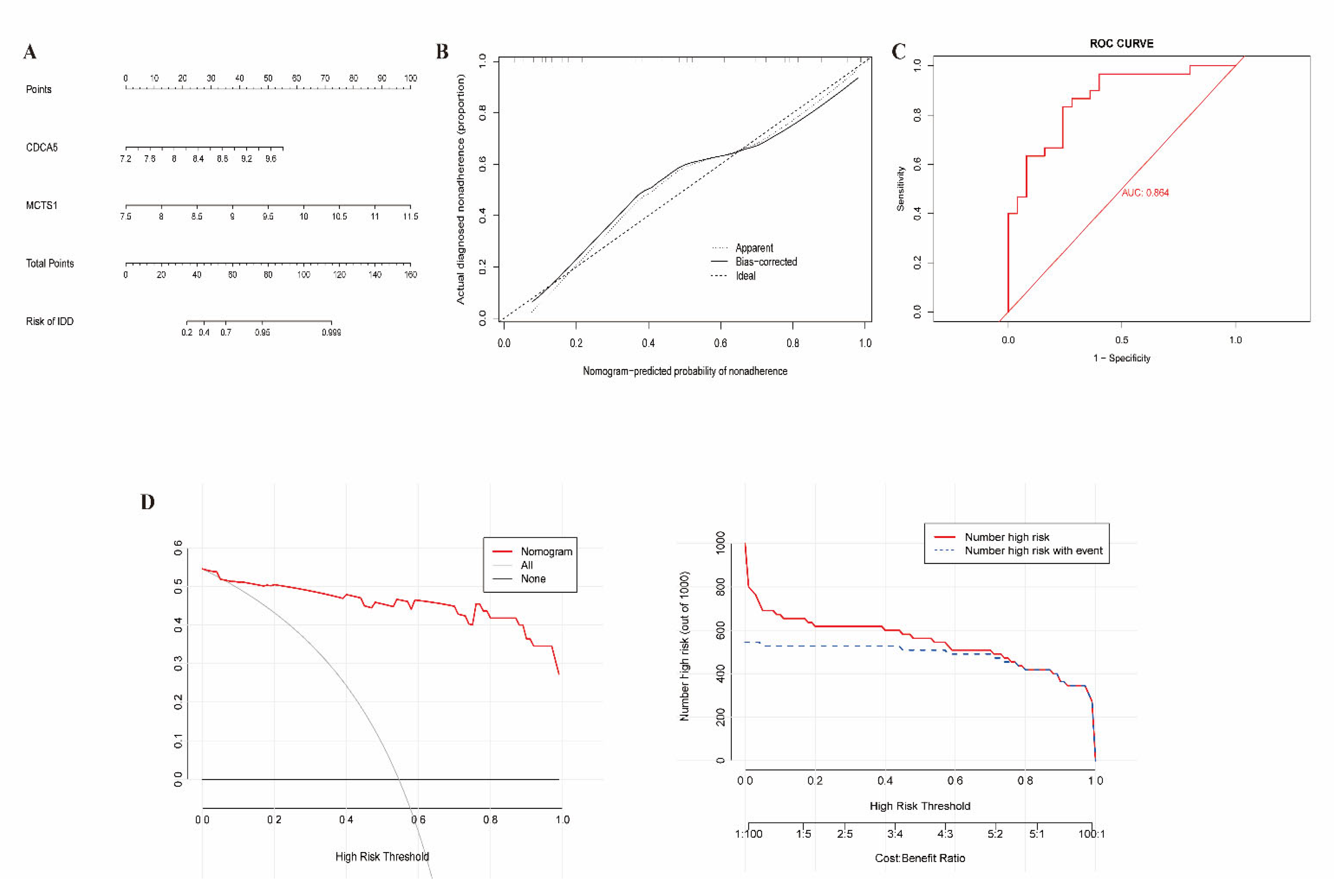
2.5. Clinical Nomogram Model
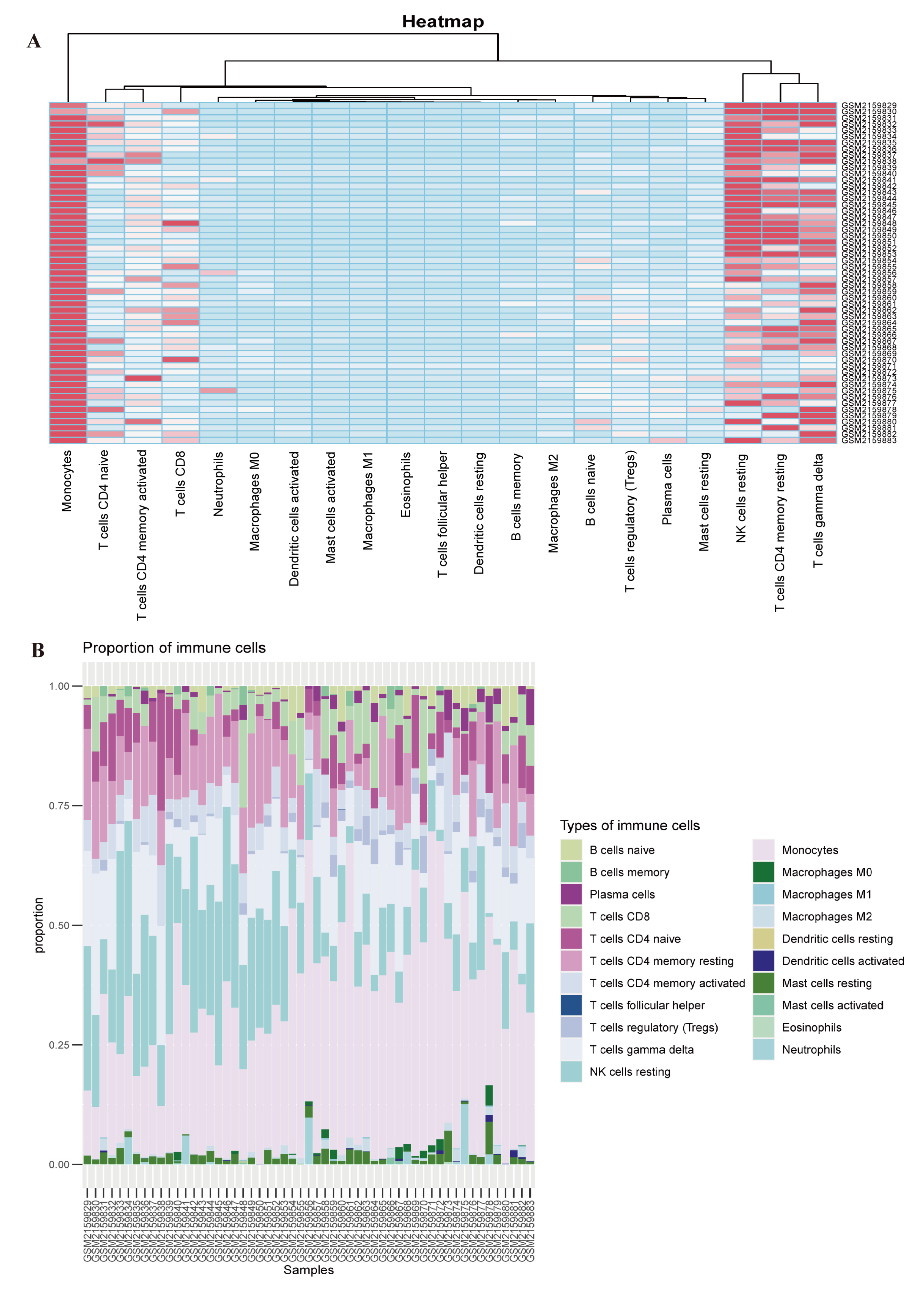
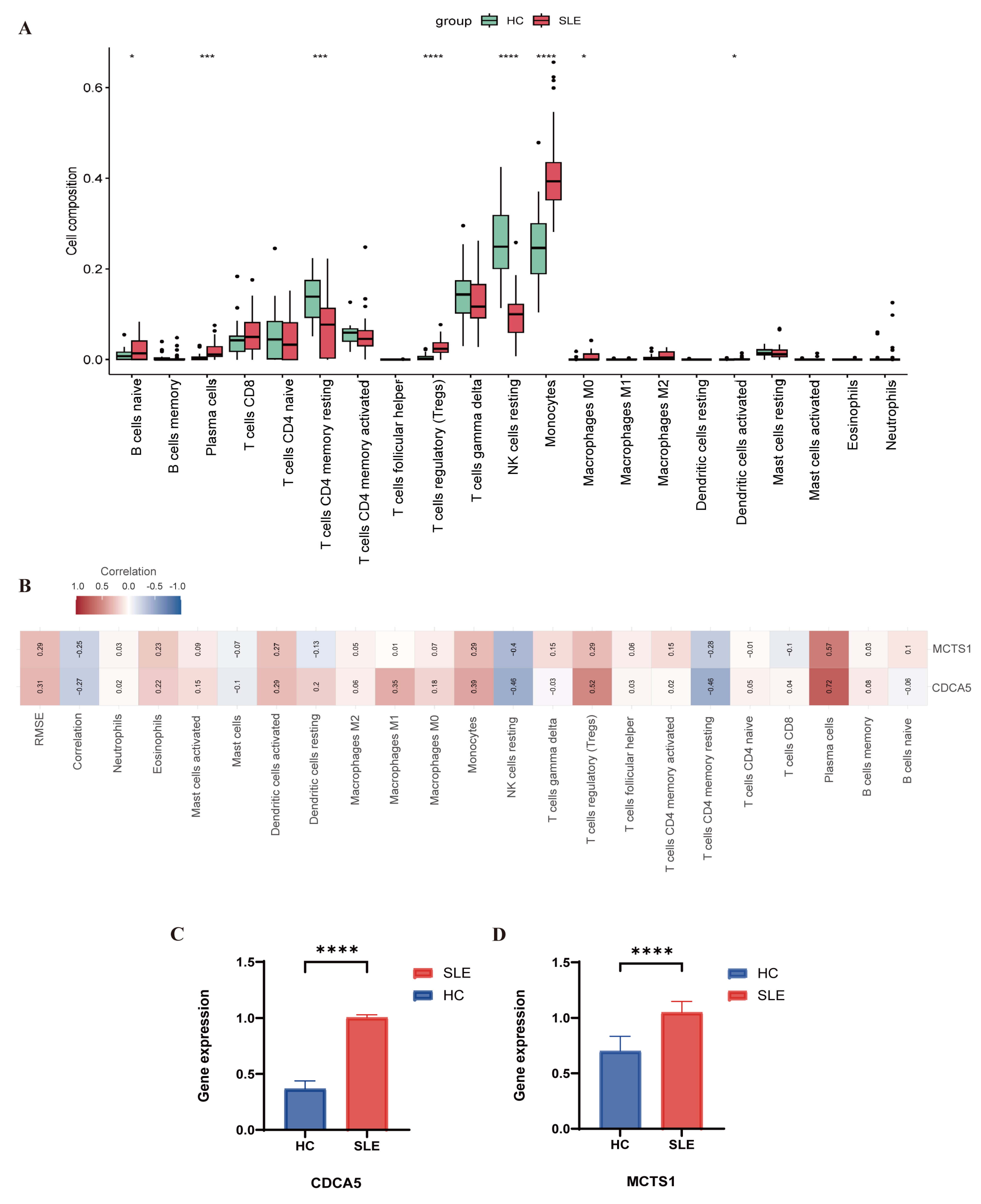
2.6. Immune Cell Infiltration Analysis
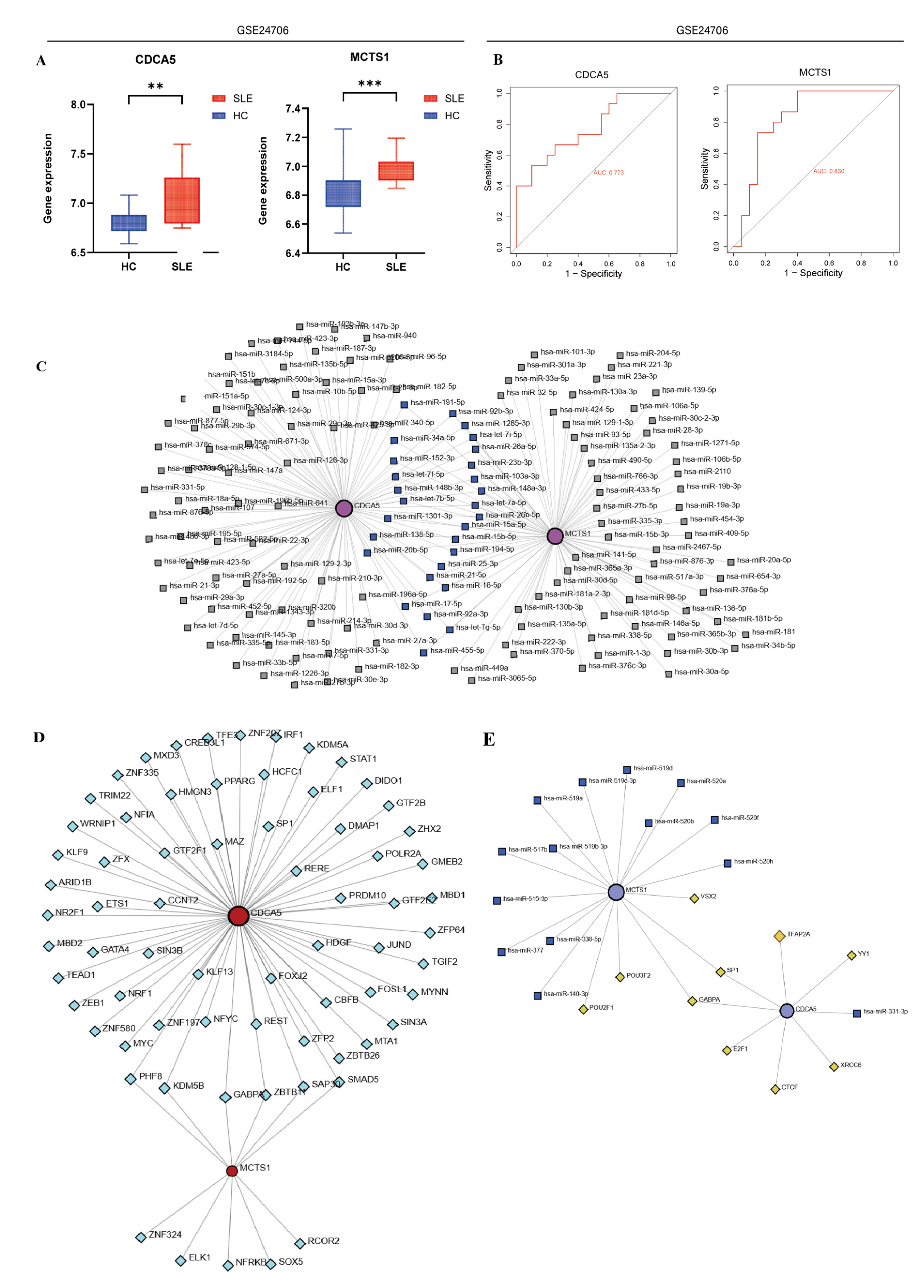
2.7. Construction of Regulatory Networks
2.8. Human Blood Specimen Collection
2.9. Quantitative PCR Assay
2.10. Statistical Analysis
3. Results
3.1. Identifications of DEGs Between SLE Patients and HCs
3.2. Functional Enrichment Analysis of Co-Associated DEGs
3.3. Identification and Validation of SLE-Associated Lactylation and Acetylation Regulators
3.4. Construction of Clinical Nomogram
3.5. Evaluation of Immune Cell Infiltration
3.6. Quantitative PCR Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dossybayeva, K.; Abdukhakimova, D.; Poddighe, D. Basophils and Systemic Lupus Erythematosus in Murine Models and Human Patients. Biology 2020, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Kaewraemruaen, C.; Ritprajak, P.; Hirankarn, N. Dendritic cells as key players in systemic lupus erythematosus. Asian Pac. J. Allergy Immunol. 2020, 38, 225–232. [Google Scholar] [CrossRef]
- Gensous, N.; Boizard-Moracchini, A.; Lazaro, E.; Richez, C.; Blanco, P. Update on the cellular pathogenesis of lupus. Curr. Opin. Rheumatol. 2021, 33, 190–196. [Google Scholar] [CrossRef]
- Dorner, T.; Furie, R. Novel paradigms in systemic lupus erythematosus. Lancet 2019, 393, 2344–2358. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Irizarry-Caro, R.A.; McDaniel, M.M.; Overcast, G.R.; Jain, V.G.; Troutman, T.D.; Pasare, C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc. Natl. Acad. Sci. USA 2020, 117, 30628–30638. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- Hagihara, H.; Shoji, H.; Otabi, H.; Toyoda, A.; Katoh, K.; Namihira, M.; Miyakawa, T. Protein lactylation induced by neural excitation. Cell Rep. 2021, 37, 109820. [Google Scholar] [CrossRef]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef]
- Caielli, S.; Cardenas, J.; de Jesus, A.A.; Baisch, J.; Walters, L.; Blanck, J.P.; Balasubramanian, P.; Stagnar, C.; Ohouo, M.; Hong, S.; et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell 2021, 184, 4464–4479.e4419. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Panther, E.; Liao, X.; Grammer, A.C.; Lipsky, P.E.; Reilly, C.M. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. [Google Scholar] [CrossRef] [PubMed]
- Farivar, S.; Shaabanpour Aghamaleki, F. Effects of Major Epigenetic Factors on Systemic Lupus Erythematosus. Iran. Biomed. J. 2018, 22, 294–302. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- An, S.; Yao, Y.; Hu, H.; Wu, J.; Li, J.; Li, L.; Wu, J.; Sun, M.; Deng, Z.; Zhang, Y.; et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. 2023, 14, 457. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Zhu, H.; Mi, W.; Luo, H.; Chen, T.; Liu, S.; Raman, I.; Zuo, X.; Li, Q.Z. Whole-genome transcription and DNA methylation analysis of peripheral blood mononuclear cells identified aberrant gene regulation pathways in systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 162. [Google Scholar] [CrossRef]
- Li, Q.Z.; Karp, D.R.; Quan, J.; Branch, V.K.; Zhou, J.; Lian, Y.; Chong, B.F.; Wakeland, E.K.; Olsen, N.J. Risk factors for ANA positivity in healthy persons. Arthritis Res. Ther. 2011, 13, R38. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]
- Sanz, H.; Valim, C.; Vegas, E.; Oller, J.M.; Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 2018, 19, 432. [Google Scholar] [CrossRef]
- Yang, C.; Delcher, C.; Shenkman, E.; Ranka, S. Machine learning approaches for predicting high cost high need patient expenditures in health care. Biomed. Eng. Online 2018, 17, 131. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Park, S.Y. Nomogram: An analogue tool to deliver digital knowledge. J. Thorac. Cardiovasc. Surg. 2018, 155, 1793. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Aringer, M. EULAR/ACR classification criteria for SLE. Semin. Arthritis Rheum. 2019, 49, S14–S17. [Google Scholar] [CrossRef]
- Ji, R.; Chen, W.; Wang, Y.; Gong, F.; Huang, S.; Zhong, M.; Liu, Z.; Chen, Y.; Ma, L.; Yang, Z.; et al. The Warburg Effect Promotes Mitochondrial Injury Regulated by Uncoupling Protein-2 in Septic Acute Kidney Injury. Shock 2021, 55, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; He, Z.; Li, Z.; Wang, Y.; Wu, N.; Sun, H.; Zhou, Z.; Hu, Q.; Cong, X. Lactylation: The novel histone modification influence on gene expression, protein function, and disease. Clin. Epigenetics 2024, 16, 72. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Ma, Q.; Ying, X.; Wang, F.; Hou, Y.; Wang, D.; Zhu, L.; Huang, J.; He, C. Histone lactylation in macrophage biology and disease: From plasticity regulation to therapeutic implications. eBioMedicine 2025, 111, 105502. [Google Scholar] [CrossRef]
- Ren, H.; Tang, Y.; Zhang, D. The emerging role of protein L-lactylation in metabolic regulation and cell signalling. Nat. Metab. 2025, 7, 647–664. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, H.; Liu, M.; Zhou, T.; Cheng, X.; Huang, W.; Cao, L. The role and mechanism of histone lactylation in health and diseases. Front. Genet. 2022, 13, 949252. [Google Scholar] [CrossRef]
- Tsukihara, S.; Akiyama, Y.; Shimada, S.; Hatano, M.; Igarashi, Y.; Taniai, T.; Tanji, Y.; Kodera, K.; Yasukawa, K.; Umeura, K.; et al. Delactylase effects of SIRT1 on a positive feedback loop involving the H19-glycolysis-histone lactylation in gastric cancer. Oncogene 2025, 44, 724–738. [Google Scholar] [CrossRef]
- Wan, L.; Zhang, H.; Liu, J.; He, Q.; Zhao, J.; Pan, C.; Zheng, K.; Tang, Y. Lactylation and human disease. Expert. Rev. Mol. Med. 2025, 27, e10. [Google Scholar] [CrossRef]
- Jog, N.R.; Wagner, C.A.; Aberle, T.; Chakravarty, E.F.; Arriens, C.; Guthridge, J.M.; James, J.A. Neutrophils isolated from systemic lupus erythematosus patients exhibit a distinct functional phenotype. Front. Immunol. 2024, 15, 1339250. [Google Scholar] [CrossRef]
- Kaveri, S.V.; Mouthon, L.; Bayry, J. Basophils and nephritis in lupus. N. Engl. J. Med. 2010, 363, 1080–1082. [Google Scholar] [CrossRef]
- Pan, Q.; Feng, Y.; Peng, Y.; Zhou, H.; Deng, Z.; Li, L.; Han, H.; Lin, J.; Shi, L.; Wang, S.; et al. Basophil Recruitment to Skin Lesions of Patients with Systemic Lupus Erythematosus Mediated by CCR1 and CCR2. Cell Physiol. Biochem. 2017, 43, 832–839. [Google Scholar] [CrossRef]
- Charles, N.; Hardwick, D.; Daugas, E.; Illei, G.G.; Rivera, J. Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat. Med. 2010, 16, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Dossybayeva, K.; Bexeitov, Y.; Mukusheva, Z.; Almukhamedova, Z.; Assylbekova, M.; Abdukhakimova, D.; Rakhimzhanova, M.; Poddighe, D. Analysis of Peripheral Blood Basophils in Pediatric Systemic Lupus Erythematosus. Diagnostics 2022, 12, 1701. [Google Scholar] [CrossRef]
- Liang, P.; Tang, Y.; Fu, S.; Lv, J.; Liu, B.; Feng, M.; Li, J.; Lai, D.; Wan, X.; Xu, A. Basophil count, a marker for disease activity in systemic lupus erythematosus. Clin. Rheumatol. 2015, 34, 891–896. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Z.; Feng, Y.; Zheng, C.; Li, F.; Sun, Z.; Xiang, M.; Zhu, J.; Chu, M.; Xu, J.; Liang, J. Identification of Biomarkers Co-Associated with Lactylation and Acetylation in Systemic Lupus Erythematosus. Biomedicines 2025, 13, 1274. https://doi.org/10.3390/biomedicines13061274
Gao Z, Feng Y, Zheng C, Li F, Sun Z, Xiang M, Zhu J, Chu M, Xu J, Liang J. Identification of Biomarkers Co-Associated with Lactylation and Acetylation in Systemic Lupus Erythematosus. Biomedicines. 2025; 13(6):1274. https://doi.org/10.3390/biomedicines13061274
Chicago/Turabian StyleGao, Zhanyan, Yang Feng, Chenghui Zheng, Fei Li, Zhan Sun, Mengmeng Xiang, Junrong Zhu, Mingyu Chu, Jinhua Xu, and Jun Liang. 2025. "Identification of Biomarkers Co-Associated with Lactylation and Acetylation in Systemic Lupus Erythematosus" Biomedicines 13, no. 6: 1274. https://doi.org/10.3390/biomedicines13061274
APA StyleGao, Z., Feng, Y., Zheng, C., Li, F., Sun, Z., Xiang, M., Zhu, J., Chu, M., Xu, J., & Liang, J. (2025). Identification of Biomarkers Co-Associated with Lactylation and Acetylation in Systemic Lupus Erythematosus. Biomedicines, 13(6), 1274. https://doi.org/10.3390/biomedicines13061274