
Emerging Epigenetic Therapies for the Treatment of Cardiac Fibrosis


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Classification and Progression of Cardiac Fibrosis
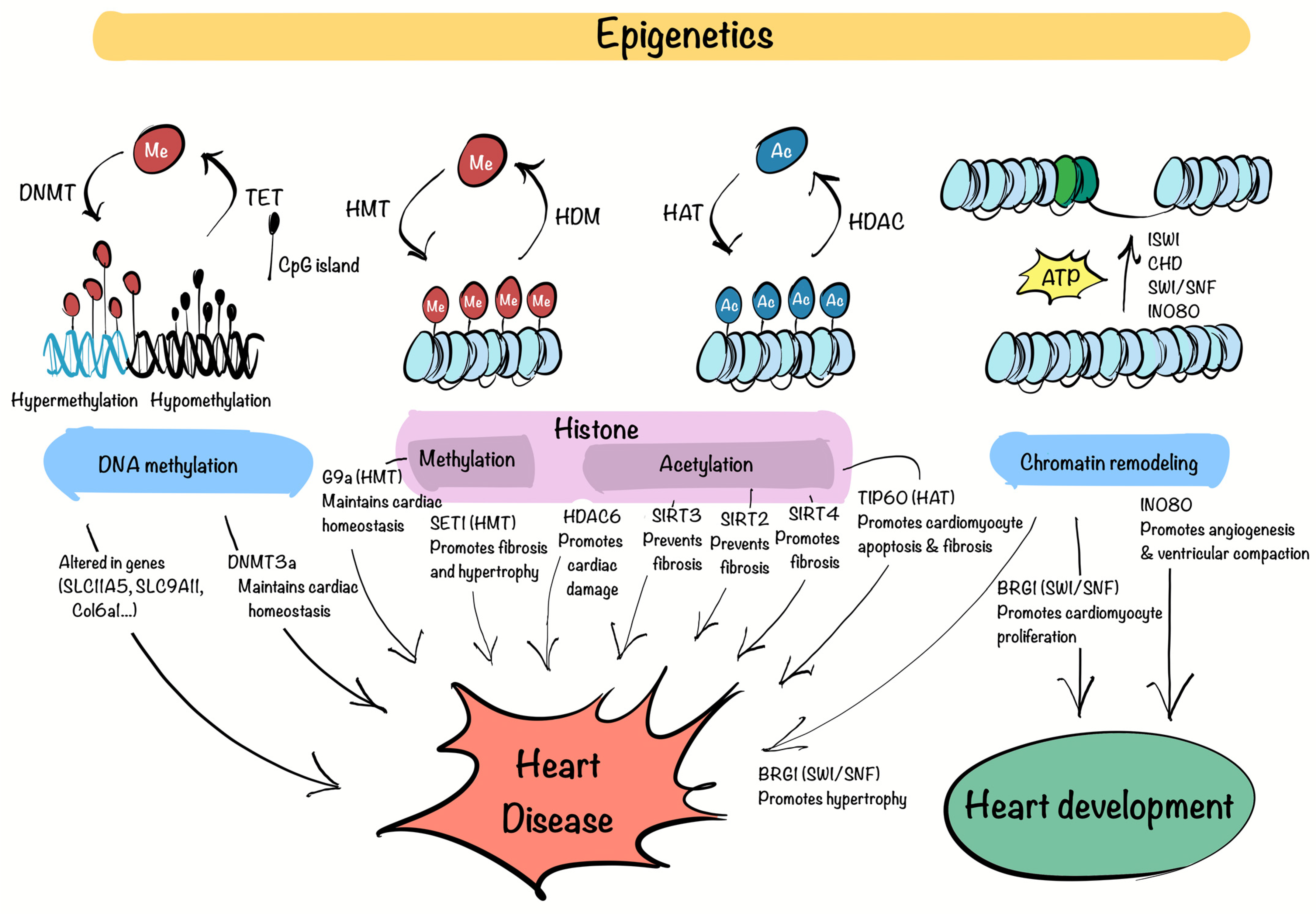
3. Chromatin Factors and Epigenetic Regulators in Cardiovascular Diseases
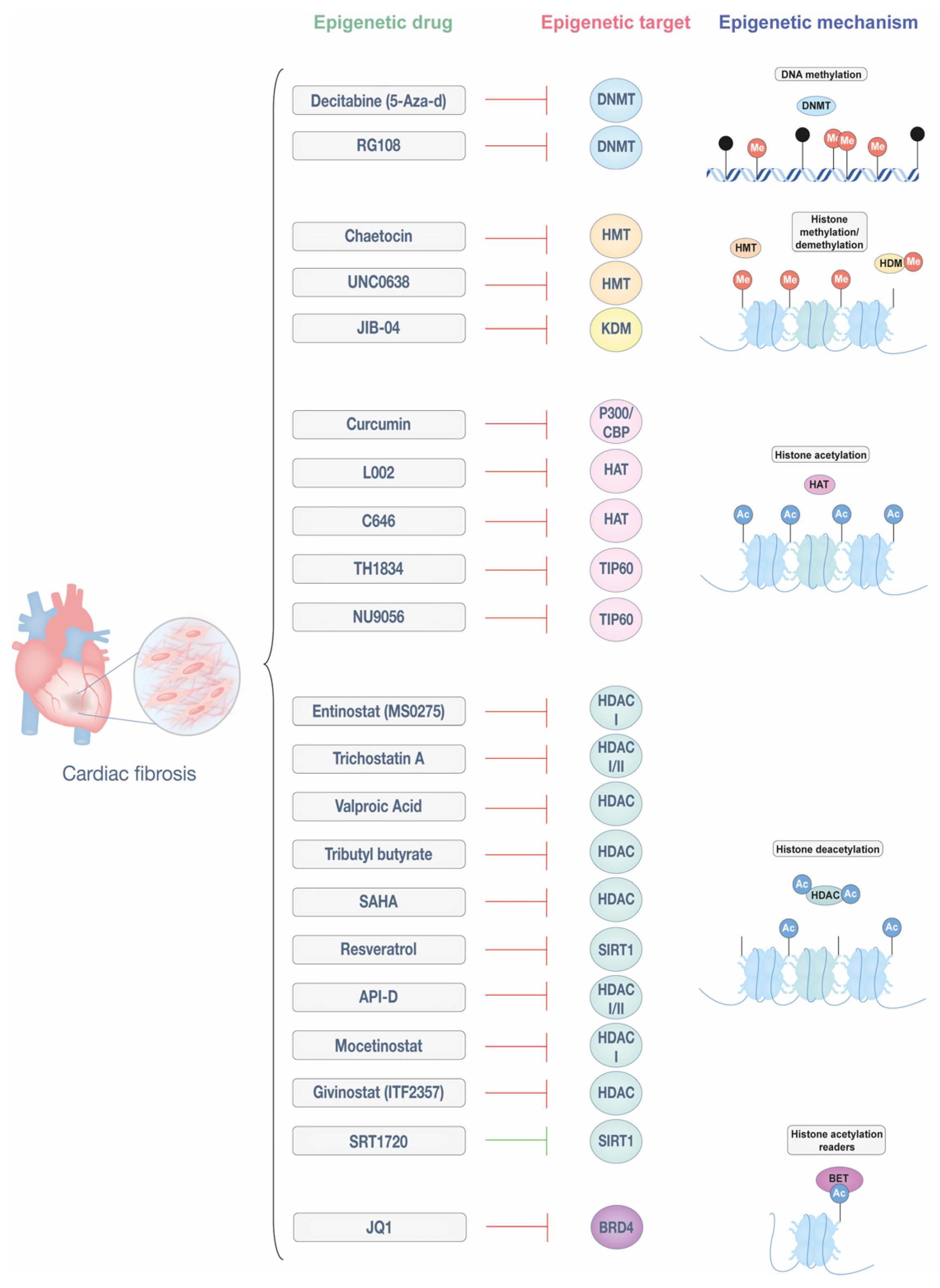
4. Epigenetic Therapies for Cardiac Fibrosis
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AF | Atrial fibrillation |
| Ang II | Angiotensin II |
| ARB | Angiotensin receptor blocker |
| ARVC | Arrhythmogenic right ventricular cardiomyopathy |
| ATAC-seq | Assay for Transposase-Accessible Chromatin-sequencing |
| ATM | Ataxia telangiectasia mutated |
| BET | Bromodomain and extra-terminal motif |
| BRD | Bromodomain |
| CF | Cardiac fibroblast |
| CpG | Cytosine-phosphodiester bond-guanine |
| CVD | Cardiovascular disease |
| DAMPS | Damage-associated molecular patterns |
| DCM | Dilated cardiomyopathy |
| DDR | DNA damage response |
| DNMT | DNA methyltransferase |
| ECM | Extracellular matrix |
| FDA | Food and Drug Administration |
| HAT | Histone acetyltransferase |
| HCM | Hypertrophic cardiomyopathy |
| HDAC | Histone deacetylase |
| HDM | Histone demethylase |
| HF | Heart failure |
| HMT | Histone methyltransferase |
| KDM | Lysine demethylase |
| LOX | Lysyl oxidase |
| MI | Myocardial infarction |
| MMP | Matrix metalloproteinase |
| MRA | Mineralocorticoid receptor antagonist |
| PDGF | Platelet-derived growth factor |
| RAAS | Renin-angiotensin-aldosterone system |
| ROS | Reactive oxygen species |
| SAM | S-adenosylmethionine |
| scRNA-seq | Single-cell RNA sequencing |
| TF | Transcription factor |
| TGF-β | Transforming growth factor beta |
References
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From Mechanisms to Medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Mensah, G.A.; Fuster, V.; Murray, C.J.L.; Roth, G.A.; Mensah, G.A.; Abate, Y.H.; Abbasian, M.; Abd-Allah, F.; Abdollahi, A.; Abdollahi, M.; et al. Global Burden of Cardiovascular Diseases and Risks, 1990–2022. J. Am. Coll. Cardiol. 2023, 82, 2350–2473. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting Metabolic Dysregulation for Fibrosis Therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, Definitions, and Functions in Health and Disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Lurje, I.; Gaisa, N.T.; Weiskirchen, R.; Tacke, F. Mechanisms of Organ Fibrosis: Emerging Concepts and Implications for Novel Treatment Strategies. Mol. Asp. Med. 2023, 92, 101191. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Isoyama, M.S.; Nitta-Komatsubara, M.Y. Acute and Chronic Adaptation to Hemodynamic Overload and Ischemia in the Aged Heart. Heart Fail. Rev. 2002, 7, 63–69. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac Fibrosis—A Short Review of Causes and Therapeutic Strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, X.; Huang, G.N. Heart Regeneration from the Whole-Organism Perspective to Single-Cell Resolution. NPJ Regen. Med. 2024, 9, 34. [Google Scholar] [CrossRef]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Purohit, S.; Tian, R. Cardiac Metabolism and Its Interactions with Contraction, Growth, and Survival of Cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil Roles in Left Ventricular Remodeling Following Myocardial Infarction. Fibrogenesis Tissue Repair 2013, 6, 11. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The Healing Myocardium Sequentially Mobilizes Two Monocyte Subsets with Divergent and Complementary Functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- González-Santamaría, J.; Villalba, M.; Busnadiego, O.; López-Olañeta, M.M.; Sandoval, P.; Snabel, J.; López-Cabrera, M.; Erler, J.T.; Hanemaaijer, R.; Lara-Pezzi, E.; et al. Matrix Cross-Linking Lysyl Oxidases Are Induced in Response to Myocardial Infarction and Promote Cardiac Dysfunction. Cardiovasc. Res. 2016, 109, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Martínez-González, J. The Role of Lysyl Oxidase Enzymes in Cardiac Function and Remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized Fibroblast Differentiated States Underlie Scar Formation in the Infarcted Mouse Heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Korantzopoulos, P.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial Fibrosis in Atrial Fibrillation: Mechanistic Insights, Diagnostic Challenges, and Emerging Therapeutic Targets. Int. J. Mol. Sci. 2024, 26, 209. [Google Scholar] [CrossRef]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef]
- Gladka, M.M.; Molenaar, B.; De Ruiter, H.; Van Der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; Van Oudenaarden, A.; Van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Romero, J.P.; Hernández, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; García-Olloqui, P.; Palacio, M.; et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2020, 30, 3149–3163.e6. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-Tissue Organization of the Fibroblast Lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Koenig, A.L.; Shchukina, I.; Amrute, J.; Andhey, P.S.; Zaitsev, K.; Lai, L.; Bajpai, G.; Bredemeyer, A.; Smith, G.; Jones, C.; et al. Single-Cell Transcriptomics Reveals Cell-Type-Specific Diversification in Human Heart Failure. Nat. Cardiovasc. Res. 2022, 1, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Chaffin, M.; Papangeli, I.; Simonson, B.; Akkad, A.D.; Hill, M.C.; Arduini, A.; Fleming, S.J.; Melanson, M.; Hayat, S.; Kost-Alimova, M.; et al. Single-Nucleus Profiling of Human Dilated and Hypertrophic Cardiomyopathy. Nature 2022, 608, 174–180. [Google Scholar] [CrossRef]
- Kuppe, C.; Ramirez Flores, R.O.; Li, Z.; Hayat, S.; Levinson, R.T.; Liao, X.; Hannani, M.T.; Tanevski, J.; Wünnemann, F.; Nagai, J.S.; et al. Spatial Multi-Omic Map of Human Myocardial Infarction. Nature 2022, 608, 766–777. [Google Scholar] [CrossRef]
- Amrute, J.M.; Lai, L.; Ma, P.; Koenig, A.L.; Kamimoto, K.; Bredemeyer, A.; Shankar, T.S.; Kuppe, C.; Kadyrov, F.F.; Schulte, L.J.; et al. Defining Cardiac Functional Recovery in End-Stage Heart Failure at Single-Cell Resolution. Nat. Cardiovasc. Res. 2023, 2, 399–416. [Google Scholar] [CrossRef]
- Fu, M.; Hua, X.; Shu, S.; Xu, X.; Zhang, H.; Peng, Z.; Mo, H.; Liu, Y.; Chen, X.; Yang, Y.; et al. Single-Cell RNA Sequencing in Donor and End-Stage Heart Failure Patients Identifies NLRP3 as a Therapeutic Target for Arrhythmogenic Right Ventricular Cardiomyopathy. BMC Med. 2024, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Aguado-Alvaro, L.P.; Garitano, N.; Esser-Skala, W.; Sayers, J.; del Valle, C.; Alameda-Serrano, D.; Mendieta-Esteban, J.; Calleja-Cervantes, M.E.; Goñi-Salaverri, A.; Zazpe, J.; et al. Identification of Epigenetic Regulators of Fibrotic Transformation in Cardiac Fibroblasts through Bulk and Single-Cell CRISPR Screens. bioRxiv 2025. bioRxiv:645873. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Kaluscha, S.; Domcke, S.; Wirbelauer, C.; Stadler, M.B.; Durdu, S.; Burger, L.; Schübeler, D. Evidence That Direct Inhibition of Transcription Factor Binding Is the Prevailing Mode of Gene and Repeat Repression by DNA Methylation. Nat. Genet. 2022, 54, 1895–1906. [Google Scholar] [CrossRef]
- Liu, R.; Wu, J.; Guo, H.; Yao, W.; Li, S.; Lu, Y.; Jia, Y.; Liang, X.; Tang, J.; Zhang, H. Post-translational Modifications of Histones: Mechanisms, Biological Functions, and Therapeutic Targets. MedComm 2023, 4, e292. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Patel, A.B.; Hopfner, K.-P.; He, Y.; Korber, P. Energy-Driven Genome Regulation by ATP-Dependent Chromatin Remodellers. Nat. Rev. Mol. Cell Biol. 2024, 25, 309–332. [Google Scholar] [CrossRef] [PubMed]
- Westerman, K.; Sebastiani, P.; Jacques, P.; Liu, S.; DeMeo, D.; Ordovás, J.M. DNA Methylation Modules Associate with Incident Cardiovascular Disease and Cumulative Risk Factor Exposure. Clin. Epigenetics 2019, 11, 142. [Google Scholar] [CrossRef]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.S.; Parsana, P.; Segrè, A.V.; et al. Genetic Effects on Gene Expression across Human Tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Luo, X.; Hu, Y.; Shen, J.; Liu, X.; Wang, T.; Li, L.; Li, J. Integrative Analysis of DNA Methylation and Gene Expression Reveals Key Molecular Signatures in Acute Myocardial Infarction. Clin. Epigenetics 2022, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Moran, B.; Collier, P.; Moravec, C.S.; Phelan, D.; Donnellan, E.; Russell-Hallinan, A.; O’Connor, D.P.; Gallagher, W.M.; Gallagher, J.; et al. Targeted DNA Methylation Profiling of Human Cardiac Tissue Reveals Novel Epigenetic Traits and Gene Deregulation Across Different Heart Failure Patient Subtypes. Circ. Heart Fail. 2019, 12, e005765. [Google Scholar] [CrossRef]
- Madsen, A.; Höppner, G.; Krause, J.; Hirt, M.N.; Laufer, S.D.; Schweizer, M.; Tan, W.L.W.; Mosqueira, D.; Anene-Nzelu, C.G.; Lim, I.; et al. An Important Role for DNMT3A-Mediated DNA Methylation in Cardiomyocyte Metabolism and Contractility. Circulation 2020, 142, 1562–1578. [Google Scholar] [CrossRef]
- Papait, R.; Serio, S.; Pagiatakis, C.; Rusconi, F.; Carullo, P.; Mazzola, M.; Salvarani, N.; Miragoli, M.; Condorelli, G. Histone Methyltransferase G9a Is Required for Cardiomyocyte Homeostasis and Hypertrophy. Circulation 2017, 136, 1233–1246. [Google Scholar] [CrossRef]
- Yu, L.; Yang, G.; Weng, X.; Liang, P.; Li, L.; Li, J.; Fan, Z.; Tian, W.; Wu, X.; Xu, H.; et al. Histone Methyltransferase SET1 Mediates Angiotensin II–Induced Endothelin-1 Transcription and Cardiac Hypertrophy in Mice. Arter. Thromb. Vasc. Biol. 2015, 35, 1207–1217. [Google Scholar] [CrossRef]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 Activity Alleviates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats: Potential Role of Peroxiredoxin 1 Acetylation and Redox Regulation. Oxid. Med. Cell Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef] [PubMed]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of Myocardial Reperfusion Injury by Ischemic Postconditioning Requires Sirtuin 3-Mediated Deacetylation of Cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.-F.; Wang, N.-Y.; Wang, X.-M.; Liang, S.-T.; Zheng, W.; Lu, Y.-B.; Zhao, X.; Hao, D.-L.; Zhang, Z.-Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Bindu, S.; Pillai, V.B.; Samant, S.; Pan, Y.; Huang, J.-Y.; Gupta, M.; Nagalingam, R.S.; Wolfgeher, D.; Verdin, E.; et al. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β. Mol. Cell. Biol. 2016, 36, 678–692. [Google Scholar] [CrossRef]
- Luo, Y.-X.; Tang, X.; An, X.-Z.; Xie, X.-M.; Chen, X.-F.; Zhao, X.; Hao, D.-L.; Chen, H.-Z.; Liu, D.-P. Sirt4 Accelerates Ang II-Induced Pathological Cardiac Hypertrophy by Inhibiting Manganese Superoxide Dismutase Activity. Eur. Heart J. 2016, 38, 1389–1398. [Google Scholar] [CrossRef]
- Sapountzi, V.; Logan, I.R.; Robson, C.N. Cellular Functions of TIP60. Int. J. Biochem. Cell Biol. 2006, 38, 1496–1509. [Google Scholar] [CrossRef] [PubMed]
- McAllister, D.; Merlo, X.; Lough, J. Characterization and Expression of the Mouse Tat Interactive Protein 60 KD (TIP60) Gene. Gene 2002, 289, 169–176. [Google Scholar] [CrossRef]
- Wang, X.; Wan, T.C.; Lauth, A.; Purdy, A.L.; Kulik, K.R.; Patterson, M.; Lough, J.W.; Auchampach, J.A. Conditional Depletion of the Acetyltransferase Tip60 Protects against the Damaging Effects of Myocardial Infarction. J. Mol. Cell. Cardiol. 2022, 163, 9–19. [Google Scholar] [CrossRef]
- Wang, X.; Lupton, C.; Lauth, A.; Wan, T.C.; Foster, P.; Patterson, M.; Auchampach, J.A.; Lough, J.W. Evidence That the Acetyltransferase Tip60 Induces the DNA Damage Response and Cell-Cycle Arrest in Neonatal Cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 155, 88–98. [Google Scholar] [CrossRef]
- Fisher, J.B.; Kim, M.-S.; Blinka, S.; Ge, Z.-D.; Wan, T.; Duris, C.; Christian, D.; Twaroski, K.; North, P.; Auchampach, J.; et al. Stress-Induced Cell-Cycle Activation in Tip60 Haploinsufficient Adult Cardiomyocytes. PLoS ONE 2012, 7, e31569. [Google Scholar] [CrossRef]
- Fisher, J.B.; Horst, A.; Wan, T.; Kim, M.-S.; Auchampach, J.; Lough, J. Depletion of Tip60 from In Vivo Cardiomyocytes Increases Myocyte Density, Followed by Cardiac Dysfunction, Myocyte Fallout and Lethality. PLoS ONE 2016, 11, e0164855. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.M.; Howard, S.; Villa del Campo, C.; Bollini, S.; Dubé, K.N.; Masters, M.; Barnette, D.N.; Rohling, M.; Sun, X.; Hankins, L.E.; et al. BRG1-SWI/SNF-Dependent Regulation of the Wt1 Transcriptional Landscape Mediates Epicardial Activity during Heart Development and Disease. Nat. Commun. 2017, 8, 16034. [Google Scholar] [CrossRef] [PubMed]
- Hota, S.K.; Bruneau, B.G. ATP-Dependent Chromatin Remodeling during Mammalian Development. Development 2016, 143, 2882–2897. [Google Scholar] [CrossRef]
- Han, P.; Hang, C.T.; Yang, J.; Chang, C.-P. Chromatin Remodeling in Cardiovascular Development and Physiology. Circ. Res. 2011, 108, 378–396. [Google Scholar] [CrossRef]
- Rhee, S.; Chung, J.I.; King, D.A.; D’amato, G.; Paik, D.T.; Duan, A.; Chang, A.; Nagelberg, D.; Sharma, B.; Jeong, Y.; et al. Endothelial Deletion of Ino80 Disrupts Coronary Angiogenesis and Causes Congenital Heart Disease. Nat. Commun. 2018, 9, 368. [Google Scholar] [CrossRef]
- Webber, M.; Jackson, S.P.; Moon, J.C.; Captur, G. Myocardial Fibrosis in Heart Failure: Anti-Fibrotic Therapies and the Role of Cardiovascular Magnetic Resonance in Drug Trials. Cardiol. Ther. 2020, 9, 363–376. [Google Scholar] [CrossRef]
- Zhi, H.; Luptak, I.; Alreja, G.; Shi, J.; Guan, J.; Metes-Kosik, N.; Joseph, J. Effects of Direct Renin Inhibition on Myocardial Fibrosis and Cardiac Fibroblast Function. PLoS ONE 2013, 8, e81612. [Google Scholar] [CrossRef] [PubMed]
- Abareshi, A.; Norouzi, F.; Asgharzadeh, F.; Beheshti, F.; Hosseini, M.; Farzadnia, M.; Khazaei, M. Effect of Angiotensin-Converting Enzyme Inhibitor on Cardiac Fibrosis and Oxidative Stress Status in Lipopolysaccharide-Induced Inflammation Model in Rats. Int. J. Prev. Med. 2017, 8, 69. [Google Scholar] [CrossRef]
- Wu, L.; Iwai, M.; Nakagami, H.; Chen, R.; Suzuki, J.; Akishita, M.; de Gasparo, M.; Horiuchi, M. Effect of Angiotensin II Type 1 Receptor Blockade on Cardiac Remodeling in Angiotensin II Type 2 Receptor Null Mice. Arter. Thromb. Vasc. Biol. 2002, 22, 49–54. [Google Scholar] [CrossRef]
- Brilla, C.G.; Matsubara, L.S.; Weber, K.T. Antifibrotic Effects of Spironolactone in Preventing Myocardial Fibrosis in Systemic Arterial Hypertension. Am. J. Cardiol. 1993, 71, A12–A16. [Google Scholar] [CrossRef]
- Chimenti, I.; Pagano, F.; Cavarretta, E.; Angelini, F.; Peruzzi, M.; Barretta, A.; Greco, E.; De Falco, E.; Marullo, A.G.M.; Sciarretta, S.; et al. Β-Blockers Treatment of Cardiac Surgery Patients Enhances Isolation and Improves Phenotype of Cardiosphere-Derived Cells. Sci. Rep. 2016, 6, 36774. [Google Scholar] [CrossRef] [PubMed]
- Dolivo, D.M.; Reed, C.R.; Gargiulo, K.A.; Rodrigues, A.E.; Galiano, R.D.; Mustoe, T.A.; Hong, S.J. Anti-Fibrotic Effects of Statin Drugs: A Review of Evidence and Mechanisms. Biochem. Pharmacol. 2023, 214, 115644. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; KG Maier, S.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming Growth Factor Beta Inhibition Increases Mortality and Left Ventricular Dilatation after Myocardial Infarction. Basic Res. Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Ding, C.; Wilson, E.; Marcus, G.M.; Olgin, J.E. Pirfenidone Mitigates Left Ventricular Fibrosis and Dysfunction after Myocardial Infarction and Reduces Arrhythmias. Heart Rhythm. 2010, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; De Raaf, M.A.; Sun, X.Q.; Kuiper, V.P.; Da Silva Gonçalves Bos, D.; Happé, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib Improves Cardiac Fibrosis but Leaves Pulmonary Vascular Remodelling Unaltered in Experimental Pulmonary Hypertension. Cardiovasc. Res. 2019, 115, 432–439. [Google Scholar] [CrossRef]
- Roger, V.L. Epidemiology of Heart Failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Dai, W.; Qiao, X.; Fang, Y.; Guo, R.; Bai, P.; Liu, S.; Li, T.; Jiang, Y.; Wei, S.; Na, Z.; et al. Epigenetics-Targeted Drugs: Current Paradigms and Future Challenges. Signal Transduct. Target. Ther. 2024, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Dasgupta, C.; Chen, M.; Zhang, K.; Buchholz, J.; Xu, Z.; Zhang, L. Inhibition of DNA Methylation Reverses Norepinephrine-Induced Cardiac Hypertrophy in Rats. Cardiovasc. Res. 2014, 101, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Horgan, S.; Neary, R.; Glezeva, N.; Tea, I.; Corrigan, N.; McDonald, K.; Ledwidge, M.; Baugh, J. Epigenetic Therapy for the Treatment of Hypertension-Induced Cardiac Hypertrophy and Fibrosis. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 127–137. [Google Scholar] [CrossRef]
- Nührenberg, T.G.; Hammann, N.; Schnick, T.; Preißl, S.; Witten, A.; Stoll, M.; Gilsbach, R.; Neumann, F.-J.; Hein, L. Cardiac Myocyte De Novo DNA Methyltransferases 3a/3b Are Dispensable for Cardiac Function and Remodeling after Chronic Pressure Overload in Mice. PLoS ONE 2015, 10, e0131019. [Google Scholar] [CrossRef]
- Stenzig, J.; Schneeberger, Y.; Löser, A.; Peters, B.S.; Schaefer, A.; Zhao, R.-R.; Ng, S.L.; Höppner, G.; Geertz, B.; Hirt, M.N.; et al. Pharmacological Inhibition of DNA Methylation Attenuates Pressure Overload-Induced Cardiac Hypertrophy in Rats. J. Mol. Cell. Cardiol. 2018, 120, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Kamimura, N.; Matsuhashi, T.; Nagai, T.; Nishiyama, T.; Endo, J.; Hishiki, T.; Nakanishi, T.; Shimizu, N.; Tanaka, H.; et al. The Histone 3 Lysine 9 Methyltransferase Inhibitor Chaetocin Improves Prognosis in a Rat Model of High Salt Diet-Induced Heart Failure. Sci. Rep. 2017, 7, 39752. [Google Scholar] [CrossRef]
- Sung, P.-H.; Luo, C.-W.; Chiang, J.Y.; Yip, H.-K. The Combination of G9a Histone Methyltransferase Inhibitors with Erythropoietin Protects Heart against Damage from Acute Myocardial Infarction. Am. J. Transl. Res. 2020, 12, 3255–3271. [Google Scholar] [PubMed]
- Zhang, Q.-J.; Tran, T.A.T.; Wang, M.; Ranek, M.J.; Kokkonen-Simon, K.M.; Gao, J.; Luo, X.; Tan, W.; Kyrychenko, V.; Liao, L.; et al. Histone Lysine Dimethyl-Demethylase KDM3A Controls Pathological Cardiac Hypertrophy and Fibrosis. Nat. Commun. 2018, 9, 5230. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Meng, X.; Yang, X.; Liu, X.; Ou-Yang, C.; Liu, C. Curcumin Administration Suppresses Collagen Synthesis in the Hearts of Rats with Experimental Diabetes. Acta Pharmacol. Sin. 2018, 39, 195–204. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Y.; Chen, Q.; Hong, T.; Zhong, Z.; He, J.; Ni, C. Curcumin Ameliorates Cardiac Fibrosis by Regulating Macrophage-Fibroblast Crosstalk via IL18-P-SMAD2/3 Signaling Pathway Inhibition. Front. Pharmacol. 2022, 12, 784041. [Google Scholar] [CrossRef]
- Liu, H.; Wang, C.; Qiao, Z.; Xu, Y. Protective Effect of Curcumin against Myocardium Injury in Ischemia Reperfusion Rats. Pharm. Biol. 2017, 55, 1144–1148. [Google Scholar] [CrossRef]
- Rai, R.; Verma, S.K.; Kim, D.; Ramirez, V.; Lux, E.; Li, C.; Sahoo, S.; Wilsbacher, L.D.; Vaughan, D.E.; Quaggin, S.E.; et al. A Novel Acetyltransferase P300 Inhibitor Ameliorates Hypertension-Associated Cardio-Renal Fibrosis. Epigenetics 2017, 12, 1004–1013. [Google Scholar] [CrossRef]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase P300 Inhibitor Reverses Hypertension-induced Cardiac Fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef]
- Wang, X.; Wan, T.C.; Kulik, K.R.; Lauth, A.; Smith, B.C.; Lough, J.W.; Auchampach, J.A. Pharmacological Inhibition of the Acetyltransferase Tip60 Mitigates Myocardial Infarction Injury. Dis. Model. Mech. 2023, 16, dmm049786. [Google Scholar] [CrossRef]
- Yakubu, J.; Pandey, A.V. Innovative Delivery Systems for Curcumin: Exploring Nanosized and Conventional Formulations. Pharmaceutics 2024, 16, 637. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Lv, M.; Li, Y. Nanotechnology-Based Drug Delivery Systems for Curcumin and Its Derivatives in the Treatment of Cardiovascular Diseases. J. Funct. Foods 2024, 122, 106476. [Google Scholar] [CrossRef]
- Freundt, J.K.; Frommeyer, G.; Spieker, T.; Wötzel, F.; Grotthoff, J.S.; Stypmann, J.; Hempel, G.; Schäfers, M.; Jacobs, A.H.; Eckardt, L.; et al. Histone Deacetylase Inhibition by Entinostat for the Prevention of Electrical and Structural Remodeling in Heart Failure. BMC Pharmacol. Toxicol. 2019, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.J.; Baarine, M.; Aune, S.E.; Li, X.; Ball, L.E.; Lemasters, J.J.; Beeson, C.C.; Chou, J.C.; Menick, D.R. HDAC1 Localizes to the Mitochondria of Cardiac Myocytes and Contributes to Early Cardiac Reperfusion Injury. J. Mol. Cell. Cardiol. 2018, 114, 309–319. [Google Scholar] [CrossRef]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; McDonald, K.S.; Higashi, Y.; Noda, M.; Chandrasekar, B. Histone Deacetyltransferase Inhibitors Trichostatin A and Mocetinostat Differentially Regulate MMP9, IL-18 and RECK Expression, and Attenuate Angiotensin II-Induced Cardiac Fibroblast Migration and Proliferation. Hypertens. Res. 2016, 39, 709–716. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Wang, L.; Zhao, J.; Zhong, Z.; Wang, Y.; Xu, J. Inhibition of Histone Deacetylases Prevents Cardiac Remodeling After Myocardial Infarction by Restoring Autophagosome Processing in Cardiac Fibroblasts. Cell. Physiol. Biochem. 2018, 49, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, X.; Zhao, Y.; Fast, L.; Zhuang, S.; Liu, P.; Cheng, G.; Zhao, T.C. Inhibition of Histone Deacetylases Preserves Myocardial Performance and Prevents Cardiac Remodeling through Stimulation of Endogenous Angiomyogenesis. J. Pharmacol. Exp. Ther. 2012, 341, 285–293. [Google Scholar] [CrossRef]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone Deacetylase Inhibition Blunts Ischemia/Reperfusion Injury by Inducing Cardiomyocyte Autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef]
- Nagata, S.; Marunouchi, T.; Tanonaka, K. Histone Deacetylase Inhibitor SAHA Treatment Prevents the Development of Heart Failure after Myocardial Infarction via an Induction of Heat-Shock Proteins in Rats. Biol. Pharm. Bull. 2019, 42, 453–461. [Google Scholar] [CrossRef]
- Lee, T.-M.; Lin, M.-S.; Chang, N.-C. Inhibition of Histone Deacetylase on Ventricular Remodeling in Infarcted Rats. Am. J. Physiol. -Heart Circ. Physiol. 2007, 293, H968–H977. [Google Scholar] [CrossRef]
- Wang, K.; Tang, R.; Wang, S.; Wang, W.; Zhang, K.; Li, J.; Li, P.; Tang, Y.-D. SAHA Could Inhibit TGF-Β1/P38 Pathway in MI-Induced Cardiac Fibrosis through DUSP4 Overexpression. Heart Vessel. 2022, 37, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.T.; Orlandin, C.B.; Bonácio, G.F.; Sanchez, E.R.; Durão, M.P.; Lataro, R.M.; Ramalho, L.N.; Silva, C.A.; Amaral, J.H.; Restini, C.B.; et al. Antioxidant Resveratrol Attenuates Cardiac Fibrosis and Reduces Matrix Metalloproteinase-2 Activity in Heart Failure Rats. Hypertension 2019, 74, Abstract P2005. [Google Scholar] [CrossRef]
- Chen, Q.; Zeng, Y.; Yang, X.; Wu, Y.; Zhang, S.; Huang, S.; Zhong, Y.; Chen, M. Resveratrol Ameliorates Myocardial Fibrosis by Regulating Sirt1/Smad3 Deacetylation Pathway in Rat Model with Dilated Cardiomyopathy. BMC Cardiovasc. Disord. 2022, 22, 17. [Google Scholar] [CrossRef]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC Class I Inhibitor, Mocetinostat, Reverses Cardiac Fibrosis in Heart Failure and Diminishes CD90+ Cardiac Myofibroblast Activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef]
- Gallo, P.; Latronico, M.V.G.; Gallo, P.; Grimaldi, S.; Borgia, F.; Todaro, M.; Jones, P.; Gallinari, P.; De Francesco, R.; Ciliberto, G.; et al. Inhibition of Class I Histone Deacetylase with an Apicidin Derivative Prevents Cardiac Hypertrophy and Failure. Cardiovasc. Res. 2008, 80, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.; Zakharova, L.; Feehery, L.; Sljukic, S.; Gaballa, M. Anti-Fibrotic Effects of Class I HDAC Inhibitor, Mocetinostat Is Associated with IL-6/Stat3 Signaling in Ischemic Heart Failure. Int. J. Mol. Sci. 2015, 16, 11482–11499. [Google Scholar] [CrossRef]
- Milan, M.; Pace, V.; Maiullari, F.; Chirivì, M.; Baci, D.; Maiullari, S.; Madaro, L.; Maccari, S.; Stati, T.; Marano, G.; et al. Givinostat Reduces Adverse Cardiac Remodeling through Regulating Fibroblasts Activation. Cell. Death Dis. 2018, 9, 108. [Google Scholar] [CrossRef]
- Ford, C.M.; Civitarese, R.A.; Bugyei-Twum, A.; Mitchell, M.; Desjardins, J.-F.; Thai, K.; Abadeh, A.; Zhang, Y.; Switzer, J.; Advani, A.; et al. The Sirt1 Activator, SRT1720, Attenuates Cardiac Hypertrophy and Fibrosis in a Rodent Pressure Overload Model. Circulation 2014, 130, A19029. [Google Scholar]
- Bugyei-Twum, A.; Ford, C.; Civitarese, R.; Seegobin, J.; Advani, S.L.; Desjardins, J.-F.; Kabir, G.; Zhang, Y.; Mitchell, M.; Switzer, J.; et al. Sirtuin 1 Activation Attenuates Cardiac Fibrosis in a Rodent Pressure Overload Model by Modifying Smad2/3 Transactivation. Cardiovasc. Res. 2018, 114, 1629–1641. [Google Scholar] [CrossRef]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic Regulation in Cardiovascular Disease: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2022, 7, 200. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, X.; Qi, Z.; Fang, Z.; Jiang, Y.; Huang, Y.; Wang, Y.; Wu, L.; Yang, Y. An Antioxidant Nanozyme for Targeted Cardiac Fibrosis Therapy Post Myocardial Infarction. J. Nanobiotechnol. 2024, 22, 760. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Zou, W.; Tian, F.; Xie, H.; Liu, A.; Liu, W.; Liu, Y.; Zhou, N.; Cai, X.; Wu, J.; et al. Inhalable Cardiac Targeting Peptide Modified Nanomedicine Prevents Pressure Overload Heart Failure in Male Mice. Nat. Commun. 2024, 15, 6058. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Y.; Li, S. The Advances in the Development of Epigenetic Modifications Therapeutic Drugs Delivery Systems. Int. J. Nanomed. 2024, 19, 10623–10637. [Google Scholar] [CrossRef]
- Papazoglou, P.; Peng, L.; Sachinidis, A. Epigenetic Mechanisms Involved in the Cardiovascular Toxicity of Anticancer Drugs. Front. Cardiovasc. Med. 2021, 8, 658900. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garitano, N.; Aguado-Alvaro, L.P.; Pelacho, B. Emerging Epigenetic Therapies for the Treatment of Cardiac Fibrosis. Biomedicines 2025, 13, 1170. https://doi.org/10.3390/biomedicines13051170
Garitano N, Aguado-Alvaro LP, Pelacho B. Emerging Epigenetic Therapies for the Treatment of Cardiac Fibrosis. Biomedicines. 2025; 13(5):1170. https://doi.org/10.3390/biomedicines13051170
Chicago/Turabian StyleGaritano, Nerea, Laura Pilar Aguado-Alvaro, and Beatriz Pelacho. 2025. "Emerging Epigenetic Therapies for the Treatment of Cardiac Fibrosis" Biomedicines 13, no. 5: 1170. https://doi.org/10.3390/biomedicines13051170
APA StyleGaritano, N., Aguado-Alvaro, L. P., & Pelacho, B. (2025). Emerging Epigenetic Therapies for the Treatment of Cardiac Fibrosis. Biomedicines, 13(5), 1170. https://doi.org/10.3390/biomedicines13051170