
Targeting Aggressive Prostate Carcinoma Cells with Mesothelin-CAR-T Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Analysis of Gene Expression Datasets Derived from PCa Patients
2.3. Cytotoxicity Assay
2.4. Tumor-Killing Assays
2.5. Protein Preparation and Western Blot Analysis
2.6. Immunohistochemistry and Immunofluorescence Stainings
2.7. RNA Preparation, cDNA Synthesis, and Quantitative Real-Time PCR
2.8. Flow Cytometry and Antibodies
2.9. Statistical Analysis
3. Results
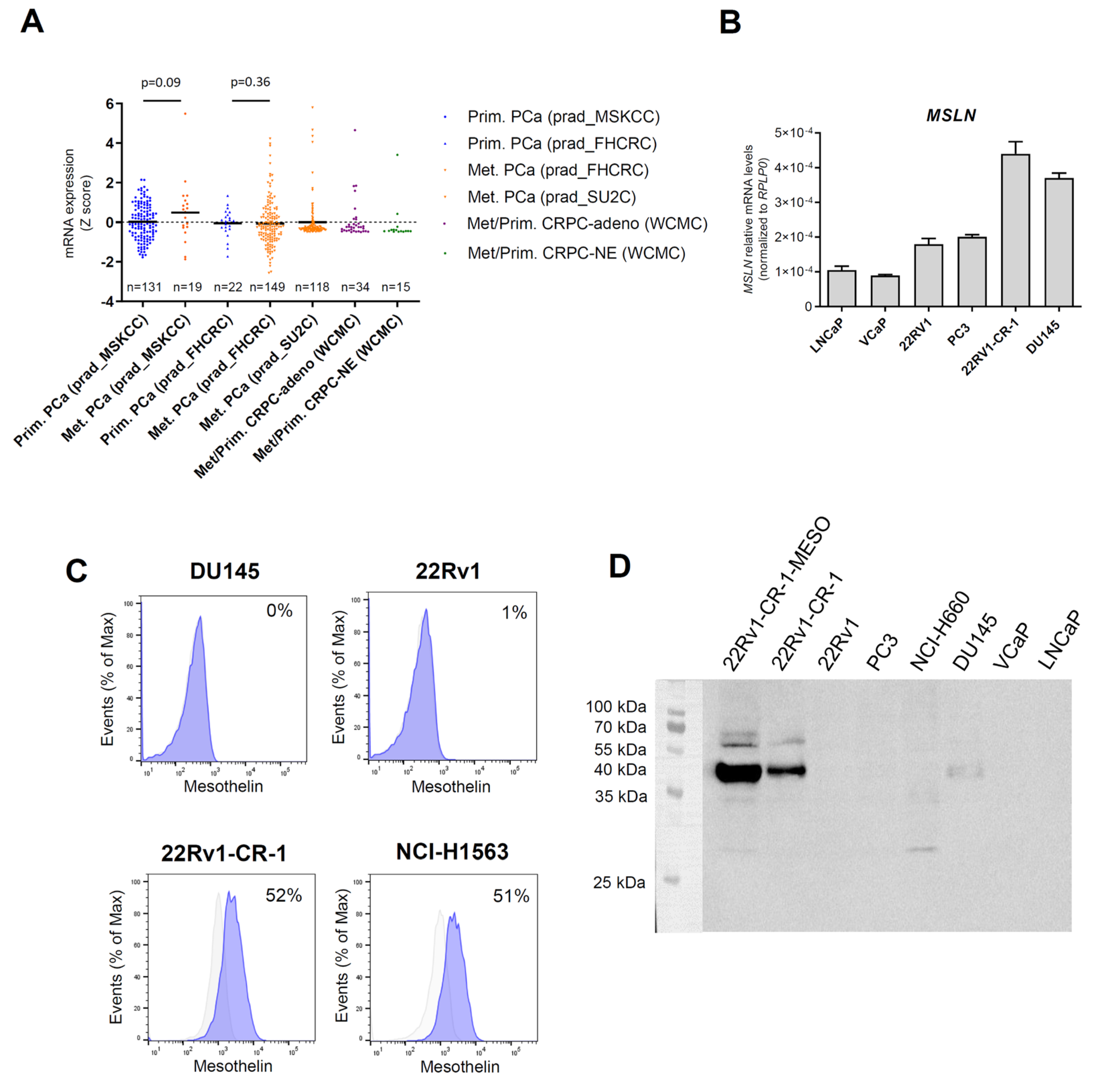
3.1. Mesothelin Expression Was Marginal in Primary Prostate Cancers but Upregulated in a Subset of Metastatic Prostate Cancers
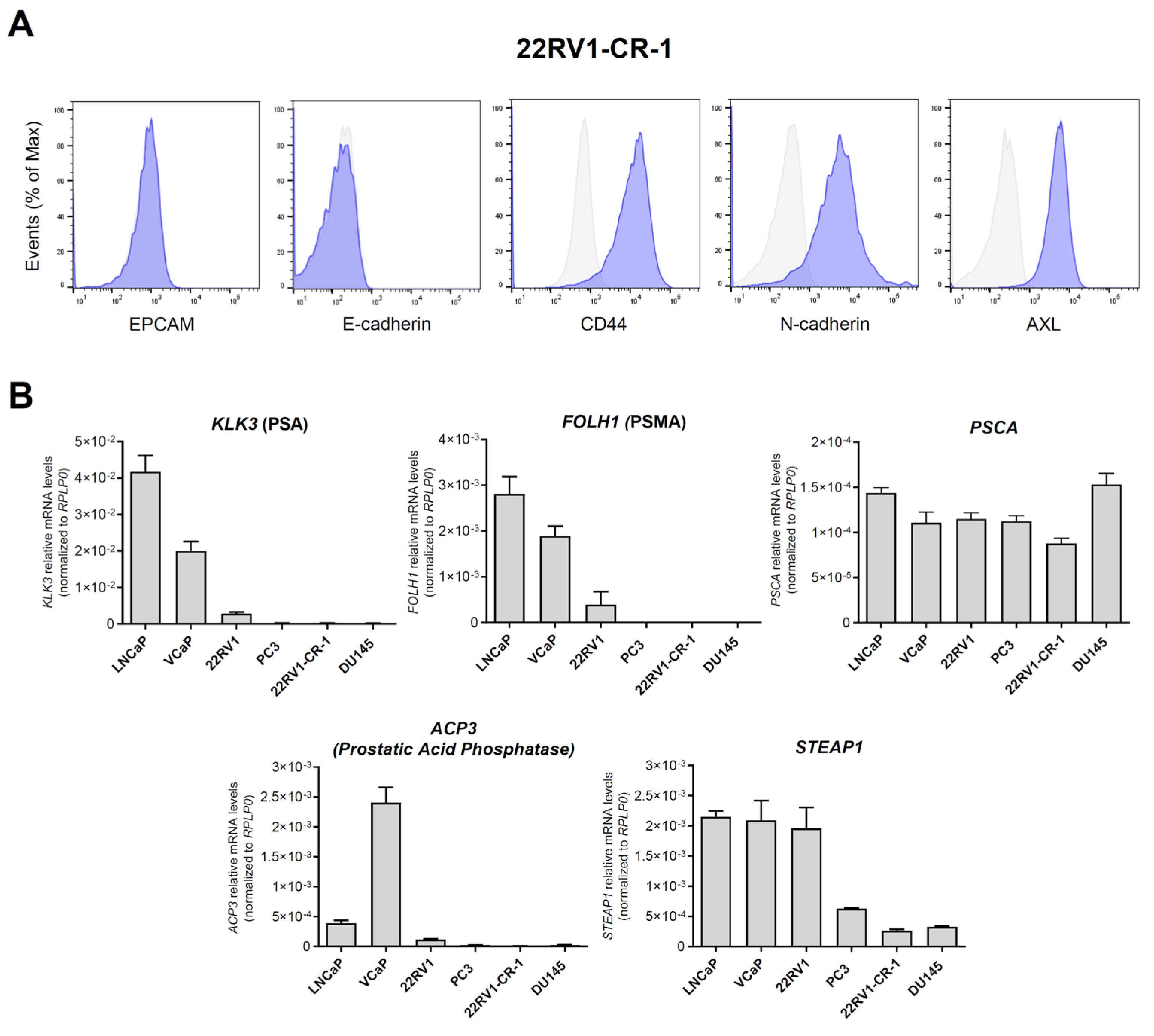
3.2. A PCa Model with Aggressive Features Endogenously Expresses Significant Amounts of Mesothelin
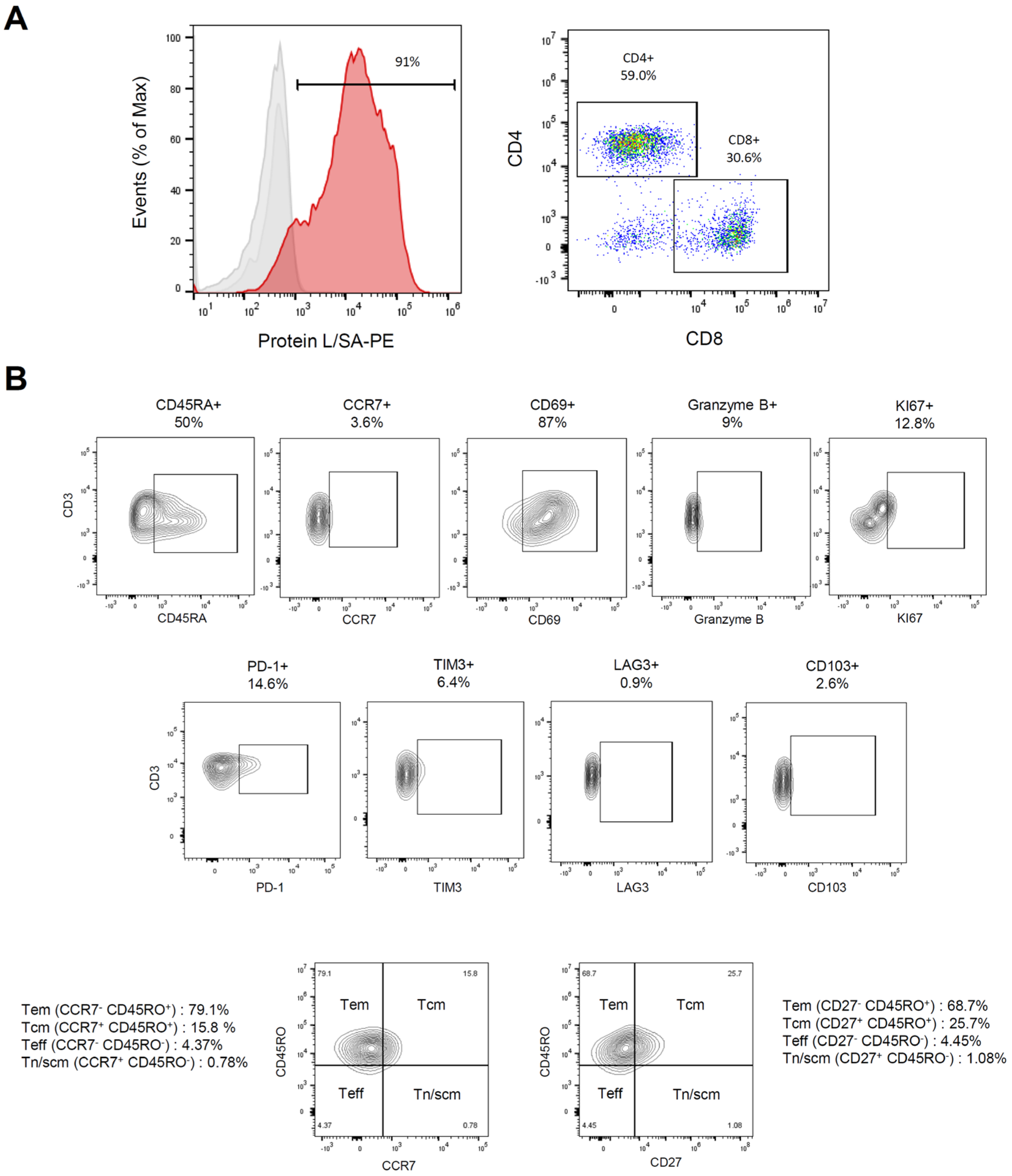
3.3. Characterization of Meso-BBζ-CART Cells
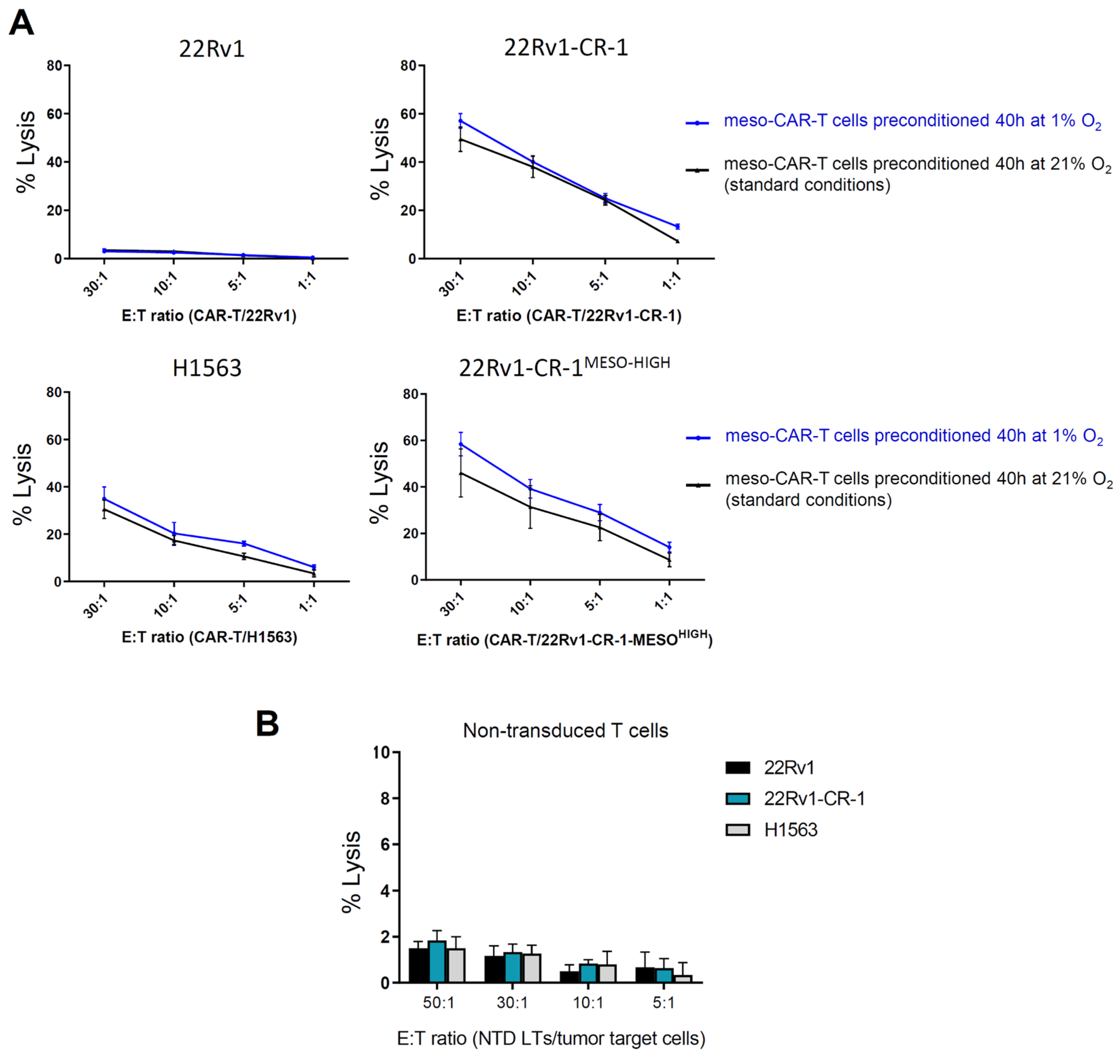
3.4. Meso-BBζ-CAR-T Cells Efficiently Kill Aggressive PCa Cells Expressing Mesothelin
3.5. Meso-BBζ-CAR-T Cells Showed Similar Cancer Killing Capacities in Normoxia and Hypoxia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Foulon, S.; Carles, J.; Roubaud, G.; McDermott, R.; Fléchon, A.; Tombal, B.; Supiot, S.; Berthold, D.; Ronchin, P.; et al. Abiraterone plus Prednisone Added to Androgen Deprivation Therapy and Docetaxel in de Novo Metastatic Castration-Sensitive Prostate Cancer (PEACE-1): A Multicentre, Open-Label, Randomised, Phase 3 Study with a 2 × 2 Factorial Design. Lancet 2022, 399, 1695–1707. [Google Scholar] [CrossRef]
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, Á.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2022, 386, 1132–1142. [Google Scholar] [CrossRef]
- Corsini, C.; Garmo, H.; Orrason, A.W.; Gedeborg, R.; Stattin, P.; Westerberg, M. Survival Trend in Individuals with De Novo Metastatic Prostate Cancer After the Introduction of Doublet Therapy. JAMA Netw. Open 2023, 6, e2336604. [Google Scholar] [CrossRef]
- Posdzich, P.; Darr, C.; Hilser, T.; Wahl, M.; Herrmann, K.; Hadaschik, B.; Grünwald, V. Metastatic Prostate Cancer—A Review of Current Treatment Options and Promising New Approaches. Cancers 2023, 15, 461. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients with Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. JCO 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E.; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with Enzalutamide versus Enzalutamide Alone in Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase 3 Trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Doger De Spéville, B.; Carles Galceran, J.; Peer, A.; Sarid, D.L.; Eigl, B.J.; Avadhani, A.; Yao, D.; Lin, V.; Wu, S.; et al. 1367P JNJ-70218902 (JNJ-902), a TMEFF2 x CD3 Bispecific Antibody, in Prostate Cancer: Initial Results from a Phase I Dose Escalation Study. Ann. Oncol. 2022, 33, S1166. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Park, J.A.; Long, A.; Guo, H.-F.; Cheung, N.-K.V. Novel Potent Anti-STEAP1 Bispecific Antibody to Redirect T Cells for Cancer Immunotherapy. J. Immunother. Cancer 2021, 9, e003114. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.-Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.-T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-Targeting TGFβ-Insensitive Armored CAR T Cells in Metastatic Castration-Resistant Prostate Cancer: A Phase 1 Trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Wolf, P.; Alzubi, J.; Gratzke, C.; Cathomen, T. The Potential of CAR T Cell Therapy for Prostate Cancer. Nat. Rev. Urol. 2021, 18, 556–571. [Google Scholar] [CrossRef]
- Kennedy, L.B.; Salama, A.K.S. A Review of Cancer Immunotherapy Toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef]
- Schepisi, G.; Cursano, M.C.; Casadei, C.; Menna, C.; Altavilla, A.; Lolli, C.; Cerchione, C.; Paganelli, G.; Santini, D.; Tonini, G.; et al. CAR-T Cell Therapy: A Potential New Strategy against Prostate Cancer. J. Immunother. Cancer 2019, 7, 258. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular Adaptation to Hypoxia through Hypoxia Inducible Factors and Beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Engelsen, A.S.T.; Buart, S.; Elsayed, W.S.; Venkatesh, G.H.; Chouaib, S. Hypoxia-Driven Intratumor Heterogeneity and Immune Evasion. Cancer Lett. 2020, 492, 1–10. [Google Scholar] [CrossRef]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. [Google Scholar] [CrossRef]
- Bigos, K.J.; Quiles, C.G.; Lunj, S.; Smith, D.J.; Krause, M.; Troost, E.G.; West, C.M.; Hoskin, P.; Choudhury, A. Tumour Response to Hypoxia: Understanding the Hypoxic Tumour Microenvironment to Improve Treatment Outcome in Solid Tumours. Front. Oncol. 2024, 14, 1331355. [Google Scholar] [CrossRef]
- Movsas, B.; Chapman, J.D.; Hanlon, A.L.; Horwitz, E.M.; Pinover, W.H.; Greenberg, R.E.; Stobbe, C.; Hanks, G.E. Hypoxia in Human Prostate Carcinoma: An Eppendorf Po2 Study. Am. J. Clin. Oncol. 2001, 24, 458–461. [Google Scholar] [CrossRef]
- Turaka, A.; Buyyounouski, M.K.; Hanlon, A.L.; Horwitz, E.M.; Greenberg, R.E.; Movsas, B. Hypoxic Prostate/Muscle Po2 Ratio Predicts for Outcome in Patients with Localized Prostate Cancer: Long-Term Results. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, e433–e439. [Google Scholar] [CrossRef]
- Hompland, T.; Hole, K.H.; Ragnum, H.B.; Aarnes, E.-K.; Vlatkovic, L.; Lie, A.K.; Patzke, S.; Brennhovd, B.; Seierstad, T.; Lyng, H. Combined MR Imaging of Oxygen Consumption and Supply Reveals Tumor Hypoxia and Aggressiveness in Prostate Cancer Patients. Cancer Res. 2018, 78, 4774–4785. [Google Scholar] [CrossRef]
- Salberg, U.B.; Skingen, V.E.; Fjeldbo, C.S.; Hompland, T.; Ragnum, H.B.; Vlatkovic, L.; Hole, K.H.; Seierstad, T.; Lyng, H. A Prognostic Hypoxia Gene Signature with Low Heterogeneity within the Dominant Tumour Lesion in Prostate Cancer Patients. Br. J. Cancer 2022, 127, 321–328. [Google Scholar] [CrossRef]
- Vuillefroy De Silly, R.; Dietrich, P.-Y.; Walker, P.R. Hypoxia and Antitumor CD8+ T Cells: An Incompatible Alliance? OncoImmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-Inducible Factors Enhance the Effector Responses of CD8+ T Cells to Persistent Antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Gropper, Y.; Feferman, T.; Shalit, T.; Salame, T.-M.; Porat, Z.; Shakhar, G. Culturing CTLs under Hypoxic Conditions Enhances Their Cytolysis and Improves Their Anti-Tumor Function. Cell Rep. 2017, 20, 2547–2555. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e5. [Google Scholar] [CrossRef]
- Perera, M.P.J.; Thomas, P.B.; Risbridger, G.P.; Taylor, R.; Azad, A.; Hofman, M.S.; Williams, E.D.; Vela, I. Chimeric Antigen Receptor T-Cell Therapy in Metastatic Castrate-Resistant Prostate Cancer. Cancers 2022, 14, 503. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRζ/CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Zuccolotto, G.; Fracasso, G.; Merlo, A.; Montagner, I.M.; Rondina, M.; Bobisse, S.; Figini, M.; Cingarlini, S.; Colombatti, M.; Zanovello, P.; et al. PSMA-Specific CAR-Engineered T Cells Eradicate Disseminated Prostate Cancer in Preclinical Models. PLoS ONE 2014, 9, e109427. [Google Scholar] [CrossRef]
- Ma, Q.; Gomes, E.M.; Lo, A.S.; Junghans, R.P. Advanced Generation Anti-prostate Specific Membrane Antigen Designer T Cells for Prostate Cancer Immunotherapy. Prostate 2014, 74, 286–296. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Zhang, Q.; Helfand, B.T.; Carneiro, B.A.; Qin, W.; Yang, X.J.; Lee, C.; Zhang, W.; Giles, F.J.; Cristofanilli, M.; Kuzel, T.M. Efficacy Against Human Prostate Cancer by Prostate-Specific Membrane Antigen-Specific, Transforming Growth Factor-β Insensitive Genetically Targeted CD8+ T-Cells Derived from Patients with Metastatic Castrate-Resistant Disease. Eur. Urol. 2018, 73, 648–652. [Google Scholar] [CrossRef]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.-Q. Adoptive T-Cell Therapy of Prostate Cancer Targeting the Cancer Stem Cell Antigen EpCAM. BMC Immunol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, A.; Cartellieri, M.; Schmitz, M.; Günes, S.; Weigle, B.; Bachmann, M.; Abken, H.; Rieber, E.P.; Temme, A. Targeting of Tumor Cells Expressing the Prostate Stem Cell Antigen (PSCA) Using Genetically Engineered T-cells. Prostate 2007, 67, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T Lymphocytes to PSCA- or PSMA Positive Prostate Cancer Cells Using the Novel Modular Chimeric Antigen Receptor Platform Technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef]
- Priceman, S.J.; Gerdts, E.A.; Tilakawardane, D.; Kennewick, K.T.; Murad, J.P.; Park, A.K.; Jeang, B.; Yamaguchi, Y.; Yang, X.; Urak, R.; et al. Co-Stimulatory Signaling Determines Tumor Antigen Sensitivity and Persistence of CAR T Cells Targeting PSCA+ Metastatic Prostate Cancer. OncoImmunology 2018, 7, e1380764. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lorvik, K.B.; Jin, Y.; Beck, C.; Sike, A.; Persiconi, I.; Kvaløy, E.; Saatcioglu, F.; Dunn, C.; Kyte, J.A. Development of STEAP1 Targeting Chimeric Antigen Receptor for Adoptive Cell Therapy against Cancer. Mol. Ther. Oncolytics 2022, 26, 189–206. [Google Scholar] [CrossRef]
- Bhatia, V.; Kamat, N.V.; Pariva, T.E.; Wu, L.-T.; Tsao, A.; Sasaki, K.; Sun, H.; Javier, G.; Nutt, S.; Coleman, I.; et al. Targeting Advanced Prostate Cancer with STEAP1 Chimeric Antigen Receptor T Cell and Tumor-Localized IL-12 Immunotherapy. Nat. Commun. 2023, 14, 2041. [Google Scholar] [CrossRef]
- Jin, Y.; Dunn, C.; Persiconi, I.; Sike, A.; Skorstad, G.; Beck, C.; Kyte, J.A. Comparative Evaluation of STEAP1 Targeting Chimeric Antigen Receptors with Different Costimulatory Domains and Spacers. Int. J. Mol. Sci. 2024, 25, 586. [Google Scholar] [CrossRef]
- Hassan, R.; Bera, T.; Pastan, I. Mesothelin. Clin. Cancer Res. 2004, 10, 3937–3942. [Google Scholar] [CrossRef]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef]
- Lv, J.; Li, P. Mesothelin as a Biomarker for Targeted Therapy. Biomark. Res. 2019, 7, 18. [Google Scholar] [CrossRef]
- Faust, J.R.; Hamill, D.; Kolb, E.A.; Gopalakrishnapillai, A.; Barwe, S.P. Mesothelin: An Immunotherapeutic Target beyond Solid Tumors. Cancers 2022, 14, 1550. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Mao, L.; Wu, M.; Liu, J.; Yu, S. Challenges of Anti-Mesothelin CAR-T-Cell Therapy. Cancers 2023, 15, 1357. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Wang, Z.; Lasota, J.; Onda, M.; Czapiewski, P.; Langfort, R.; Rys, J.; Szpor, J.; Waloszczyk, P.; Okoń, K.; et al. Comprehensive Immunohistochemical Study of Mesothelin (MSLN) Using Different Monoclonal Antibodies 5B2 and MN-1 in 1562 Tumors with Evaluation of Its Prognostic Value in Malignant Pleural Mesothelioma. Oncotarget 2017, 8, 26744–26754. [Google Scholar] [CrossRef]
- Weidemann, S.; Gagelmann, P.; Gorbokon, N.; Lennartz, M.; Menz, A.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Blessin, N.C.; Fraune, C.; et al. Mesothelin Expression in Human Tumors: A Tissue Microarray Study on 12,679 Tumors. Biomedicines 2021, 9, 397. [Google Scholar] [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple Injections of Electroporated Autologous T Cells Expressing a Chimeric Antigen Receptor Mediate Regression of Human Disseminated Tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef]
- Terry, S.; El-Sayed, I.Y.; Destouches, D.; Maille, P.; Nicolaiew, N.; Ploussard, G.; Semprez, F.; Pimpie, C.; Beltran, H.; Londono-Vallejo, A.; et al. CRIPTO Overexpression Promotes Mesenchymal Differentiation in Prostate Carcinoma Cells through Parallel Regulation of AKT and FGFR Activities. Oncotarget 2015, 6, 11994–12008. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of Microarray Data Using Z Score Transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef]
- Cheadle, C.; Cho-Chung, Y.S.; Becker, K.G.; Vawter, M.P. Application of Z-Score Transformation to Affymetrix Data. Appl. Bioinform. 2003, 2, 209–217. [Google Scholar]
- Thomas, J.G.; Olson, J.M.; Tapscott, S.J.; Zhao, L.P. An Efficient and Robust Statistical Modeling Approach to Discover Differentially Expressed Genes Using Genomic Expression Profiles. Genome Res. 2001, 11, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Lorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisition of Tumor Cell Phenotypic Diversity along the EMT Spectrum under Hypoxic Pressure: Consequences on Susceptibility to Cell-Mediated Cytotoxicity. Oncoimmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.-C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of Non-ETS Gene Fusions in Human Prostate Cancer Using next-Generation RNA Sequencing. Genome Res 2011, 21, 56–67. [Google Scholar] [CrossRef]
- Terry, S.; Maille, P.; Baaddi, H.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Ceraline, J.; Firlej, V.; Beltran, H.; Allory, Y.; et al. Cross Modulation between the Androgen Receptor Axis and Protocadherin-PC in Mediating Neuroendocrine Transdifferentiation and Therapeutic Resistance of Prostate Cancer. Neoplasia 2013, 15, 761–772. [Google Scholar] [CrossRef]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The Multifaceted Role of the Embryonic Gene Cripto-1 in Cancer, Stem Cells and Epithelial-Mesenchymal Transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef]
- Arnouk, H.; Yum, G.; Shah, D. Cripto-1 as a Key Factor in Tumor Progression, Epithelial to Mesenchymal Transition and Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 9280. [Google Scholar] [CrossRef]
- van der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB Costimulation Promotes CAR T Cell Survival through Noncanonical NF-κB Signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef]
- Zheng, Z.; Chinnasamy, N.; Morgan, R.A. Protein L: A Novel Reagent for the Detection of Chimeric Antigen Receptor (CAR) Expression by Flow Cytometry. J. Transl. Med. 2012, 10, 29. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Aggen, D.H.; Schietinger, A.; Schreiber, H.; Kranz, D.M. A Sensitivity Scale for Targeting T Cells with Chimeric Antigen Receptors (CARs) and Bispecific T-Cell Engagers (BiTEs). OncoImmunology 2012, 1, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.J.; Wang, X.; Wang, Z.; Zhao, L.; Li, S.; Zhang, Z.; Wei, X.; Yun, H.; Choi, S.; Liu, Z.; et al. Switchable CAR-T Cells Outperformed Traditional Antibody-Redirected Therapeutics Targeting Breast Cancers. ACS Synth. Biol. 2021, 10, 1176–1183. [Google Scholar] [CrossRef]
- Zoni, E.; Chen, L.; Karkampouna, S.; Granchi, Z.; Verhoef, E.I.; La Manna, F.; Kelber, J.; Pelger, R.C.M.; Henry, M.D.; Snaar-Jagalska, E.; et al. CRIPTO and Its Signaling Partner GRP78 Drive the Metastatic Phenotype in Human Osteotropic Prostate Cancer. Oncogene 2017, 36, 4739–4749. [Google Scholar] [CrossRef]
- Rodrigues Sousa, E.; De Brot, S.; Zoni, E.; Zeinali, S.; Brunello, A.; Scarpa, M.; De Menna, M.; La Manna, F.; Abey Alexander, A.; Klima, I.; et al. CRIPTO’s Multifaceted Role in Driving Aggressive Prostate Cancer Unveiled by in Vivo, Organoid, and Patient Data. Oncogene 2025, 44, 462–475. [Google Scholar] [CrossRef]
- He, X.; Wang, L.; Riedel, H.; Wang, K.; Yang, Y.; Dinu, C.Z.; Rojanasakul, Y. Mesothelin Promotes Epithelial-to-Mesenchymal Transition and Tumorigenicity of Human Lung Cancer and Mesothelioma Cells. Mol. Cancer 2017, 16, 63. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing Anti-Tumour Efficacy with Immunotherapy Combinations. Lancet 2021, 397, 1010–1022. [Google Scholar] [CrossRef]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H.; Rostami, S.; Jalil, A.T.; Al-Gazally, M.E.; Alsaikhan, F.; Rizaev, J.A.; Mohammad, T.A.M.; et al. CAR-T Cell Combination Therapy: The next Revolution in Cancer Treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef]
- Uslu, U.; Castelli, S.; June, C.H. CAR T Cell Combination Therapies to Treat Cancer. Cancer Cell 2024, 42, 1319–1325. [Google Scholar] [CrossRef]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-Targeted CAR-T Cell Therapy for Solid Tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Haas, A.R.; Golden, R.J.; Litzky, L.A.; Engels, B.; Zhao, L.; Xu, F.; Taraszka, J.A.; Ramones, M.; Granda, B.; Chang, W.-J.; et al. Two Cases of Severe Pulmonary Toxicity from Highly Active Mesothelin-Directed CAR T Cells. Mol. Ther. 2023, 31, 2309–2325. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Testas de Folmont, A.; Fauvel, A.; Vacherot, F.; Soyeux, P.; Abdou, A.; Chouaib, S.; Terry, S. Targeting Aggressive Prostate Carcinoma Cells with Mesothelin-CAR-T Cells. Biomedicines 2025, 13, 1215. https://doi.org/10.3390/biomedicines13051215
de Testas de Folmont A, Fauvel A, Vacherot F, Soyeux P, Abdou A, Chouaib S, Terry S. Targeting Aggressive Prostate Carcinoma Cells with Mesothelin-CAR-T Cells. Biomedicines. 2025; 13(5):1215. https://doi.org/10.3390/biomedicines13051215
Chicago/Turabian Stylede Testas de Folmont, Apolline, Angèle Fauvel, Francis Vacherot, Pascale Soyeux, Abdérémane Abdou, Salem Chouaib, and Stéphane Terry. 2025. "Targeting Aggressive Prostate Carcinoma Cells with Mesothelin-CAR-T Cells" Biomedicines 13, no. 5: 1215. https://doi.org/10.3390/biomedicines13051215
APA Stylede Testas de Folmont, A., Fauvel, A., Vacherot, F., Soyeux, P., Abdou, A., Chouaib, S., & Terry, S. (2025). Targeting Aggressive Prostate Carcinoma Cells with Mesothelin-CAR-T Cells. Biomedicines, 13(5), 1215. https://doi.org/10.3390/biomedicines13051215